

Abstract
Kinesin is an ATP-driven microtubule (MT)-based motor fundamental to organelle transport. Although a number of kinesin crystal structures have been solved, the structural evidence for coupling between the bound nucleotide and the conformation of kinesin is elusive. In addition, the structural basis of the MT-induced ATPase activity of kinesin is not clear because of the absence of the MT in the structure. Here, we report cryo-electron microscopy structures of the monomeric kinesin KIF1A–MT complex in two nucleotide states at about 10 Å resolution, sufficient to reveal the secondary structure. These high-resolution maps visualized clear structural changes that suggest a mechanical pathway from the nucleotide to the neck linker via the motor core rotation. In addition, new nucleotide binding pocket conformations are observed that are different from X-ray crystallographic structures; it is closed in the 5′-adenylyl-imidodiphosphate state, but open in the ADP state. These results suggest a structural model of biased diffusion movement of monomeric kinesin motor.
Keywords: cryo-electron microscopy, kinesin, microtubule
Introduction
Kinesin is a mechanochemical enzyme that converts the chemical energy derived from ATP hydrolysis to mechanical energy to produce force along the microtubule (MT) (Hirokawa, 1998). During the mechanochemical cycle of kinesin, MTs serve not only as mechanical tracks but also as regulators of ATP hydrolysis.
Current structural models of kinesin motility depend mostly on X-ray crystallographic structures obtained in the absence of MTs. Thus, a number of fundamental questions remain unanswered. First, X-ray crystal structures indicate that the bound nucleotide does not fully control kinesin conformation in the absence of MTs. Presumed ATP-like structures were observed, even when ADP was bound (Sack et al, 1997; Sindelar et al, 2002; Garcia-Saez et al, 2004; Yan et al, 2004), and a presumed ADP structure was observed with an ATP analog (Ogawa et al, 2004). Therefore, the nucleotide and kinesin structural states are not tightly coupled in the absence of MTs. Secondly, when a single kinesin motor domain is attached to a bead, it randomly diffuses along the MT, and binding to the MT is biased toward the plus-end (Okada et al, 2003; Kamei et al, 2005). Recent data also support that the biased diffusion plays important roles in the two-headed kinesin movement (Carter and Cross, 2005; Taniguchi et al, 2005). However, the structural basis of the biased diffusion is yet to be established. Third, MTs play two opposing roles in the kinesin ATPase cycle: they act as a nucleotide exchange factor (NEF) by opening its nucleotide binding pocket (NBP) to accelerate ADP release from kinesin (Hackney, 1988), and as an NTPase-activating protein (NAP) by closing of the NBP to activate ATP hydrolysis (Ma and Taylor, 1997). Currently, the structural basis of the opposing dual roles of MTs remains to be elucidated.
In this study, we use cryo-electron microscopy (cryo-EM) to visualize high-resolution structures of the KIF1A–microtubule complex (KIF1A–MT complex) in the presence of a nonhydrolyzable ATP analog 5′-adenylyl-imidodiphosphate (AMPPNP) and ADP. The KIF1A–MT complexes display clear structural changes between the two nucleotide states, including a 20° rotation of the catalytic core, docking/undocking of the neck linker, and piston-like movement of the switch II helix. In addition, new NBP conformations are observed that are different from X-ray crystallographic structures; NBP is closed in the AMPPNP state, but open in the ADP state. This finding suggests that NEF and NAP activities depend on rotation of the kinesin catalytic core. On the basis of these results, we propose a structural model of KIF1A thermal ratchet movement.
Results
High-resolution cryo-EM analysis
We have extended the resolution of the monomeric kinesin KIF1A–MT complexes to 11 Å for the ADP state, and 10 Å for the AMPPNP state (Figure 1A). The ADP complex represents the structure immediately prior to ADP release, since MTs stimulate this rate-limiting step. The AMPPNP complex represents the structure just before ATP hydrolysis.
Figure 1.
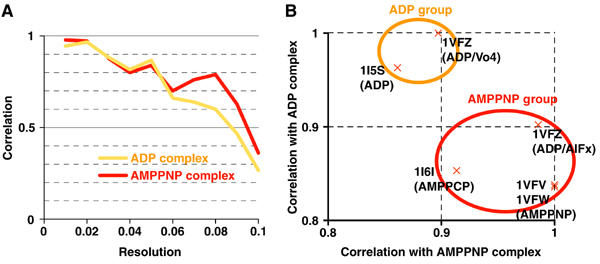
Evaluation of resolution of maps and docking of atomic models. (A) Fourier shell correlations between the KIF1A–MT complex density maps and atomic models generated from the crystal structures of KIF1A and tubulin. (B) Correlation between the KIF1A–MT complex density maps and KIF1A atomic models. Each cross represents correlation values of the atomic model specified by PDB ID. Correlation values were normalized by the maximum value among the atomic models.
High-resolution analysis was achieved by introducing several technical improvements. Specifically, cryo-EM images were recorded at lower defocus, different helical classes of MTs (14–16 protofilament MTs) were combined, and newly developed helical image analysis programs were employed to correct image distortions (Metlagel et al, 2006).
MT structures do not change upon motor binding
Examination of the MT portion of the cryo-EM density maps disclosed a wealth of structural details and ensured that the maps can resolve secondary structures. For example, Figure 3A and B depicts well-resolved tubulin α-helices with comparable appearance to those of the bare MT at 8 Å resolution (Li et al, 2002). The MT structure of the KIF1A–MT complexes is mostly consistent with tubulin atomic models (Nogales et al, 1998; Lowe et al, 2001) except for a small shift of helix H3 in α-tubulin (Figure 3B, arrowhead), indicating that motor binding has minor effects on tubulin. Although Krebs et al (2004) reported increased density near the C-terminus of α-tubulin helix H7 upon motor binding, our high-resolution maps showed no such movement.
Figure 3.
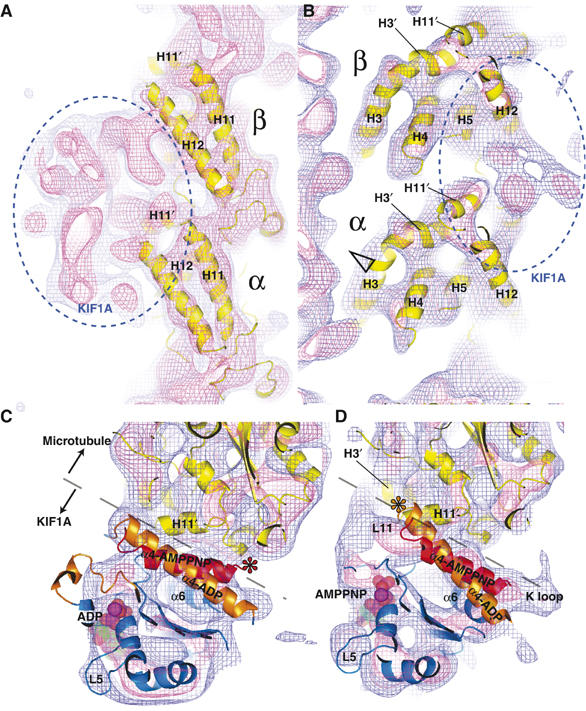
Detailed views of the 3D reconstructions of the KIF1A–MT complex. (A) Right and (B) left sides of the MT protofilament outer surface in the AMPPNP complex showing the similarity between the tubulin atomic model and map. Blue and pink chickenwires represent 0.7σ and 1.5σ of the density map, respectively. Solvent-exposed helices (H3–5, H11 and H12) are well resolved. A small shift of α-tubulin helix H3 is indicated with an open arrowhead, possibly as a result of interactions with loop L11 of KIF1A. (C, D) Top view of the switch II helix from the plus-end of the MT (C) in the ADP and (D) AMPPNP states. Both ADP (orange) and AMPPNP (red) KIF1A switch II helices are fitted into the complexes. The interface between KIF1A and MT is depicted with dashed lines. Asterisks indicate the possible points of steric clashes between KIF1A and tubulin atomic models.
Fitting KIF1A atomic models into cryo-EM density maps
To interpret the KIF1A structure in the maps, we initially selected the best-fit KIF1A atomic model for each map. Since the conformations of kinesin and bound nucleotide do not always correlate with each other, we fitted all the available atomic models of KIF1A into the KIF1A–MT complexes and measured the similarities by the correlations between atomic models and cryo-EM density maps. As expected, atomic models were classified into ADP and AMPPNP groups (Figure 1B). The switch II helix and neck linker comprise the main differences between the two groups that contribute to the correlation values. In the following analyses, we used the ‘ADP' atomic model (PDB ID: 1I5S) for the ADP KIF1A–MT complex, and the ‘AMPPNP' atomic model (1VFV) for the AMPPNP KIF1A–MT complex.
Switch-controlled rotation of the kinesin core
In view of the above fitting of the KIF1A atomic models, the most evident overall change between the two-nucleotide states involves rotation of the kinesin core. The core is ‘tilted' in the ADP state (Figure 2A), while ‘upright' in the AMPPNP state (Figure 2B). The rotation axis penetrates near KIF1A loop L8 at an angle of 23° to the MT axis (Figure 2D and F). The kinesin core rotates 20.6° around the axis. As a result, the helix α6, whose C-terminal end is the base of the neck linker, shifts more than 5 Å (Figure 5). As discussed below, this 5 Å-shift may play a key role in kinesin movement.
Figure 2.
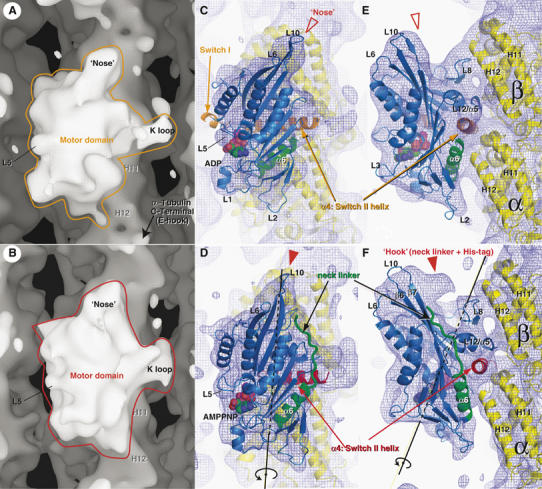
Cryo-EM maps of KIF1A–MT complexes in the ADP (A, C, E) and AMPPNP states (B, D, F). (A, B) Isosurface representation of KIF1A–MT complexes. (C–F) Fitting of the X-ray crystal structures into cryo-EM maps. MTs are shown with their plus-end up and assignment of α- and β-tubulin is based on that of Krebs et al (2004). The blue chickenwires are contoured at 0.7σ of the density map, with a mesh size of 1 Å. A 20.6° rotation around the axis shown in (D) and (F) explains conformational changes of the kinesin core from the ADP state to the AMPPNP state.
Figure 5.
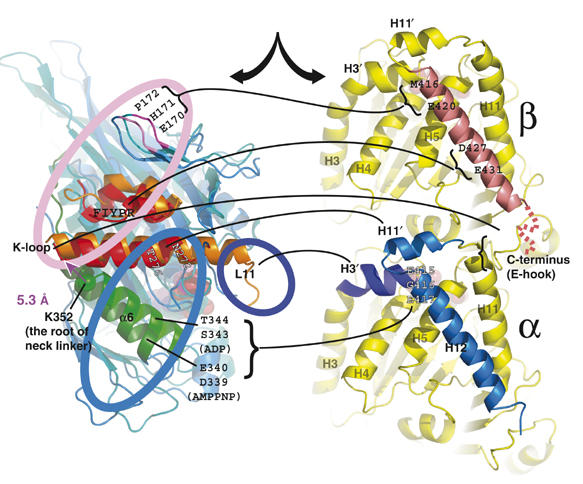
MT-centric superposition of the ADP and AMPPNP KIF1A crystal structures from KIF1A–MT complexes, presenting interactions between KIF1A residues and tubulin. Interacting residues observed in ADP (orange), AMPPNP (red) or both complexes (black) are depicted with lines connecting to the corresponding tubulin-binding site.
During the rotation, the loop L8 of KIF1A works as the fulcrum point interacting with the N-terminal side of the β-tubulin helix H12. The residues involved in binding include E170, H171 and P172 of KIF1A, and N416 and E420 of β-tubulin (Figure 5A). E170 is conserved among plus-end directed kinesins, and its mutations either block detachment from the MT (Klumpp et al, 2004) or lower MT-activated ATPase activity (Woehlke et al, 1997).
The switch II helix stays fixed to the MT
While the core rotates, the switch II helix of KIF1A remains fixed to the MT. The switch II helix is located at the center of the KIF1A MT-binding interface. Based on similarity to GTP-binding proteins, the helix is proposed to function as a ‘relay' that forms a bridge between the nucleotide and neck linker. Previous X-ray crystallographic studies on KIF1A by our group reveled marked conformational changes of the switch II helix relative to the core (Kikkawa et al, 2001; Nitta et al, 2004). However, the conformations of the switch II helix in KIF1A–MT complexes were unclear.
To establish the structure of the switch II helix in the KIF1A–MT complex, the complex is viewed from the MT plus-end, and both the ADP and AMPPNP KIF1A atomic models are superimposed for comparison (Figure 3C and D). As shown in Figure 3C, the density map of the ADP KIF1A–MT complex encloses the switch II helix of the ADP KIF1A model better than that of the ATP KIF1A model. Similarly, the AMPPNP KIF1A model fits better to the AMPPNP KIF1A–MT complex (Figure 3D). If a different type of KIF1A crystal structure is fitted into the KIF1A–MT complex, the switch II helix does not fit into the density, and has a steric clash with the tubulin model (Figure 3C and D, asterisks). Consistent with a previous prediction (Kikkawa et al, 2001), the relative arrangement between the switch II helix and the MT is not significantly altered (Figures 2E, F and 4A). Hence, from the MT centric view, the switch II helix remains fixed, while the motor core rotates relative to the MT.
Figure 4.
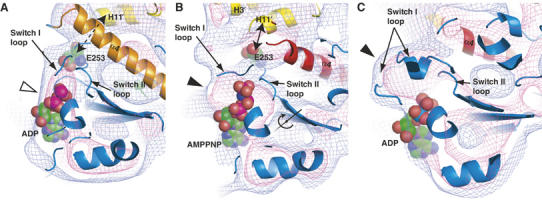
Experimental and simulated density maps surrounding NBP. (A, B) Slab view around the KIF1A NBP. (A) ADP is mostly exposed to solvent (open arrowhead), while (B) the phosphate portion of AMPPNP is enclosed by density (filled arrowhead). Nucleotides are surrounded by switch I and switch II loops. In the AMPPNP state, a well-conserved E253 (illustrated with a spacefill model) is in direct contact with α-tubulin helix H11′. The displacement of E253 is explained by rotation of the core. The rotation axis of the core is shown in (B). (C) Simulated 10 Å density map of KCBP (PDB ID:1SDM) displaying a closed conformation. Blue and pink chickenwires represent 0.7σ and 1.5σ, respectively.
KIF1A helix α6–MT interface
The KIF1A helix α6 interacts with the helix H12 of α-tubulin and its movement may be associated with MT binding and unbinding of KIF1A. In the ADP state, S343 and T344 of KIF1A helix α6 interact with E415, E420 and E423 of α-tubulin helix H12. Therefore, the interface of KIF1A is generally positively charged, and the tubulin interface is negatively charged. In the AMPPNP state, rotation of the core shifted the interface to the previous turn of the helix α6, and D339 and E340 of KIF1A interact with the same MT residues as in the ADP state. These two amino acids are the only negatively charged residues in the MT-facing surface of kinesin, and are highly conserved among the plus-end directed kinesin motors. It is generally known that kinesin strongly binds to MTs in the ATP state. Surprisingly, however, the repulsive force between the two molecules appears strengthened in the AMPPNP KIF1A–MT complex (Figure 5).
These changes in interaction are intriguing in explaining the unbinding of KIF1A after ATP hydrolysis. The crystal structure shows that the MT-interacting loops, L11 and L12, are shortened in the ADP/Pi state (Nitta et al, 2004). This shortening may weaken the binding between L11-α4-L12, while interactions between L8 and H12 remain unchanged. Therefore, the repulsive force between α6 and H12 should release the KIF1A core toward the plus end. If the highly conserved E340 was mutated in conventional kinesin, both Km MT and Kcat were decreased to about 0.3 of those of wild type (Woehlke et al, 1997). This phenotype is explained by assuming that the mutant kinesin is locked into the ATP binding state on MTs with higher affinity, and cannot dissociate ADP from the nucleotide-binding pocket.
Neck linker docking/undocking
High-resolution maps were also employed to directly visualize docking/undocking of the neck linker to the core, which was postulated as a power stroke of kinesin (Vale and Milligan, 2000).
In ATP-like kinesin molecules solved by X-ray crystallography, the neck linker is docked along the core β-sheet that forms a ‘nose'-like structure. The nose of the kinesin core is wider in the AMPPNP KIF1A–MT complexes (Figure 2B and D) compared to that in the ADP state (Figure 2A and C), clearly representing the docked-neck linker. Furthermore, the tip of the nose is hooked when viewed from the right in the AMPPNP state (Figure 2F, filled arrowhead), while the nose does not display a hooked tip in the ADP state (Figure 2E, open arrowhead). This ‘hook' is not filled with the fitted atomic model (C340). The hook comprises an extra 11 amino acids and the six-histidine tag present only in the recombinant KIF1A protein (C351) used for cryo-EM. In the ADP state, the neck linker containing the extra amino acids does not appear in the density map, possibly due to a disordered state. These differences collectively show that the neck linker is docked in the AMPPNP state, but undocked in the ADP state.
From the kinesin-centric view, movement of the switch II helix explains the docking/undocking of the neck linker: nucleotide changes the conformation of the switch II helix and, as a result, the neck linker docks or undocks from the core. In contrast, from the MT-centric view, the nucleotide and the neck linker are mechanically connected via the motor core. Nucleotide exchange at the left side induces the rotation of the motor core. As the helix α6 is a part of the core, this rotation shifts the helix α6 relative to the switch II helix. In the ADP state, the C-terminal end of the helix α6 points toward the switch II helix, which occludes docking of the neck linker to the core (Figure 2E). In the AMPPNP state, the helix α6 has an offset to the switch II helix, allowing docking of the neck linker (Figure 2F, neck linker colored green). This ATP-induced neck linker docking may, in part, drive plus-end-directed kinesin motility (Rice et al, 1999; Vale and Milligan, 2000).
Open/closed states of the nucleotide-binding pocket
We examined the density maps surrounding the nucleotide to determine the effects of MT binding to the NBP, in view of the fact that MTs act as both NEF and NAP for kinesin. In the ADP KIF1A–MT complex, the left side of ADP was exposed to solvent, showing an ‘open state' (Figure 4A). Notably, the switch I loop of the fitted atomic model is outside the density (Figure 3C), suggesting that MT alters the switch I conformation and opens the NBP.
On the other hand, in the AMPPNP KIF1A–MT complex, the triphosphate moiety is buried into the density, while the adenine ring of the nucleotide is still exposed to solvent (Figure 4B). This structure surrounding the triphosphate is similar to the ‘phosphate tube' identified in myosins (Lawson et al, 2004) and non-motile kinesin-like calmodulin-binding protein (KCBP; Figure 4C) (Vinogradova et al, 2004). Accordingly, we predict that the switch I loop covers the left side of the nucleotide. This arrangement may create an environment suitable for ATP hydrolysis by proper positioning of a water molecule for nucleophilic attack on the ATP γ-phosphate (Naber et al, 2003). Hydrolyzed ATP would release phosphate from the ‘back door' of the phosphate tube, thus contributing to the detachment of kinesin from MT (Nitta et al, 2004).
Technically, it is somewhat surprising that the open/closed conformations of switch I (residues 205–218, molecular weight of about 1.5 kDa) are distinguishable at 10–11 Å resolution. Detailed examination of the density maps shows that it seems possible to detect small differences down to 1 kDa. For example, the maps clearly illustrate the neck linker (Figure 2C and D; molecular weight=1.3 kDa) and loop L5 (Figures 2A–D, 3C and D; 1 kDa).
The switch II loop connects MT with the nucleotide-binding pocket
Next, we attempted to identify a kinesin element that senses MTs and sends signals to the NBP. Judging from the fitted atomic models, the switch II loop, L11, may link the two elements. In the AMPPNP KIF1A–MT complex, a conserved residue, E253 (LAGSE), in the switch II loop interacts with helix H11′ of α-tubulin (Figure 4B). In contrast, E253 is further away from H11′ in the ADP state (Figure 4A). Therefore, it is likely that E253 acts as an MT sensor for the ATP state and is required for closing the NBP. The idea is supported by corresponding conventional kinesin mutants that bind to ATP and undergo neck linker docking but display very low ATPase activity (Rice et al, 1999; Yun et al, 2001). In these mutants, due to the lack of MT-sensing E253, NBP may not be closed thus ATP hydrolysis is impaired.
Discussion
In summary, we have simultaneously visualized nucleotide-dependent conformational changes of KIF1A, including those of the switch II helix, the neck linker, and the core rotation. These structures demonstrate that the nucleotide and the structural states are tightly coupled in the presence of MTs. Although the movement of the neck linker is observed in kinesin–MT complexes (Rice et al, 1999; Sindelar et al, 2002), the correlation between the switch II and the neck linker has been unclear. From our new structures, it becomes clear that, from the MT-centric view, neck linker docking is regulated through the kinesin core, while loop L8 and switch II helix are fixed to the MT.
Core rotation may regulate NEF/NAP activity
A comparison of the two KIF1A–MT complexes and existing KIF1A crystal structures suggests that the NEF and NAP activities of MTs can be defined by the tilted/upright rotation states of the motor core. In the absence of MTs, the NBP of kinesin is not fully open, since ADP release is slow (Hackney, 1988; Ma and Taylor, 1997). In our ADP KIF1A–MT complex, NBP is open and the conformation of the switch I loop differs from that in the ADP KIF1A crystal structure. Since the presence of MT constitutes the only difference between the two, it is reasonable to assume that MT binding with a tilted conformation is required for the open state. On the other hand, closing of NBP would require direct contact between the switch II loop and MT, which is unique to the upright conformation. Thus, the two opposing NEF/NAP activities of MT may be explained as follows: the tilted kinesin core induces NEF activity, while the upright core stimulates NAP activity.
A structural model for single-headed kinesin thermal ratchet movement
Based on the hypothetical core rotation-controlled NEF/NAP activities, we propose a structural model for KIF1A force generation, which we call ‘biased-capturing' model. To explain the movement of KIF1A, we have assumed an anisotropic potential in the weak binding ADP state, which may be important for the directional movement of a minimal single-headed KIF1A construct (Okada and Hirokawa, 1999). Additionally, we propose that ADP release is biased towards the MT plus-end.
When single-headed kinesins are tethered to a bead (Okada et al, 2003; Kamei et al, 2005), the motor randomly diffuses along MT in the ADP state. If the motor proceeds to the forward binding site, backward tension rotates the core clockwise (Figure 6A). On the other hand, if the motor diffuses to the backward binding site, forward tension will induce counter-clockwise rotation. In view of our structural data, which suggests that only clockwise rotation (‘tilted' conformation) opens NBP, ADP release would be ‘biased' toward the forward binding site. KIF1A should then ‘capture' the forward MT binding site by subsequent transition from the weak to strong binding state.
Figure 6.
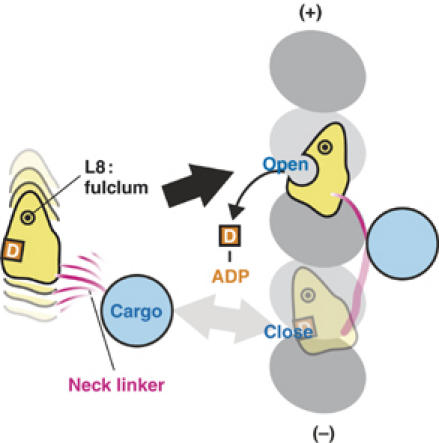
‘Biased-capturing' model of monomeric kinesins. The model explains the structural basis of plus-end biased binding of monomeric kinesin motors. The monomeric kinesin motor domain is linked to a bead, and the linker determines the direction of rotation of the motor core. The core rotate clockwise only when the motor moves to the forward binding site, which allows ADP release and strong binding of kinesin to the MT.
The core rotation in other kinesins
Similar, but smaller, degrees of rotations were observed for other kinesins. For Unc104, the rotation of the catalytic core is at most 5° between the AMPPNP and ADP states (Al-Bassam et al, 2003). For conventional kinesin, a 12° rotation is observed between the nucleotide-free and AMPPNP state (Skiniotis et al, 2004). These different degrees of rotations from ours are explained by the tilted rotation axis and resolution of density maps.
First, the rotation around the tilted axis appears smaller from outside of MT. According to our new high-resolution maps, the rotation axis is at an angle of 23° to the MT axis (Figure 2F). In the above two papers and our previous study (Kikkawa et al, 2001), the rotations were estimated only around the radial direction. In fact, if the new 20.6° rotation is seem from the outside of MT, it appears approximately 10° (Figure 2C and D).
Second, it is difficult to determine rotations accurately from low-resolution maps, especially when the shape of the molecule is symmetric. To demonstrate it, we simulated 25 Å resolution density maps of the human kinesin–MT complex based on our results. The simulated density maps were quite similar to those obtained by other groups (Al-Bassam et al, 2003; Skiniotis et al, 2004). Although the ellipsoid shape of kinesin core from outside (Figure 7A and B) helps to identify the rotation, the symmetric shape viewed from the top (Figure 7C and D) makes it difficult to identify the rotation around the tilted axis. As a result, the core rotation can appear smaller than the actual rotation at lower resolutions.
Figure 7.
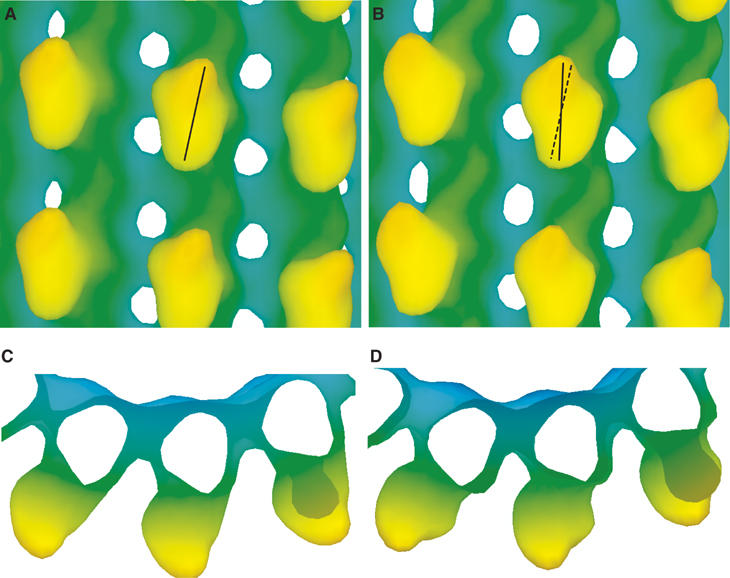
Simulated 25 Å resolution density maps of the human kinesin–microtubule complexes, (A, C) in the ADP state and (B, D) in the AMPPNP complex. The atomic models of the complexes were constructed based on our docking results derived from the KIF1A–microtubule complex.
For the above reasons, the core rotations observed by other groups agree with our current results within experimental error. Although high-resolution studies on conventional kinesin–microtubule complex is required to support the generality of the core rotation, considering the highly conserved amino-acid sequences between KIF1A and conventional kinesin, it is likely that conventional kinesin can also take the conformations similar to KIF1A during the chemo-mechanical cycle.
Structural model for dimeric conventional kinesin movement
If the core rotation is general among kinesins, the biased-capturing mechanism may also play a pivotal role in the hand-over-hand movement of dimeric conventional kinesin. Similar to KIF1A, the tethered head may diffuse randomly between the front and back of the anchoring head. The tension applied to the neck linker would influence the rotational state of the core and subsequently regulate the ATP cycle. Accumulating evidence supports that the tension applied to neck linker regulates ADP release (Uemura and Ishiwata, 2003; Higuchi et al, 2004) or ATP hydrolysis (Hackney et al, 2003; Hackney, 2005). However, the pathway between the neck linker and NBP is unclear. Our model proposes that the kinesin core as a possible pathway between the neck linker and NBP. To support this model, further structural studies of conventional kinesin under tension are required.
Materials and methods
Sample preparation
Samples of MTs decorated by C351 in the presence of 2 mM ADP or 2 mM AMPPNP were prepared as described (Kikkawa et al, 2000, 2001). Cryo-EM images were taken with a defocus range of 11 000–33 000 Å, using a JEM-2010F electron microscope equipped with a field emission gun (JEOL, Tokyo).
Image analysis
Images were processed with a script system, Ruby-Helix (Metlagel et al, 2006). KIF1A–MT complex images were straightened iteratively by locating the tube center from the phase residuals of near and far side layer lines. The length of each helical repeat was precisely determined by maximizing the intensity of the 40 Å layer line. CTF effects including astigmatism were corrected based on defocus values determined by CTFFIND3 (Mindell and Grigorieff, 2003). Different helical classes of MTs were converted to a standard 15-protofilament MT and averaged in reciprocal space (Yonekura and Toyoshima, 2000). Final maps were calculated from the best 90 data sets representing approximately 80 000 asymmetric units.
Fitting atomic models
Atomic models were fitted into maps as rigid-bodies using the CoLoRes program (Chacon and Wriggers, 2002). Fitted atomic models were used to estimate the amplitude-scaling factor of the cryo-EM data for each resolution shell. Effective resolution (11 and 10 Å for the ADP and AMPPNP KIF1A–MT complexes, respectively) was determined by Fourier shell correlation between the fitted atomic model and experimental data (van Heel, 1986; Figure 1A). Since the crystal structures are not precisely the same in the KIF1A–MT complex, the resolution determined by this method is conservative. The rotation axis between the ADP and AMPPNP states was derived from the rotation matrix determined with LSQMAN (Kleywegt and Jones, 1994). Threshold values (0.7σ) for the isosurface representations were selected to enclose the tubulin atomic model.
Simulated density maps
We initially aligned the atomic models in the helical lattice and projected them to generate simulated images (Figure 4C). Next, 3D maps were calculated from simulated images using the same image analysis method as for experimental cryo-EM images.
Atomic models of the KIF1A–MT complex
Atomic coordinates have been deposited in the Protein Data Bank under numbers 2HXF and 2HXH.
Acknowledgments
We specially thank H Yajima for collecting ADP KIF1A–MT complex images, T Oda for image analysis and Dr W Wriggers (University of Texas, Houston) for providing the CoLoRes program. We also thank Drs R Nitta, EP Sablin (UCSF) and L Lum (University of Texas, Southwestern) for insightful discussions and critical reading of the manuscript. This work was supported by grants from the Human Frontier Science Program and the Welch Foundation to MK and a Special Grant-in-Aid from the Ministry of Education, Culture, Sport, Science and Technology of the Ministry of Education, Culture, Sports, Science and Technology of Japan to NH.
References
- Al-Bassam J, Cui Y, Klopfenstein D, Carragher BO, Vale RD, Milligan RA (2003) Distinct conformations of the kinesin Unc104 neck regulate a monomer to dimer motor transition. J Cell Biol 163: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter NJ, Cross RA (2005) Mechanics of the kinesin step. Nature 435: 308–312 [DOI] [PubMed] [Google Scholar]
- Chacon P, Wriggers W (2002) Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol 317: 375–384 [DOI] [PubMed] [Google Scholar]
- Garcia-Saez I, Yen T, Wade RH, Kozielski F (2004) Crystal structure of the motor domain of the human kinetochore protein CENP-E. J Mol Biol 340: 1107–1116 [DOI] [PubMed] [Google Scholar]
- Hackney DD (1988) Kinesin ATPase: rate-limiting ADP release. Proc Natl Acad Sci USA 85: 6314–6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD (2005) The tethered motor domain of a kinesin–microtubule complex catalyzes reversible synthesis of bound ATP. Proc Natl Acad Sci USA 102: 18338–18343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD, Stock MF, Moore J, Patterson RA (2003) Modulation of kinesin half-site ADP release and kinetic processivity by a spacer between the head groups. Biochemistry 42: 12011–12018 [DOI] [PubMed] [Google Scholar]
- Higuchi H, Bronner CE, Park HW, Endow SA (2004) Rapid double 8-nm steps by a kinesin mutant. EMBO J 23: 2993–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N (1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519–526 [DOI] [PubMed] [Google Scholar]
- Kamei T, Kakuta S, Higuchi H (2005) Biased binding of single molecules and continuous movement of multiple molecules of truncated single-headed kinesin. Biophys J 88: 2068–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa M, Okada Y, Hirokawa N (2000) 15 Å resolution model of the monomeric kinesin motor, KIF1A. Cell 100: 241–252 [DOI] [PubMed] [Google Scholar]
- Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N (2001) Switch-based mechanism of kinesin motors. Nature 411: 439–445 [DOI] [PubMed] [Google Scholar]
- Kleywegt G, Jones T (1994) A super position. CCP4/ESF-EACBM Newsletter on Protein Crystallography. Vol. 31, pp 9–14
- Klumpp LM, Hoenger A, Gilbert SP (2004) Kinesin's second step. Proc Natl Acad Sci USA 101: 3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs A, Goldie KN, Hoenger A (2004) Complex formation with kinesin motor domains affects the structure of microtubules. J Mol Biol 335: 139–153 [DOI] [PubMed] [Google Scholar]
- Lawson JD, Pate E, Rayment I, Yount RG (2004) Molecular dynamics analysis of structural factors influencing back door pi release in myosin. Biophys J 86: 3794–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, DeRosier DJ, Nicholson WV, Nogales E, Downing KH (2002) Microtubule structure at 8 Å resolution. Structure 10: 1317–1328 [DOI] [PubMed] [Google Scholar]
- Lowe J, Li H, Downing KH, Nogales E (2001) Refined structure of alpha beta-tubulin at 3.5 Å resolution. J Mol Biol 313: 1045–1057 [DOI] [PubMed] [Google Scholar]
- Ma YZ, Taylor EW (1997) Kinetic mechanism of a monomeric kinesin construct. J Biol Chem 272: 717–723 [DOI] [PubMed] [Google Scholar]
- Metlagel Z, Kikkawa YS, Kikkawa M (2006) Ruby-helix: an implementation of helical image processing based on object-oriented scripting language. J Struc Biol (in press) [DOI] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142: 334–347 [DOI] [PubMed] [Google Scholar]
- Naber N, Minehardt TJ, Rice S, Chen X, Grammer J, Matuska M, Vale RD, Kollman PA, Car R, Yount RG, Cooke R, Pate E (2003) Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science 300: 798–801 [DOI] [PubMed] [Google Scholar]
- Nitta R, Kikkawa M, Okada Y, Hirokawa N (2004) KIF1A alternately uses two loops to bind microtubules. Science 305: 678–683 [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391: 199–203 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Nitta R, Okada Y, Hirokawa N (2004) A common mechanism for microtubule destabilizers-M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell 116: 591–602 [DOI] [PubMed] [Google Scholar]
- Okada Y, Higuchi H, Hirokawa N (2003) Processivity of the single-headed kinesin KIF1A through biased binding to tubulin. Nature 424: 574–577 [DOI] [PubMed] [Google Scholar]
- Okada Y, Hirokawa N (1999) A processive single-headed motor: kinesin superfamily protein KIF1A. Science 283: 1152–1157 [DOI] [PubMed] [Google Scholar]
- Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD (1999) A structural change in the kinesin motor protein that drives motility. Nature 402: 778–784 [DOI] [PubMed] [Google Scholar]
- Sack S, Muller J, Marx A, Thormahlen M, Mandelkow EM, Brady ST, Mandelkow E (1997) X-ray structure of motor and neck domains from rat brain kinesin. Biochemistry 36: 16155–16165 [DOI] [PubMed] [Google Scholar]
- Sindelar CV, Budny MJ, Rice S, Naber N, Fletterick R, Cooke R (2002) Two conformations in the human kinesin power stroke defined by X-ray crystallography and EPR spectroscopy. Nat Struct Biol 9: 844–848 [DOI] [PubMed] [Google Scholar]
- Skiniotis G, Cochran JC, Muller J, Mandelkow E, Gilbert SP, Hoenger A (2004) Modulation of kinesin binding by the C-termini of tubulin. EMBO J 23: 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Nishiyama M, Ishii Y, Yanagida T (2005) Entropy rectifies the Brownian steps of kinesin. Nat Chem Biol 1: 342–347 [DOI] [PubMed] [Google Scholar]
- Uemura S, Ishiwata S (2003) Loading direction regulates the affinity of ADP for kinesin. Nat Struct Biol 10: 308–311 [DOI] [PubMed] [Google Scholar]
- Vale RD, Milligan RA (2000) The way things move: looking under the hood of molecular motor proteins. Science 288: 88–95 [DOI] [PubMed] [Google Scholar]
- van Heel M (1986) Similarity measures between images. Ultramicroscopy 21: 95–100 [Google Scholar]
- Vinogradova MV, Reddy VS, Reddy AS, Sablin EP, Fletterick RJ (2004) Crystal structure of kinesin regulated by Ca(2+)-calmodulin. J Biol Chem 279: 23504–23509 [DOI] [PubMed] [Google Scholar]
- Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD (1997) Microtubule interaction site of the kinesin motor. Cell 90: 207–216 [DOI] [PubMed] [Google Scholar]
- Yan Y, Sardana V, Xu B, Homnick C, Halczenko W, Buser CA, Schaber M, Hartman GD, Huber HE, Kuo LC (2004) Inhibition of a mitotic motor protein: where, how, and conformational consequences. J Mol Biol 335: 547–554 [DOI] [PubMed] [Google Scholar]
- Yonekura K, Toyoshima C (2000) Structure determination of tubular crystals of membrane proteins. II. Averaging of tubular crystals of different helical classes. Ultramicroscopy 84: 15–28 [DOI] [PubMed] [Google Scholar]
- Yun M, Zhang X, Park CG, Park HW, Endow SA (2001) A structural pathway for activation of the kinesin motor ATPase. EMBO J 20: 2611–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]