

Abstract
The epidermal growth factor receptor (EGFR) frequently associates with cancer and already serves as a target for therapy. We report that inflammatory cytokines and ultraviolet (UV) irradiation respectively induce transient or sustained phosphorylation of EGFR. Subsequently, EGFR internalizes via a Clathrin-mediated process. In cytokine-stimulated cells, EGFR recycles back to the cell surface, whereas in irradiated cells it arrests in Rab5-containing endosomes. Under both conditions, receptor internalization is instigated by the p38 stress-induced kinase. The underlying mechanism entails phosphorylation of EGFR at a short segment (amino acids 1002–1022) containing multiple serines and threonines, as well as phosphorylation of two Rab5 effectors, EEA1 and GDI. Like UV irradiation, a chemotherapeutic agent activates p38 and accelerates receptor internalization. We demonstrate that abrogating EGFR internalization reduces the efficacy of chemotherapy-induced cell death. Hence, by preventing EGFR-mediated survival signaling, the internalization route we uncovered enhances the cytotoxic effect of drugs like cis-platinum, which may underlie interactions between chemotherapy and EGFR-targeting drugs.
Keywords: chemotherapy, EGFR, endocytosis, UV, TNF
Introduction
Epidermal growth factor receptor (EGFR) (also called ErbB-1) is a member of the ErbB family of receptor tyrosine kinases (RTKs). Overexpression, gene amplification and mutations of EGFR have been identified in multiple types of human tumors, including cancers of the breast, head and neck, lung and ovary (Yarden and Sliwkowski, 2001). In addition, autocrine loops involving transforming growth factor alpha (TGF-α), as well as other ErbB ligands, frequently evolve in human carcinomas. For these reasons, several classes of anti-EGFR therapies have been developed, including monoclonal antireceptor antibodies and small molecule kinase inhibitors (Baselga and Arteaga, 2005). Interestingly, combinations of Cetuximab, a monoclonal anti-EGFR antibody, and cytotoxic drugs sensitize tumor cells to cell killing (Baselga et al, 1993; Prewett et al, 2002). Likewise, in vitro as well as in vivo studies of Gefitinib (ZD1839, Iressa®), an EGFR-specific kinase inhibitor, demonstrated an enhanced cytotoxic effect when combined with certain chemotherapeutic agents (Ciardiello et al, 2000; Sirotnak et al, 2000), but mechanisms underlying chemo-sensitization of tumors by EGFR-targeted molecules remain poorly understood.
We have previously shown that the inhibitory activity of anti-ErbB monoclonal antibodies (Friedman et al, 2005), as well as ErbB-specific kinase inhibitors (Citri et al, 2002), may be attributed to the ability of these drugs to translocate ErbB molecules away from the plasma membrane, the cellular compartment from which they instigate both mitogenic and prosurvival signals. Indeed, an universal regulatory step in RTK signaling involves a two-step endocytosis process (Wiley, 2003; Marmor and Yarden, 2004): A rapid ligand-dependent internalization step removes activated receptors from the cell membrane and enclaves them in endosomes. In the second step, internalized RTK molecules are either targeted to lysosomes, where they undergo degradation, or they recycle back to the plasma membrane. EGFR sorting for lysosomal degradation requires receptor auto-phosphorylation, which recruits an ubiquitin E3 ligase responsible for ubiquitinylation of EGFR (Levkowitz et al, 1999).
In addition to activation by their cognate ligands, ErbB proteins are transmodulated by several other molecules. For example, cellular stress conditions, such as treatment with inflammatory cytokines, oxidative stress, as well as ultraviolet (UV) irradiation, induce either tyrosine phosphorylation (Rosette and Karin, 1996; Hirota et al, 2001; Benhar et al, 2002; Ravid et al, 2002) or serine/threonine phosphorylation of EGFR (Bird and Saklatvala, 1990). Along with EGFR phosphorylation, cytokines and UV irradiation cause aggregation and redistribution of surface-associated receptor molecules (Rosette and Karin, 1996; Hirota et al, 2001), but the mechanisms and identity of the protein kinases involved remained unknown. Here, we report that under physiological stress conditions, as well as upon treatment with a chemotherapeutic drug, EGFR undergoes phosphorylation by the p38 mitogen activated protein kinase (MAPK) pathway and internalizes into endosomes. Further, by removing EGFR from the cell surface, p38 blocks a major route of growth factor-mediated evasion from drug-induced cytotoxicity.
Results
TNF- induces transient, ubiquitin-independent, internalization of EGFR
Previous studies demonstrated a cross-talk between tumor necrosis factor (TNF) signaling and EGFR, and raised the possibility that EGFR internalization is involved (Bird and Saklatvala, 1989, 1990; Hirota et al, 2001). To quantitatively measure EGFR internalization, we labeled EGFR at the cell surface using a cleavable biotin, and then measured the extent of translocation of the biotinylated EGFR to internal cellular compartments. Preliminary experiments using an immobilized avidin indicated that most surface EGFR underwent biotinylation. Figures 1A and B shows that upon treatment of cells with TNF-α, a significant fraction of surface-associated EGFR underwent internalization. The transient nature of EGFR behavior could be due to receptor degradation, or to recycling. Therefore, we used radioactive EGF and measured EGFR downregulation from the cell surface. As expected, treating cells with EGF resulted in persistent and extensive receptor downregulation (Figure 1C). In contrast, TNF-α induced a rapid and transient decrease in the level of surface exposed EGFR, followed by a reappearance of EGFR at the cell surface. To verify the possibility that receptor reappearance is due to recycling, we used monensin, a known recycling inhibitor (Figure 1D). An internalized, biotinylated EGFR was readily detectable following 20 min of treatment with TNF-α but it disappeared at 60 min. Treatment with TNF-α in the presence of monensinn enabled continuous intracellular accumulation of EGFR, which indicates that TNF-α-induced receptor internalization is followed by rapid recycling back to the cell surface. By using immunofluorescence, we confirmed that TNF-α induces translocation of EGFR from the plasma membrane to the cytoplasm, where the receptor coincides with a punctate pattern of staining (Figure 1E). Taken together, these observations indicate that TNF-α induces transient endocytosis of EGFR, which is followed by recycling of the internalized receptors.
Figure 1.
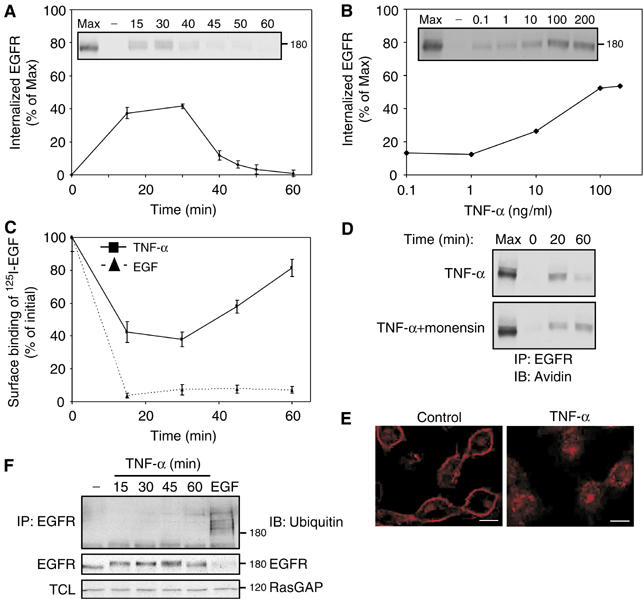
Cytotoxic cytokines induce transient internalization of EGFR with no associated ubiquitinylation or degradation. (A) Following surface biotinylation, HeLa cells were treated with TNF-α (100 ng/ml) for the indicated time intervals. Subsequently, surface-accessible biotin was cleaved, cells were lysed, and EGFR immunoprecipitated and detected in blots using streptavidin-HRP (inset). Quantification of signals corresponding to internalized receptor molecules is shown, relative to maximal surface labeling (Max), measured in the absence of a biotin cleavage step. Bars represent standard deviation values of three identical experiments. (B) Cells were treated for 30 min as in (A), except that increasing concentrations of TNF-α were used. (C) Downregulation of EGFR was tested in HeLa cells, which were treated with TNF-α or with EGF (100 ng/ml, each) for the indicated time intervals. Bars represent standard deviations of triplicate determinations. (D) HeLa cells were left untreated, or pretreated with monensin (100 μM; 15 min). Cells were then incubated with a cleavable biotin, followed by treatment with TNF-α (100 ng/ml) in the absence or presence of monensin, prior to analysis. (E) HeLa cells were left untreated or treated for 30 min with TNF-α (100 ng/ml). Cells were then fixed, permeabilized with methanol and stained with an anti-EGFR antibody, followed by an anti-mouse Cy3 antibody. Confocal microscopy images were taken from a middle section of the cell. Bars, 10 μm. (F) HeLa cells were left untreated, treated with TNF-α (100 ng/ml) for the indicated time intervals, or treated with EGF (10 ng/ml; 5 min). EGFR immunoprecipitates (IP) or total cell lysates (TCL) were immunoblotted (IB) using the indicated antibodies.
Upon binding with EGF, EGFR undergoes rapid internalization, which is associated with receptor ubiquitinylation and culminates in lysosomal degradation (Marmor and Yarden, 2004). To verify that under stress conditions EGFR recycles, rather than undergoes degradation, we treated cells with TNF-α or with interleukin-1 (IL-1), a cytokine that shares with TNF-α various signaling features, but detected neither receptor degradation nor ubiquitinylation (Figure 1F and data not shown). Altogether, the results presented in Figure 1 portray a consistent picture of cytokine-induced transient internalization followed by recycling of EGFR, which is remarkably different from EGF-induced receptor internalization and sorting for intracellular degradation.
Persistent internalization of EGFR following UV irradiation
To further explore mechanisms involved in stress-induced transregulation of EGFR, we tested the effect of UV irradiation, a process entailing receptor trafficking (Rosette and Karin, 1996; Oksvold et al, 2002). Similar to the response of EGFR to inflammatory cytokines, UV irradiation induced rapid, but persistent, internalization of EGFR, as revealed by using the cleavable biotin internalization assay (Figure 2A). The persistent nature of UV-induced effects was reflected also in a receptor downregulation assay using 125I-EGF (Figure 2B). Interestingly, the extent and kinetics of UV-induced downregulation were comparable to those induced by EGF and higher than the corresponding transient effects of TNF-α (Figure 1C). Using immunofluorescence staining of EGFR, we obtained additional evidence in support of UV-induced receptor internalization (Figure 2C), in line with previous reports (Rosette and Karin, 1996; Oksvold et al, 2002). Notably, the punctuate pattern of staining of EGFR following UV irradiation was similar to the pattern seen after stimulation with either EGF or TNF-α (Figure 1E).
Figure 2.
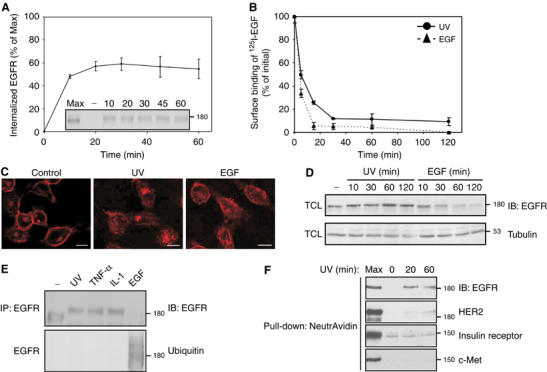
UV induces rapid internalization and covalent modification of EGFR. (A) Following surface biotinylation, HeLa cells were UV irradiated and let to recover at 37°C for the indicated time intervals. EGFR internalization was tested as in Figure 1A. Bars represent standard deviations of three repeats. (B) HeLa cells were UV irradiated, or treated with EGF (100 ng/ml), for the indicated time intervals. Downregulation of EGFR was determined as described in the legend to Figure 1C. (C) HeLa cells were left untreated, UV irradiated (and recovered for 20 min), or treated with EGF (10 ng/ml; 5 min) and EGFR visualized by using immunofluorescence. Bars, 10 μm. (D) HeLa cells were UV irradiated, or treated with EGF (10 ng/ml), for the indicated time intervals. EGFR expression level was then determined using immunoblotting of total cell lysates (TCL). (E) HeLa cells were UV irradiated (followed by 20 min recovery), or treated with TNF-α (100 ng/ml; 30 min), IL-1 (10 ng/ml; 30 min) or EGF (10 ng/ml; 5 min). EGFR was analyzed with the indicated antibodies. (F) Following surface biotinylation, HeLa cells were UV irradiated (100 J/m2) and let to recover at 37°C for the indicated time intervals. Subsequently, surface-accessible biotin was cleaved, and cells were lysed. Biotinylated proteins were pulled-down using NeutrAvidin beads, resolved by electrophoresis, and blotted with the indicated antibodies. Max, maximal surface labeling measured in the absence of a biotin cleavage step.
Immunoblotting analysis for EGFR confirmed rapid, EGF-induced receptor degradation, but no similar disappearance of EGFR was noted following UV irradiation (Figure 2D; Oksvold et al, 2002). As expected, neither UV irradiation nor treatment with IL-1 or with TNF-α induced receptor ubiquitinylation (Figure 2E). Interestingly, although UV irradiation caused robust internalization of EGFR and weaker endocytosis of the homologous protein HER2/ErbB-2, c-Met and the insulin-receptor, were not affected by UV (Figure 2F). In conclusion, UV irradiation specifically enhances internalization of EGFR, but unlike EGF-induced endocytosis, this stress-induced pathway entails no ubiquitinylation and leads to intracellular arrest.
EGFR internalization under stress conditions involves receptor phosphorylation
The results presented in Figure 2E clearly show that both cytokine- and UV-induced stress associate with retarded electrophoretic mobility of EGFR molecules. Previous analysis of UV-treated EGFR detected a similar effect, which was attributed to conformational, rather than covalent alterations (Oksvold et al, 2002, 2004). It is notable that several previous reports documented UV-induced phopshorylation of EGFR on tyrosine residues, presumably due to an autocrine loop induced upon irradiation of certain cell lines (Fischer et al, 2004). The results shown in Figure 3A excluded the possibility that the retarded gel mobility of EGFR of HeLa cells is due to tyrosine phosphorylation, but they left open phosphorylation of other residues. To explore the underlying mechanism and avoid the transient nature of cytokine-induced transmodulation, we followed the kinetics of UV-induced mobility shift. This analysis detected a time-dependent progressive up-shift, which initiated at 5 min after termination of irradiation and persisted for several hours (Figure 3B). Likewise, we found that UV irradiation at 10 J/m2 or higher doses induced a detectable mobility shift, but lower doses were ineffective (Supplementary Figure 1). The delayed kinetics of the shift suggested a preceding cascade of cellular events ignited by external stress. Therefore, we revisited possible phosphorylation of EGFR. To this end, we treated immunoprecipitated EGFR molecules with alkaline phosphatase, an enzyme that removes phosphoryl moieties. Figure 3C indicates that UV-induced EGFR modification is due, perhaps solely, to receptor phosphorylation, because its appearance was completely abolished upon treatment with an alkaline phosphatase. Taken together, the delayed and progressive appearance of the gel mobility shift, along with the effect of dephosphorylation, implies that EGFR is multiply phosphorylated on serine and/or threonine residues upon exposure to stress.
Figure 3.
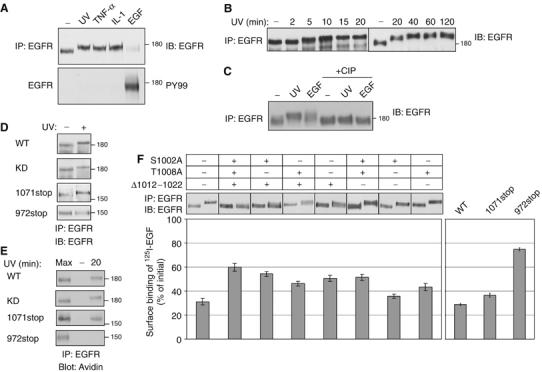
Stress induces phosphorylation at the carboxyl-tail of EGFR. (A) HeLa cells were UV irradiated (20 min), or treated with TNF-α (100 ng/ml; 30 min), IL-1 (10 ng/ml; 30 min) or EGF (10 ng/ml; 5 min). EGFR immunoprecipitates were probed with anti-EGFR and anti-phosphotyrosine (PY99) antibodies. (B) HeLa cells were UV irradiated, allowed to recover for the indicated time intervals, and immunoprecipitated EGFR analyzed. (C) HeLa cells were left untreated, UV irradiated (followed by 20 min recovery), or treated with EGF (10 ng/ml; 5 min). Following immunoprecipitation, EGFR was incubated for 30 min at 37°C without or with calf intestinal alkaline phosphatase (CIP; 20 U). (D) HeLa cells stably expressing EGFR-specific siRNA, were transfected with plasmids encoding the indicated EGFR mutants bearing silent mutations at the siRNA target sequence. After 48 h later, cells were UV irradiated, or left untreated, and let to recover at 37°C for 20 min prior to analysis. (E) Surface biotinylated cells expressing EGFR-specific siRNA, along with the respective EGFR mutant, were left untreated (lanes labeled −), or UV irradiated, and allowed to recover for 20 min at 37°C. Internalized EGFR was detected with streptavidin-HRP. For control, cell extracts were analyzed immediately following UV irradiation (lanes labeled Max). (F) HeLa cells expressing EGFR-specific siRNA were transfected with plasmids encoding the indicated EGFR mutants were untreated or treated with UV irradiation and extracts were analyzed as in (D). Following irradiation, sister cultures were incubated for 90 min on ice with a radiolabeled EGF. EGF binding results were calculated after subtraction of nonspecific binding. Bars represent standard deviation values of quadruplets.
Stress-induced phosphorylation at the carboxyl-terminal region of EGFR is essential for receptor internalization
In line with our conclusion that stress conditions elicit serine/threonine, rather than tyrosine phosphorylation, a kinase-inactive mutant of EGFR (KD), which undergoes no autophosphorylation on tyrosine residues, exhibited normal UV-induced internalization and gel mobility shift (Figures 3D and E). To determine whether the relatively hydrophilic carboxyl terminal tail of EGFR serves as the site of stress-induced phosphorylation, we examined two EGFR truncation mutants lacking portions of the carboxyl-terminal tail. The mutants were ectopically expressed in HeLa cells, whose endogenous EGFR has been stably knocked-down by using RNA interference (data not shown). Ectopic expression of the truncated receptors bearing silent mutations (to overcome the effect of siRNA) indicated that one mutant (1071stop; an EGFR truncated at amino acid 1071) retained UV-induced gel mobility shift, whereas the more extensively truncated version (972stop) lost the ability to undergo modification (Figure 3D). Importantly, the appearance of gel-mobility shifts correlated with stress-induced internalization of EGFR: whereas 1071stop displayed UV-induced internalization, the mutant lacking most of the noncatalytic portion of the cytoplasmic domain, namely 972stop, exhibited no stress-induced internalization in the cleavable biotin assay (Figure 3E).
In an effort to precisely map individual phosphorylation sites, we separately mutated multiple well-characterized sites of EGFR modification, as well as several potential targets of the p38 stress-induced MAPK (see below). In this exhaustive screen (26 mutants) only receptors mutated at serine 1002 or threonine 1008, as well as a mutant lacking a stretch of sites located within 1012–1022, exhibited partial gel mobility shifts and an impaired UV-induced internalization. Hence, we constructed and tested a triple mutant (S1002A/T1008A/Δ1012–1022-EGFR), which displayed no mobility shift and a severely impaired UV-induced downregulation (Figure 3F). Importantly, more extensive loss of UV-induced downregulation was observed with the 972stop truncation mutant, but this short mutant lost EGF-induced downregulation, whereas the triple mutant retained ligand-induced endocytosis (data not shown). In conclusion, stress-induced phosphorylation of EGFR occurs on several serine and threonine residues located in the carboxyl-terminal tail of the receptor, primarily within a stretch of 21 amino acids (1002–1022). Moreover, phosphorylation within this stretch of EGFR is essential for stress-induced internalization.
Stress-induced EGFR phosphorylation and internalization are mediated by p38MAPK
To identify the pathway involved in stress-induced phosphorylation and subsequent internalization of EGFR, we employed a panel of specific kinase inhibitors (Figure 4A). Neither an inhibitor of the phosphoinositide 3′-kinase nor antagonists of Src-family kinases, the Jun-N-terminal kinase and MEK1/2 detectably inhibited stress-induced phosphorylation of EGFR, as revealed by using the gel mobility shift assay. In contrast, inhibition of p38 MAPK by using SB202190 completely abolished the gel mobility shift exhibited by EGFR isolated from either UV-irradiated (Figure 4A) or from cytokine-treated cells (Figure 4B). Because p38 phosphorylates serine/threonine residues of a variety of substrate proteins under stress conditions, these findings suggest that p38-mediated phosphorylation of EGFR enables subsequent receptor internalization. To address this model, we tested the effect of p38 inhibition on stress-induced internalization of EGFR (Figures 4C and D). SB202190 significantly reduced both the persistent internalization of EGFR upon UV irradiation and the transient cytokine-induced receptor internalization. In contrast, SB202190 exerted only a minimal inhibitory effect on EGF-induced downregulation of EGFR (Figure 4E). In conclusion, the use of pharmacological agents suggests that p38 MAPK is the enzyme involved in EGFR phosphorylation, as well as internalization, following exposure of cells to various stress-inducing conditions.
Figure 4.
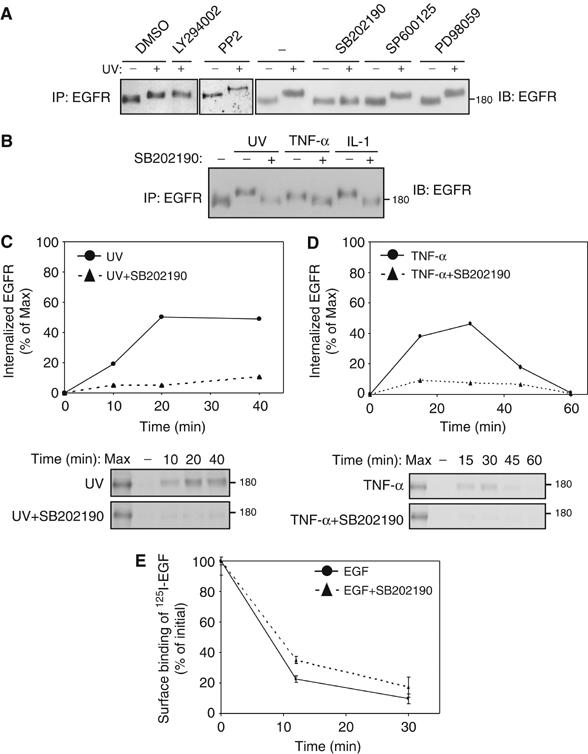
Phosphorylation by p38 mediates stress-induced internalization. (A) HeLa cells were left untreated (−) or treated for 30 min with solvent (dimetheyl sulfoxide, DMSO, at 0.1% vol/vol) or with the indicated inhibitors (LY294002, 0.1 μM; PP2, 10 μM; SB202190, 10 μM; SP600125, 5 μM or PD98059, 25 μM). Thereafter, cells were left untreated, or UV irradiated, and let to recover for 20 min at 37°C prior to analysis of EGFR. (B) HeLa cells, which were pretreated with SB202190 (10 μM) as indicated, were left untreated, UV irradiated, treated with TNF-α (100 ng/ml; 30 min), or with IL-1 (10 ng/ml, 30 min), in the absence or presence of SB202190. Immunoblot analysis of immunoprecipitated EGFR was used to detect gel mobility shifts. (C, D) HeLa cells were left untreated or pretreated with SB202190, as indicated. Following surface biotinylation, cells were UV irradiated, or treated with TNF-α, and let to recover at 37°C for the indicated time intervals, in the absence or presence of SB202190, respectively. Internalized biotinylated EGFR was determined using blotting of EGFR immunoprecipitates (lower panels) and quantification of the signals (upper panel). (E) HeLa cells were untreated or pretreated with SB202190 for 30 min. Thereafter, cells were incubated with EGF (100 ng/ml) for the indicated time intervals in the absence or presence of SB202190. Receptor downregulation was analyzed as in Figure 1C.
The kinetics of p38 activation explain why cytokines induce transient EGFR internalization whereas UV irradiation arrests internalized EGFRs
Unlike EGF-induced endocytosis, stress-induced internalization results in receptor recycling, when the stimulant is a cytokine, or it arrests EGFR in intracellular vesicles, when the stimulant is irradiation. To resolve possible involvement of p38 in transient versus persistent internalization of EGFR, we compared the patterns of p38 activation by using antibodies directed to the active, phosphorylated form of the kinase (Figure 5A). This analysis revealed transient p38 activation peaking at 15 min after addition of TNF-α, but persistent stimulation that lasted more than 1 h after cessation of UV irradiation. In accordance, the gel mobility shift exhibited by EGFR extracted from UV-treated cells persisted for at least 60 min, but it disappeared 45 min after exposure of cells to TNF-α, in parallel to p38 inactivation.
Figure 5.
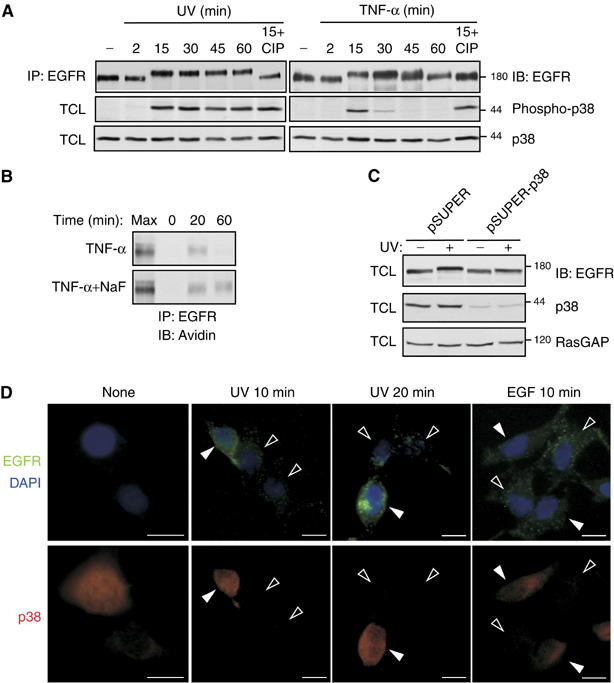
p38 is essential for stress-induced internalization of EGFR. (A) HeLa cells were treated with TNF-α (100 ng/ml), or recovered from a UV pulse, for the indicated time intervals, and extracts were analyzed using anti-EGFR and anti-phospho-p38 antibodies. (B) HeLa cells were left untreated or pretreated with NaF (5 mM; 30 min). Cells were then incubated with a cleavable biotin, followed by treatment with TNF-α (100 ng/ml) for the indicated time intervals in the absence or presence of NaF. Internalization of EGFR was then analyzed as in Figure 1A. (C) HeLa cells were transfected with a pBabe-Puro selection plasmid, along with an empty pSUPER vector or two pSUPER-p38 plasmids encoding p38 siRNAs. Following selection with puromycin (2 μg/ml; 48 h), cells were left untreated, or UV irradiated, and let to recover at 37°C for 20 min. Total cell lysates (TCL) were analyzed with the indicated antibodies. (D) HeLa cells were transfected with plasmids encoding p38-specific siRNAs. After 48 h, cells were placed on ice and incubated with an anti-EGFR murine antibody for one hour. Cells were then washed, UV irradiated and let to recover at 37°C for the indicated time intervals. Alternatively, cells were incubated for 10 min at 37°C in the absence or presence of EGF (100 ng/ml). Cells were then placed on ice, washed, and surface-bound antibody stripped with ice-cold, dilute acetic acid, followed by cell fixation. Cells were then stained with an anti-p38 antibody followed by an anti-rabbit Cy3 (red), anti-mouse Cy2 antibodies (green), or 4,6-diamidino-2-phenylindole (DAPI; blue). Signals were visualized using fluorescence microscopy. Open and closed arrowheads indicate transfected and nontransfected cells, respectively.
A similar correlation between receptor phosphorylation and p38 activation was noted when the dose of UV irradiation was varied from 0.5 to 100 J/m2 (Supplementary Figure 1). This result and the differential kinetics of p38 activation imply that the transient internalization, which follows TNF-α treatment, is due to rapid dephosphorylation (and inactivation) of the kinase, which seems inhibited in UV-irradiated cells. To test this model, we examined the effect of several phosphatase inhibitors on TNF-α-induced mobility shift of EGFR (Supplementary Figure 2). In this assay, NaF (unlike other drugs) inhibited the time-dependent reversal of EGFR mobility shift and enabled persistent activation of p38 by TNF- α. Moreover, in NaF-treated cells, TNF-α-induced internalization of EGFR was uncoupled from a subsequent recycling step (Figure 5B), thereby confirming the notion that intracellular retention of EGFR is mediated by an active p38 MAPK.
Knockdown of p38 expression suppresses stress-induced EGFR phosphorylation and internalization
To firmly confirm the involvement of p38 in EGFR phosphorylation, as well as in subsequent stress-induced internalization, we knocked-down p38 expression using two specific siRNA sequences cloned in an expression plasmid. The efficiency of p38 knockdown was confirmed on whole-cell lysates using an anti-p38 antibody (Figure 5C). Notably, concomitant with downregulation of p38 expression, we observed partial disappearance of the gel mobility shift, an indication for reduced phosphorylation of EGFR. Due to a reduction in substrate adhesiveness of siRNA-transfected cells, we developed an anti-EGFR antibody-based alternative to the biotin internalization assay. Immunofluorescent staining for p38 (red) and internalized EGFRs (green) confirmed that no receptor internalization occurred in cells that were left untreated at 37°C (Figure 5D). On the other hand, UV-irradiated cells demonstrated that knockdown of p38 expression (open arrowheads) significantly reduced internalization of EGFRs (Figure 5D; UV 10 min and UV 20 min). As expected, cells that underwent no transfection, and therefore normally expressed p38, displayed extensive internalization of EGFR (filled arrowheads). In contrast with UV-induced receptor internalization, EGF-induced endocytosis of EGFR was not affected in cells lacking p38 expression (Figure 5D; EGF 10 min, open arrowheads versus filled arrowhead). In conclusion, knockdown of p38 expression using RNA interference indicated that this MAP kinase is indispensable for stress-induced EGFR phosphorylation and internalization.
Rab5 effector proteins and Clathrin are involved in stress-induced internalization of EGFR
Two major internalization pathways, a Clathrin-mediated route and a pathway associated with Caveolin, enable EGFR internalization under different cellular conditions (Wiley, 2003). Rab5, a marker for early endosomes, was used to monitor Clathrin-dependent internalization, and Caveolin-1 was used to detect internalization from Caveolin-coated membrane micro-domains. In untreated cells, transfected Rab5-green fluorescence protein (GFP) was localized to internal endosomes, unlike EGFR, which was localized to the plasma membrane (Figure 6A). Following UV irradiation, Rab5 assumed a more punctuate pattern of distribution, while, as expected, EGFR translocated to internal vesicles. Importantly, the merged images showed significant co-localization of the internalized EGFR with Rab5-labeled early endosomes (insets). To examine whether the Clathrin-dependent pathway is the sole route facilitating EGFR internalization upon stress, we also transfected cells with Caveolin-1-mRFP. Following UV irradiation, immunostaining of EGFR revealed no co-localization of internalized EGFR with Caveolin-1-containing vesicles (Figure 6B). Hence, we concluded that stress-induced EGFR internalization is mediated by the Clathrin-dependent pathway, and it sorts internalized EGFR to early endosomes.
Figure 6.
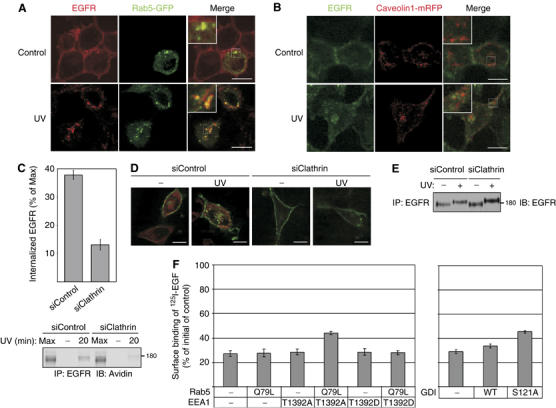
UV-induced internalization of EGFR requires Clathrin and directs the receptor to Rab5-containing vesicles. (A, B) HeLa cells were transfected with plasmids encoding Rab5-GFP (A) or Caveolin1-mRFP (B). After 48 h after transfection, cells were left untreated, or UV irradiated, and let to recover at 37°C for 20 min. Cells were then fixed and stained with an anti-EGFR antibody followed by anti-mouse Cy3 (A) or Cy2 (B) antibodies. Images were taken using confocal microscopy. Bars, 10 μm. Insets show magnifications of the squared areas. (C) HeLa cells were transfected with control (siControl) or Clathrin-specific (siClathrin) siRNA oligonucleotides. After 48 h, cells were subjected to surface biotinylation and UV irradiation, and let to recover at 37°C for 20 min (lanes labeled 20) or analyzed directly (−). Following cleavage of extracellularly accessible biotin, EGFR immunoprecipitation and blotting with streptavidin-HRP (lower panel; Max indicates signals obtained in the absence of a biotin cleavage step), the signals of internalized EGFR-biotin were quantified (upper panel; bars represent s.d. of three experiments). (D) HeLa cells were transfected with a GFP-EGFR expression vector, and 24 h later they were transfected with the indicated siRNA oligonucleotides. After 48 h, cells were left untreated or UV irradiated, and let to recover at 37°C for 20 min. Cells were then fixed and stained with an anti-Clathrin antibody followed by a Cy3-labeled anti-mouse antibody (red). Bars, 10 μm. (E) At 48 h following transfection with the indicated siRNA oligonucleotides, HeLa cells were UV irradiated, or left untreated, and then incubated for 20 min at 37°C prior to analysis. (F) HeLa cells stably expressing an EGFR-specific siRNA were transfected with a plasmid encoding an EGFR bearing silent mutations at the siRNA target sequence, along with the indicated Rab5, EEA1 and GDI wild type or mutant constructs. After 48 h, cells were left untreated, or treated with UV irradiation and let to recover at 37°C for 20 min. Cells were then incubated on ice with a radiolabeled EGF. Following several washes, cells were lysed and surface binding of 125I-EGF was determined. Surface binding of 125I-EGF was calculated relative to ligand binding of the control. Bars represent standard deviation values of quadruplets.
To confirm the involvement of Clathrin in stress-induced EGFR internalization, we used siRNA oligonucleotides designed to specifically knockdown expression of the Clathrin heavy chain (Supplementary Figure 3A). Using biotinylation of surface-localized EGFR, we found that Clathrin is essential for effective UV irradiation-induced internalization of EGFR (Figure 6C): although incomplete, knockdown of Clathrin expression resulted in significant inhibition of EGFR internalization following UV irradiation (37.8±1.6% of surface EGFR internalized in control cells, whereas 13.0±1.9% of surface receptors underwent internalization in cells transfected with Clathrin siRNA). An immunostaining analysis confirmed this conclusion: while cells transfected with control siRNA displayed robust internalization of EGFR upon UV irradiation, EGFR was retained at the plasma membrane of cells transfected with Clathrin-specific siRNA oligonucleotides (Figure 6D). Further, using Clathrin siRNA we determined that stress-induced EGFR phosphorylation precedes internalization (Figure 6E). In conclusion, under stress conditions, EGFR undergoes phosphorylation by p38 and then internalizes via the Clathrin-dependent pathway, which targets the endocytosed receptors to Rab5-containing endosomes.
The small GTPase Rab5 is not only a marker for early endosomes; through its effector proteins, such as the early endosmal autoantigen 1 (EEA1), Rabenosyn-5 and the Rab GDP dissociation inhibitor (GDI), it plays a driving role in receptor endocytosis. Because EEA1 phosphorylation by p38 MAPK (at threonine 1392) instigates constitutive internalization of G-protein coupled receptors (Mace et al, 2005), we tested the possibility that EEA1, in addition to EGFR phosphorylation by p38, contributes to UV-induced EGFR internalization. As predicted, a phosphorylation defective mutant of EEA1, T1392A, partly inhibited UV-induced internalization of EGFR, but only on the background of an active mutant of Rab5 (Q79L; Figure 6F). Notably, the phosphorylation-mimicking T1392D mutant exerted no effect in this assay. Similar to EEA1, p38-mediated phosphorylation of GDI (at serine 121) promotes endocytosis (Cavalli et al, 2001), an observation we confirmed with EGFR using the S121A mutant and UV-irradiated HeLa cells (Figure 6F).
Taken together, the internalization defect of p38-resistant mutants of EGFR (Figure 3) and the relatively small inhibitory effects of mutant Rab5 effectors (Figure 6F) suggest that p38 impacts EGFR internalization through a dual mechanism. To leave out the primary mechanism involving direct phosphorylation of EGFR, we used a composite mutant, S1002A/T1008A/Δ1012–1022-EGFR (see Figure 3F), which is not phosphorylated upon stress and exhibits 30% less internalization as compared to WT-EGFR. Ectopic expression of GDI/S121A or EEA1/T1392A showed that the mutants reduced internalization of the p38-insensitive EGFR by 11 or 15%, respectively (Supplementary Figure 3B). When co-expressed, they reduced S1002A/T1008A/Δ1012–1022-EGFR internalization by 25%. Overall, internalization of the S1002A/T1008A/Δ1012–1022 mutant was inhibited to the extent observed with the 972stop mutant of EGFR, which undergoes no phosphorylation or internalization upon cellular stress. This observation reinforces the existence of a dual mechanism of p38-mediated regulation of EGFR.
Chemotherapy associated with p38 stimulation induces EGFR internalization, thereby augmenting cytotoxicity
Because under stress conditions EGFR is removed from the cell surface, we predicted a concomitant reduction in EGF-induced signaling and cell survival. This prediction was tested in UV-irradiated HeLa cells (Figure 7A). As expected, a clear reduction in EGF-induced phosphorylation of both EGFR and Shc was noted in irradiated cells, in support of stress-induced desensitization of survival signals. To examine relations between p38-induced internalization of EGFR and survival, we used SW480 colon cancer cells, which express high levels of EGFR and depend on autocrine TGF-α secretion (Ciardiello et al, 2001). It is notable that p38 MAPK has been implicated in the cytotoxic effects of certain chemotherapeutic agents, such as cisplatinum (CDDP) and related drugs (Losa et al, 2003). When tested on SW480 cells, CDDP induced sustained activation (albeit delayed) of p38 and facilitated EGFR modification (Figure 7B). This modification was abolished in cells that were simultaneously treated with CDDP (or UV) and SB202190 (Figure 7C), confirming a role for p38. To overcome the deleterious effects of CDDP on cell attachment to the substrate, we measured EGFR downregulation using flow cytometry. This assay revealed that the extent of CDDP-induced downregulation (41%: Figure 7D, upper part) is comparable to that of UV-induced internalization (39%, light gray). In line with p38 involvement, SB202190 significantly reduced this effect of CDDP (to 6%). Taken together, these results show that a widely used chemotherapeutic agent, in similarity to UV irradiation and inflammatory cytokines, induces p38-dependnet EGFR phosphorylation and internalization.
Figure 7.
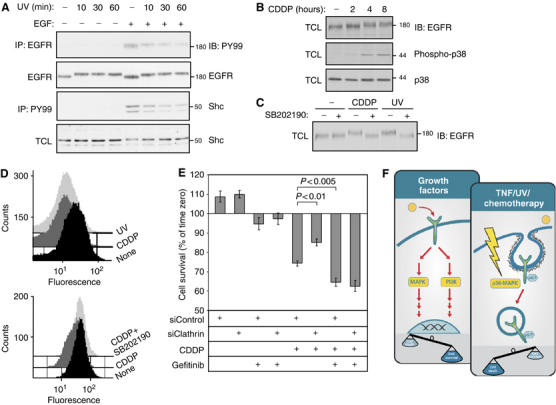
CDDP induces p38-dependent internalization of EGFR, thereby augmenting chemotherapy-induced cell death. (A) HeLa cells were exposed to UV irradiation and let to recover for the indicated time intervals, prior to treatment with EGF (20 ng/ml; 5 min) and immunoblot analysis of immunoprecipitates and total cell lysates (TCL), as indicated. (B) SW480 cells were treated with CDDP (50 μg/ml) for the indicated time intervals, prior to analysis of TCL with the indicated antibodies. (C) SW480 cells were left untreated, treated with CDDP (50 μg/ml, 6 h), or irradiated with UV (100J/m2, followed by 20 min recovery), in the presence or absence of SB202190 (10 μM), as indicated. TCL were analyzed by immunoblotting. (D) SW480 cells were left untreated, UV irradiated, or treated with CDDP (50 μg/ml, 6 h) in the absence or presence of SB202190 (10 μM). Cells were fixed prior to labeling of surface EGFR with an anti-EGFR antibody, followed by a fluorescently labeled secondary antibody. The histograms present surface EGFR signals collected from 50 000 cells using a FACSscan flow-cytometer. (E) SW480 cells were plated in a 96-well plate. Following transfection with siRNA oligonucleotides (siControl or siClathrin), cells were left untreated or pretreated with Gefitinib (5 μM) for 24 h. Cells were then left untreated or treated with CDDP (50 μg/ml) in the absence or presence of Gefitinib for 10 h. Cell viability was measured using the MTT assay. The histogram presents cell viability relative to the viability signal obtained prior to treatment. Bars represent standard deviation values; P-values are indicated. (F) A model contrasting growth factor-mediated survival signaling (through pathways like MAPK and phosphoinositide 3′ kinase; PI3K) and stress-induced cytotoxic pathways. The latter are stimulated by cytokines, UV irradiation and chemotherapy (e.g., CDDP), and they involve activation of p38 MAPK, followed by serine/threonine phosphorylation of EGFR and internalization into Rab5-containing endosomes. We propose that removal of EGFR and other receptors from the cell surface helps evade survival signaling and enhances stress-induced cell death.
Because EGFR-targeting drugs, such as kinase inhibitors and monoclonal antibodies, often sensitize tumors to chemotherapeutic agents like CDDP (Baselga et al, 2005 and references therein), we addressed the possibility that chemotherapy-induced, p38-mediated internalization of EGFR is involved in the response of tumor cells to chemotherapy. To this end, we employed a Clathrin-specific siRNA that blocks EGFR internalization. In cells transfected with control siRNA, CDDP treatment for 10 h reduced cell survival by 25.7±1.4% (Figure 7E). In contrast, Clathrin siRNA significantly limited cytotoxicity of the chemotherapeutic agent (14.8±1.8% reduction in cell survival; P<0.01). Presumably, the Clathrin-specific siRNA prevented p38-induced EGFR internalization, which enabled SW480 cells to benefit from self-production of TGF-α and partially evade drug-induced death. To test this notion we employed Gefitinib, an EGFR-specific kinase inhibitor. Consistent with previous reports (Ciardiello et al, 2000; Sirotnak et al, 2000), Gefitinib augmented the cytotoxic effect of the chemotherapeutic agent, and as predicted, it abolished the protective effect of Clathrin-specific siRNAs (P<0.005; Figure 7E). Taken together, the results presented in Figure 7 reveal a drug-induced pathway of EGFR internalization and attribute to this mechanism an important part in augmenting the cytotoxic effect of chemotherapeutic agents that activate p38 MAPK.
Discussion
The present study unravels a previously unrecognized EGFR transregulatory mechanism that involves p38 MAPK. This stress-induced MAPK, or a downstream kinase like MAPKAP-2, phosphorylates EGFR, thereby triggering receptor internalization. Upon inactivation of p38, EGFR undergoes rapid recycling (Figures 1, 5A and B). Thus, transient internalization of EGFR occurs in cytokine-stimulated cells, but chronic stimulation of p38, for example in cells treated with certain cytotoxic drugs, arrests EGFR in endosomes (see model in Figure 7F).
Transregulatory mechanisms involving EGF-like ligands commonly lead to enhanced tyrosine phosphorylation of EGFR, which ultimately leads to receptor degradation (Prenzel et al, 2001). In striking contrast, processes that entail receptor phosphorylation in trans evade the degradative fate. For example, although oxidative stress promotes tyrosine phosphorylation of EGFR, the c-Cbl docking site undergoes no phosphorylation and hence no subsequent ubiquitinylation and receptor degradation take place (Ravid et al, 2002). Likewise, PKC mediated transphosphorylation inhibits EGF-induced ubiquitinylation and degradation of EGFR, but concomitantly internalizes EGFR into recycling endosomes (Bao et al, 2000). Our data identify p38 MAPK as a stress- and cytokine-induced protein kinase responsible for both transphosphorylation of EGFR and for subsequent receptor internalization. Two recent reports support this notion: EGFR internalization upon treatment of cells with the antibiotic anisomycin (Vergarajauregui et al, 2006) or with CDDP (Winograd-Katz and Levitzki, 2006) has been attributed to a mechanism involving p38.
In aggregate, our results portray the following sequence of events that follow exposure of cells to stress conditions (see model in Figure 7F): stimulation of p38 MAPK leads to phosphorylation of EGFR on multiple serine and threonine sites located within a short segment of EGFR (residues 1002–1022; Figure 3). Because a Clathrin-specific siRNA inhibited EGFR internalization, we concluded that phosphorylation mediated by p38 instigates rapid receptor internalization via a Clathrin-dependent pathway. The underlying mechanism appears to be dual: because stress-induced internalization of a receptor mutated at the multiple phosphorylation segment is severely impaired (Figure 3F), we assume that p38-phosphorylated EGFRs are recognized by an unknown sorting protein that recruits them to early endosomes. A secondary mechanism involves two or more Rab5 effector proteins (Figure 6F; Supplementary Figure 3). The underlying mechanism may involve formation of a GDI:Rab5 complex (Cavalli et al, 2001) and phosphorylation of the endosomal protein EEA1, an event necessary for constitutive internalization of opioid receptors (Mace et al, 2005). Internalized receptors arrest in a Rab5-containing vesicular compartment, presumably early endosomes (Figure 6A). Nevertheless, as soon as p38 is inactivated, the internalized receptors undergo dephosphorylation and recycle back to the cell surface (Figures 1 and 5).
This model is consistent with the ability of chemotherapy to impact on EGFR in living cells. CDDP and other derivatives of platinum potently stimulate p38 MAPK in epithelial cells (Figure 7B; Losa et al, 2003; Winograd-Katz and Levitzki, 2006) to induce a phosphorylation-dependent EGFR gel mobility shift (Figure 7C), and enhance receptor internalization (Figure 7D). Treatment of platinum refractory metastatic squamous cell carcinoma of the head and neck with a combination of an antibody to EGFR and platinum chemotherapy revealed a chemosensitizing effect in patients (Baselga et al, 2005). In terms of our data (Figure 7E), enhanced tumor chemosensitivity may be due to a double blockade of escape routes: along with DNA-damaging effects, CDDP induces internalization of an important receptor for growth and survival factors, as well as its major partner, HER2/ErbB-2 (Figure 2F). When chemotherapy is combined with kinase inhibitors, receptors remaining at the cell surface are catalytically inhibited, thereby blocking escape from cell death. Alternatively, when antireceptor antibodies (e.g., Erbitux and Herceptin) are combined with chemotherapy, the antibodies internalize the remaining receptors through an apparently distinct route of endocytosis, which involves formation of large antibody–receptor complexes at the cell surface (Maier et al, 1991; Friedman et al, 2005). If proved and extended to other signaling pathways, this model may offer ways to predict optimal drug combinations and scheduling.
Materials and methods
Cell lines and transfections
Transfection of subconfluent HeLa and SW480 cultures was carried out using the calcium phosphate method or by using Oligofectamine (Gibco BRL, Grand Island, NY). For selection of HeLa cells stably expressing EGFR-specific siRNA, cells were co-transfected with pBabe-Puro vector and selected in puromycin-containing medium (1.5 μg/ml). Cells were starved in serum-free medium for 12 h prior to all experiments. All treatments were carried out in starvation medium.
Cleavable biotin internalization assay
Cells were washed once with phosphate-buffered saline (PBS) and incubated with Sulfo-NHS-S-S-Biotin (0.5 mg/ml; Pierce, Rockford, IL) in PBS for 20 min at 22°C. Excess biotin was quenched with 15 mM glycine. Following treatments, cells were placed on ice, washed, and residual surface biotin was removed by washing three times, for 8 min each, with ice-cold glutathione cleavage solution (50 mM glutathione, 75 mM NaCl, 1 mM EDTA, 1% albumin and 0.75% 10N NaOH). Cells were then washed three times, lysed and EGFR immunoprecipitated.
Protein immunoprecipitation and immunoblotting analyses
Cells were washed once with ice-cold PBS, harvested in solubilization buffer (see buffer compositions in Citri et al, 2002) and lysates cleared by centrifugation (14 000 r.p.m., 20 min). For immunoprecipitation, cleared lysates were incubated for 90 min at 4°C with antibodies precoupled to anti-mouse IgG-agarose beads. Immunoprecipitation and immunoblotting analyses were performed as described (Bao et al, 2000).
Receptor downregulation assays
See Supplementary data.
Immunofluorescence
Cells grown on coverslips were treated as indicated, and then fixed with paraformaldehyde (3%) and permeabilized with methanol. Alternatively, when p38 was stained, cells were permeabilized for 10 min at 22°C with PBS containing 0.2% Triton X-100 and 1% albumin. For labeling, coverslips were incubated for 1 h at 22°C with the indicated primary and secondary antibodies. Confocal microscopy was performed using a Zeiss Axiovert 100 TV microscope (Oberkochen, Germany).
UV irradiation of living cells
Cells were washed once with PBS, and then exposed to UV irradiation using a UVC lamp (15W, Philips). Time of exposure was calculated to achieve the intensity of 100J/m2 using an UVC energy probe. Following irradiation, cells were allowed to recover as indicated in DME medium, prewarmed to 37°C.
Alkaline phosphatase treatment
Following immunoprecipitation of EGFR, beads were incubated with alkaline phosphatase (20 U; New England BioLabs, Beverly, MA) in the presence of 10 mM MgCl2 at 37°C for 30 min. Beads were then washed once before addition of gel sample buffer.
MTT viability assay
Viability assays using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were performed in sextuplets as described (Bao et al, 2000).
Reagents
See Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Information
Acknowledgments
We thank members of our group for useful insights and Drs S Lev, R Pagano, A Nebreda and J Gruenberg for plasmids. Our laboratory is supported by research grants from Minerva, the Israel Cancer Research Fund, the German Israel Foundation, the Prostate Cancer Foundation and the National Cancer Institute (grant CA72981). YY is the incumbent of the Harold and Zelda Goldenberg Professorial Chair.
References
- Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, Yarden Y (2000) Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J Biol Chem 275: 26178–26186 [DOI] [PubMed] [Google Scholar]
- Baselga J, Arteaga CL (2005) Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol 23: 2445–2459 [DOI] [PubMed] [Google Scholar]
- Baselga J, Norton L, Masui H, Pandiella A, Coplan K, Miller WH Jr, Mendelsohn J (1993) Antitumor effects of doxorubicin in combination with anti-epidermal growth factor receptor monoclonal antibodies. J Natl Cancer Inst 85: 1327–1333 [DOI] [PubMed] [Google Scholar]
- Baselga J, Trigo JM, Bourhis J, Tortochaux J, Cortes-Funes H, Hitt R, Gascon P, Amellal N, Harstrick A, Eckardt A (2005) Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol 23: 5568–5577 [DOI] [PubMed] [Google Scholar]
- Benhar M, Engelberg D, Levitzki A (2002) Cisplatin-induced activation of the EGF receptor. Oncogene 21: 8723–8731 [DOI] [PubMed] [Google Scholar]
- Bird TA, Saklatvala J (1989) IL-1 and TNF transmodulate epidermal growth factor receptors by a protein kinase C-independent mechanism. J Immunol 142: 126–133 [PubMed] [Google Scholar]
- Bird TA, Saklatvala J (1990) Down-modulation of epidermal growth factor affinity in fibroblasts treated with interleukin 1 or tumor necrosis factor is associated with phosphorylation at a site other than threonine 654. J Biol Chem 265: 235–240 [PubMed] [Google Scholar]
- Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J (2001) The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol Cell 7: 421–432 [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR, Tortora G (2001) Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 7: 1459–1465 [PubMed] [Google Scholar]
- Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G (2000) Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 6: 2053–2063 [PubMed] [Google Scholar]
- Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, Patterson C, Neckers L, Fry DW, Yarden Y (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 21: 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer OM, Hart S, Gschwind A, Prenzel N, Ullrich A (2004) Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol Cell Biol 24: 5172–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LM, Rinon A, Schechter B, Lyass L, Lavi S, Bacus SS, Sela M, Yarden Y (2005) Synergistic down-regulation of receptor tyrosine kinases by combinations of mAbs: implications for cancer immunotherapy. Proc Natl Acad Sci USA 102: 1915–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Murata M, Itoh T, Yodoi J, Fukuda K (2001) Redox-sensitive transactivation of epidermal growth factor receptor by tumor necrosis factor confers the NF-kappa B activation. J Biol Chem 276: 25953–25958 [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4: 1029–1040 [DOI] [PubMed] [Google Scholar]
- Losa JH, Parada Cobo C, Viniegra JG, Sanchez-Arevalo Lobo VJ, Ramon y Cajal S, Sanchez-Prieto R (2003) Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene 22: 3998–4006 [DOI] [PubMed] [Google Scholar]
- Mace G, Miaczynska M, Zerial M, Nebreda AR (2005) Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid receptor endocytosis. EMBO J 24: 3235–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LA, Xu FJ, Hester S, Boyer CM, McKenzie S, Bruskin AM, Argon Y, Bast RC Jr (1991) Requirements for the internalization of a murine monoclonal antibody directed against the HER-2/neu gene product c-erbB-2. Cancer Res 51: 5361–5369 [PubMed] [Google Scholar]
- Marmor MD, Yarden Y (2004) Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23: 2057–2070 [DOI] [PubMed] [Google Scholar]
- Oksvold MP, Huitfeldt HS, Ostvold AC, Skarpen E (2002) UV induces tyrosine kinase-independent internalisation and endosome arrest of the EGF receptor. J Cell Sci 115: 793–803 [DOI] [PubMed] [Google Scholar]
- Oksvold MP, Thien CB, Widerberg J, Chantry A, Huitfeldt HS, Langdon WY (2004) UV-radiation-induced internalization of the epidermal growth factor receptor requires distinct serine and tyrosine residues in the cytoplasmic carboxy-terminal domain. Radiat Res 161: 685–691 [DOI] [PubMed] [Google Scholar]
- Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A (2001) The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer 8: 11–31 [DOI] [PubMed] [Google Scholar]
- Prewett MC, Hooper AT, Bassi R, Ellis LM, Waksal HW, Hicklin DJ (2002) Enhanced antitumor activity of anti-epidermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xenografts. Clin Cancer Res 8: 994–1003 [PubMed] [Google Scholar]
- Ravid T, Sweeney C, Gee P, Carraway KLr, Goldkorn T (2002) Epidermal growth factor receptor activation under oxidative stress fails to promote c-Cbl mediated down-regulation. J Biol Chem 277: 31214–31219 [DOI] [PubMed] [Google Scholar]
- Rosette C, Karin M (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274: 1194–1197, 0036-8075 [DOI] [PubMed] [Google Scholar]
- Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG (2000) Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res 6: 4885–4892 [PubMed] [Google Scholar]
- Vergarajauregui S, San Miguel A, Puertollano R (2006) Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic 7: 686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley HS (2003) Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res 284: 78–88 [DOI] [PubMed] [Google Scholar]
- Winograd-Katz SE, Levitzki A (2006) Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene advance online publication 19 June 2006; doi:10.1038/sj.onc.1209737 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Information