

Abstract
Objective:
To determine whether sodium butyrate (NaB), a major short-chain fatty acid produced in the human gut by bacterial fermentation of dietary fiber, enhances transforming growth factor (TGF)-β signaling and potentiates its tumor suppressor activity in the gut.
Summary Background Data:
The molecular mechanisms by which dietary fiber decreases the risk of colon cancers are poorly characterized. TGF-β is an important tumor suppressor in the gut and has many similar biologic activities as NaB. Therefore, we hypothesized that the chemo-preventive effects of NaB are mediated in part by enhancing TGF-β signaling and its tumor suppressor function in the gut.
Methods:
The effects of NaB on Smad3 expression in rat intestinal epithelial (RIE-1) cells and 6 human colon cancer cell lines were examined. The effects of NaB on TGF-β-induced Smad3 phosphorylation and plasminogen activator inhibitor-1 (PAI-1) and cyclooxygenase-2 (COX-2) gene expression were also examined in RIE-1 cells. Finally, the effects of NaB and TGF-β on anchorage-independent growth were examined in Akt-transformed RIE-1 cells.
Results:
NaB induced Smad3 in RIE-1 cells and in 4 human colon cancer cell lines. NaB enhanced TGF-β-induced Smad3 phosphorylation and potentiated TGF-β-induced PAI-1 expression. NaB and TGF-β synergistically inhibited anchorage-independent growth of Akt-transformed RIE-1 cells.
Conclusions:
These results demonstrate that NaB induces Smad3 and potentiates TGF-β signaling and its tumor suppressor activity in gut epithelial cells. Our data reveal a novel molecular mechanism that may explain in part the beneficial effects of dietary fiber in decreasing the risk of colon cancers.
This study found that butyrate, a short-chain fatty acid, induced Smad3 and potentiated TGF-β signaling and its tumor suppressor activity in gut epithelial cells. These results reveal a novel molecular mechanism that may explain in part the beneficial effects of dietary fiber in decreasing the risk of colon cancers.
Colon cancer is the second leading cause of cancer death in the United States with a combined yearly mortality rate of 55,000.1 Surgical resection is an effective treatment of localized disease, achieving a 5-year survival rate of 90%; however, many patients present with advanced disease resulting in an overall 5-year survival rate of approximately 60%. Furthermore, as the U.S. population ages, the number of hospital admissions for colorectal cancer is projected to double over the next 50 years.2
Dietary fiber is an important part of a healthy diet and has been defined as nondigestible plant materials composed predominantly of nonstarch polysaccharides and nonpolysaccharides. Early epidemiologic observations brought the notion that consumption of large amounts of high-fiber foods was a major factor lowering the risk of colon cancer.3 This has been confirmed in the last 3 years by 3 large prospective epidemiologic studies.4–6 By definition, all of the consumed fiber arrives at the colon. The fate of fiber that reaches the colon depends on its chemical characteristics and on the colonic microflora. Highly fermentable fiber sources, such as fruits and vegetables, produce a high amount of short-chain fatty acids (SCFAs) including butyrate, which has been shown experimentally to prevent colon cancer development.7 Poorly fermentable fiber, such as those from wheat bran, results in lower levels of SCFAs compared with highly fermentable fiber but may decrease the concentrations of pro-carcinogens and carcinogens in the fecal stream by increasing the bulk stool volume. Butyrate is a potent inhibitor of histone deacetylases (HDACs)8 and has been shown to suppress growth and induce differentiation of a variety of cell lines, including colon carcinoma cells.9 By inhibition of the activities of multiple HDACs, butyrate can induce expression of specific genes that elicit extensive cellular morphologic and metabolic changes, such as growth arrest, differentiation, and apoptosis.
Transforming growth factor beta (TGF-β) is expressed in the gut epithelium and serves as an important negative regulator of the proliferation of enterocytes and colonocytes. During colon carcinogenesis, TGF-β serves as an important tumor suppressor by inhibiting cellular proliferation and inducing apoptosis.10,11 However, most colon cancer cells are resistant to the tumor suppressor activities of TGF-β by acquiring defects of various components of the TGF-β signaling pathway. For example, TGF-β type I receptor,12 type II receptor,13 Smad2,14,15 and Smad416–18 have been shown to be either mutated or down-regulated in human colorectal cancers. TGF-β signals through its binding to a cell surface receptor complex, which subsequently phosphorylates Smad2 and Smad3. The phosphorylated Smad2 or Smad3 form a heteromeric complex with Smad4, which translocates into nucleus and regulates transcription of target genes.19,20
Even though butyrate has been reported to modulate the expression of number of proteins involved in biologic processes that may contribute to the development of colon cancer, the effects of butyrate on TGF-β signaling pathway are not known. Since butyrate and TGF-β share many similar biologic activities and both appear to prevent the development of colon cancer, we formed the hypothesis that the chemo-prevention effects of butyrate are mediated in part by enhancing TGF-β signaling and its tumor suppressor function in the gut. We have designed a series of experiments to begin testing this novel hypothesis.
MATERIALS AND METHODS
Reagents
Sodium butyrate (NaB, Sigma, MO) was dissolved in PBS and TGF-β1 (Pharmingen, CA) was diluted in vehicle (0.1% BSA, 4 mmol/L HCl). Reagents were stored at −20°C in small aliquots.
Cell Culture
RIE-1 cell line (a gift from Dr. Kenneth D. Brown, Cambridge Research Station, Babraham, Cambridge, UK) has morphologic and biologic characteristics of intestinal crypt cells.21 This cell line does not have transformed phenotypes in culture and has been used by many laboratories as a model of normal gut epithelial cells. RIE-1 cells were maintained as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM, Mediatech Inc.) supplemented with 5% dialyzed fetal bovine serum (dFBS, Invitrogen). RIE-1/mAkt cells were generated as described previously11 and were maintained in DMEM supplemented with 5% dFBS and 0.4 mg/mL G418. Human colon cancer cell lines HT-29, Caco-2, and DLD-1 (from American Type Culture Collection) were maintained in DMEM supplemented with 10% FBS; MEM (Mediatech Inc.) supplemented with 10% FBS, 1× nonessential amino acid (NEAA, Sigma), 1× sodium pyruvate (Sigma); RPMI1640 (Mediatech Inc.) supplemented with 10% FBS, respectively. KM12C, KM12L4A, and KM20 (provided by Dr. I. J. Fidler, M.D. Anderson Cancer Center, Houston, TX) were maintained in MEM supplemented with 10% FBS, 1X NEAA, 1× sodium pyruvate, and 2× MEM vitamin solution (Invitrogen). All cell lines were grown at 37°C in a humidified incubator at 5% CO2. Cells were grown to subconfluence and split every 3 to 4 days.
Western Blotting
Total proteins were prepared from cells using the 1× cell lysis buffer (Cell Signaling Technology, Inc.) supplemented with 1 mmol/L PMSF. Proteins (30 μg) were resolved by SDS-PAGE and immunoblotted with antibodies against Smad2 (a gift from Dr. Carl-Henrik Heldin, Ludwig Institute for Cancer Research, Uppsala, Sweden), Smad3 (Invitrogen), phosphorylated Smad3 (a gift from Dr. Edward B. Leof, Mayo Clinic, Rochester, MN), and β-actin (Sigma) followed by an appropriate horseradish-peroxidase-conjugated secondary antibody (BioRad). Luminol substrate PicoWest (Pierce Biotechnology Inc.) was used to detect target proteins which were visualized on x-ray films (Eastman Kodak Company). The signals were quantified by densitometric analysis (Kodak 1D).
Real-Time Quantitative RT-PCR
RNA was extracted from cells using RNAqueous Kit (Ambion) and used in detecting Smad2, Smad3, PAI-1, COX-2, and β-actin mRNA expression by real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR). First, cDNA was synthesized using Retroscript cDNA synthesis kit (Ambion). Next, PCR was performed with a Prism 7700 Sequence Detector (Perkin Elmer/Applied Biosystems Division) as described previously using the TaqMan PCR core reagent kit and the appropriate forward and reverse primers and probes (Applied Biosystems).22,23
Soft Agar Assay
Soft agar assays were performed as previously described24 with modification. Briefly, 1.6 × 105 cells were mixed with SeaPlaque agarose (BioWhittaker) at a final concentration of 0.4% in DMEM and overlaid onto a 0.8% agarose layer in 60-mm plates. Plates were incubated for 14 days, and colonies from 5 microscope fields of each plate were counted under a light microscope at 100× magnification. The average colony number per field from duplicate plates was calculated and expressed as mean ± SEM.
Statistical Analysis
Data were expressed as mean ± SEM. Differences among groups were analyzed by analysis of variance with Tukey-Kramer multiple comparisons test, P < 0.05 is considered significant. Experiments were repeated at least twice with similar results.
RESULTS
NaB Induces Smad3 Expression in Intestinal Epithelial Cells
We used a nontransformed cell line (RIE-1) as an in vitro model for examining TGF-β signaling in the gut since this cell line contains intact TGF-β receptors and Smad proteins. We first examined whether NaB induced Smad2 and Smad3 expression in the gut, since these proteins mediate many of the biologic effects of TGF-β. Results from time-course experiments showed that NaB did not enhance the expression of Smad2 but induced Smad3 expression (Fig. 1A). Densitometric analyses shown in Figure 1B revealed that NaB induced Smad3 at all time points examined with maximum induction of 2.93-fold occurring at 24 hours after treatment. Smad2 protein level showed a 0.3-fold reduction at the early time points and no changes at later time points. Next, we performed dose-curve experiments and found that NaB induced Smad3 protein expression in a dose-dependent fashion after 24 hours of treatment (Fig. 2A). Maximum induction of Smad3 was obtained using a dosage of 5 mmol/L of NaB (Fig. 2B). We then examined whether NaB induced Smad2 and Smad3 mRNA expression by real-time quantitative RT-PCR. NaB did not induce Smad2 mRNA expression (Fig. 3A) but gradually induced Smad3 mRNA expression (Fig. 3B). Maximum induction of Smad3 mRNA occurred at 48 hours after treatment. Smad2 mRNA was suppressed (10%–30%) by NaB treatment. These results paralleled those of Smad2 and Smad3 protein expression (Fig. 1) and suggest that NaB induces only a subset of TGF-β signaling proteins in gut epithelial cells.
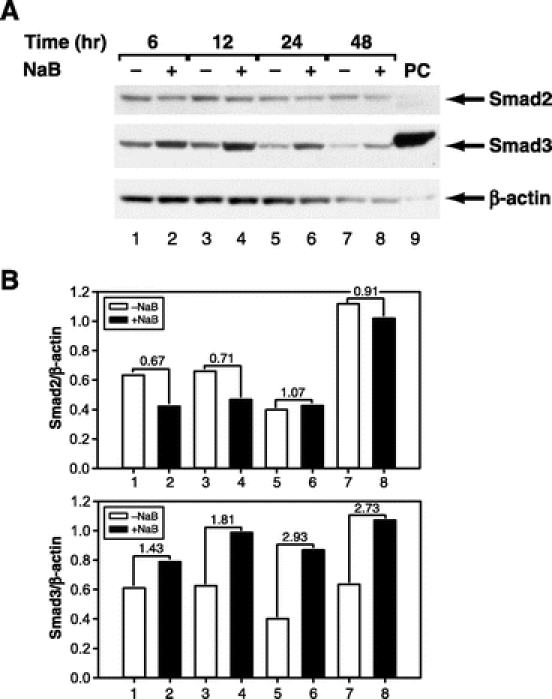
FIGURE 1. NaB induced expression of Smad3 protein in a time-dependent fashion. A, RIE-1 cells were treated without and with NaB (5 mmol/L) for the indicated duration. Whole cell lysates were prepared and Smad protein expression was determined by Western blotting using either anti-Smad2 or Smad3 antibodies. Equal loading of proteins was confirmed by stripping and reprobing the blots with an anti-β-actin antibody. Cell lysates from RIE-1 cells overexpressing human Flag-tagged Smad3 was loaded in lane 9 and served as positive control (PC) for Smad3. B, Densitometric quantification of Smad2 and Smad3 proteins from Western blotting was performed and expressed as ratios of Smad/β-actin. The number between NaB-treated and untreated groups indicates fold change.
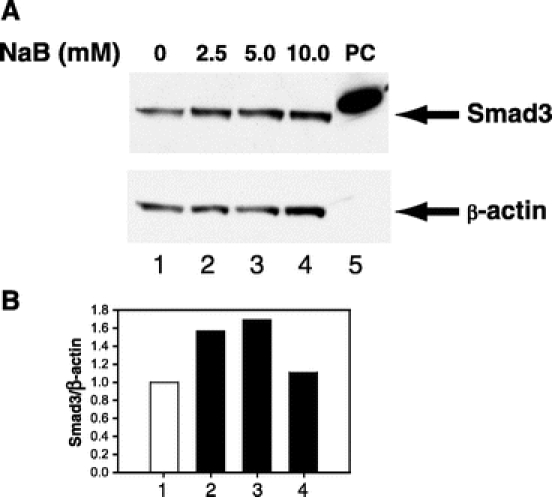
FIGURE 2. NaB induced expression of Smad3 protein in a dose-dependent fashion. A, RIE-1 cells were treated with NaB (0–10 mmol/L) for 24 hours. Whole cell lysates were prepared and Smad3 protein expression was determined by Western blotting using an anti-Smad3 antibody. Equal loading of proteins was confirmed by stripping and reprobing the blot with an anti-β-actin antibody. Cell lysates from RIE-1 cells overexpressing human Flag-tagged Smad3 was loaded in lane 5 and served as positive control (PC) for Smad3. B, Densitometric quantification of Smad3 protein from Western blotting was performed and expressed as ratios of Smad3/β-actin.
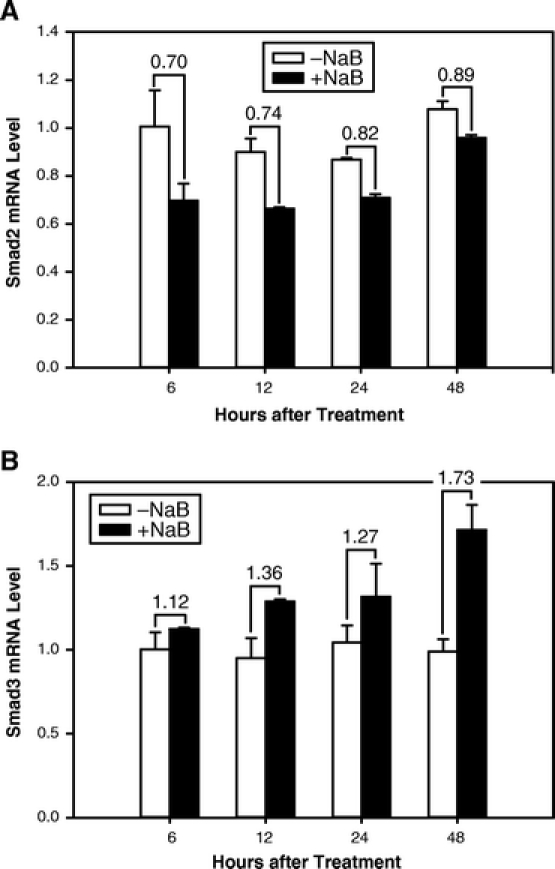
FIGURE 3. NaB induced expression of Smad3 mRNA. RIE-1 cells were treated without and with NaB (5 mmol/L) for the indicated duration. Total RNA was extracted, and mRNA levels of Smad2 (A) and Smad3 (B) were determined by real-time quantitative RT-PCR and normalized to β-actin mRNA levels. The number between NaB-treated and untreated groups indicates fold change.
NaB Induces Smad3 Expression in Human Colon Cancer Cells
NaB has been shown to inhibit growth and stimulate apoptosis in colon cancer cells, suggesting that it has a tumor suppressor activities.25,26 Therefore, we examined whether NaB also stimulated Smad3 expression in 6 human colon cancer cell lines. Treatment with NaB for 24 hours resulted in the induction of Smad3 expression in 4 out of 6 human colon cancer cell lines (HT-29, KM12C, KM12L4A, and KM20) (Fig. 4). These results suggest that NaB stimulates Smad3 expression not only in normal gut epithelial cells, but also in human colon cancer cells.
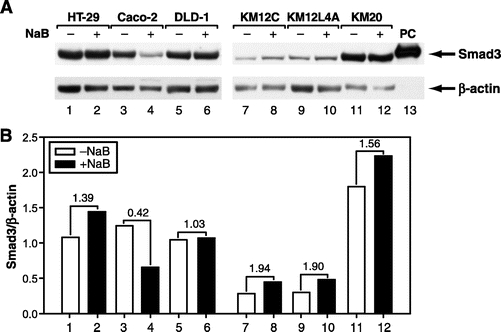
FIGURE 4. NaB induced expression of Smad3 protein in human colon cancer cell lines. A, Six human colon cancer cell lines (HT-29, Caco-2, DLD-1, KM12C, KM12L4A, and KM20) were treated without or with NaB (5 mmol/L) for 24 hours. Whole cell lysates were prepared and Smad3 protein expression was determined by Western blotting using an anti-Smad3 antibody. Equal loading of proteins was confirmed by stripping and reprobing the blot with an anti-β-actin antibody. Cell lysates from RIE-1 cells overexpressing human Flag-tagged Smad3 was loaded in lane 13 and served as positive control (PC) for Smad3. B, Densitometric quantification of Smad3 protein from Western blotting was performed and expressed as ratios of Smad3/β-actin. The number between NaB-treated and untreated groups indicates fold change.
NaB Enhances TGF-β Signaling in Intestinal Epithelial Cells
Smad3 protein is maintained in an unphosphorylated, inactive state in the cytoplasm. However, when TGF-β binds and activates its receptor complex, Smad3 is activated by phosphorylation. Since NaB enhances Smad3 expression, we examined whether NaB also enhanced Smad3 phosphorylation after TGF-β stimulation. As expected, cells treated with TGF-β alone had a 5.4-fold induction in phospho-Smad3 expression (Fig. 5). However, pretreatment with NaB, resulted in a 7-fold increase in TGF-β-induced phospho-Smad3 level compared with untreated control (Fig. 5). These results showed that NaB enhanced TGF-β-induced activation of Smad3.
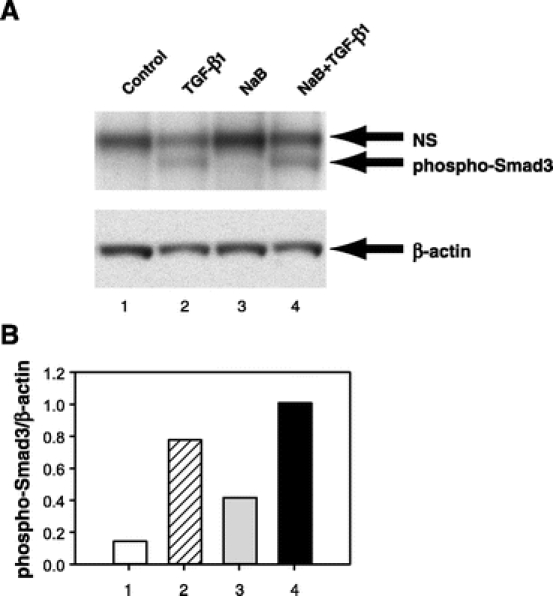
FIGURE 5. NaB enhanced TGF-β-induced Smad3 phosphorylation. A, RIE-1 cells were divided into 4 groups: 1) control group was pretreated with PBS (the dilution buffer for NaB) for 12 hours followed by treatment with vehicle for TGF-β1 (4 mmol/L HCl, 0.1% BSA) for 1 hour, 2) TGF-β1 group was pretreated with PBS followed by treatment with TGF-β1 (40 pmol/L), 3) NaB group was pretreated with NaB (5 mmol/L) followed by treatment with vehicle, and 4) NaB+TGF-β1 group was pretreated with NaB followed by treatment with TGF-β1. Whole cell lysates were prepared and phospho-Smad3 protein expression was determined by Western blotting using an antiphospho-Smad3 antibody. Nonspecific band (NS) was also detected by the antiphospho-Smad3 antibody. Equal loading of proteins was confirmed by stripping and reprobing the blot with an anti-β-actin antibody. B, Densitometric quantification of phospho-Smad3 protein from Western blotting was performed and expressed as ratios of phospho-Smad3/β-actin.
Next, we examined whether NaB enhanced TGF-β-mediated gene expression. We examined the expression of 2 well-characterized target genes of TGF-β, plasminogen activator inhibitor-1 (PAI-1) and cyclooxygenase-2 (COX-2) by real-time quantitative RT-PCR.27,28 PAI-1 is an important regulator of extracellular matrix,29 while COX-2 is the rate-limiting enzyme in the conversion of arachidonic acid to prostaglandins and other eicosanoids. As expected, treatment with TGF-β alone caused a 5-fold induction of PAI-1 mRNA (Fig. 6A). NaB pretreatment also induced PAI-1 mRNA expression to a similar magnitude as TGF-β. Importantly, TGF-β treatment of RIE-1 cells preincubated with NaB resulted in a synergistic induction of PAI-1 mRNA to greater than 25-fold compared with untreated control at both 4 and 12 hours after TGF-β treatment (Fig. 6A). This potentiation effect of NaB and TGF-β on PAI-1 expression was not seen with COX-2 (Fig. 6B). Treatment with TGF-β alone induced COX-2 expression 3- to 4-fold. NaB pretreatment attenuated TGF-β-induced COX-2 expression at 4 hours and had no effect on TGF-β-induced COX-2 expression at 12 hours (Fig. 6B). These results suggest that NaB potentiates TGF-β-induced gene expression only in a subset of target genes.
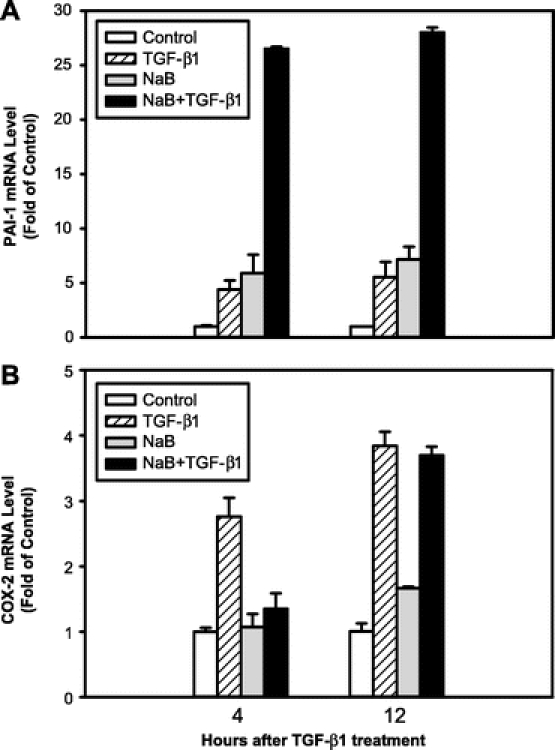
FIGURE 6. NaB potentiated TGF-β-induced PAI-1 mRNA expression. RIE-1 cells were divided into 4 groups: 1) control group was pretreated with PBS (the dilution buffer for NaB) for 12 hours followed by treatment with vehicle for TGF-β1 (0.1% BSA, 4 mmol/L HCl), 2) TGF-β1 group was pretreated with PBS followed by treatment with TGF-β1 (40 pmol/L), 3) NaB group was pretreated with NaB (5 mmol/L) followed by treatment with vehicle, and 4) NaB+TGF-β1 group was pretreated with NaB followed by treatment with TGF-β1. Total RNA was extracted at 4 and 12 hours after TGF-β1 treatment. PAI-1 (A) and COX-2 (B) mRNA levels were determined by real-time quantitative RT-PCR and normalized to β-actin mRNA levels. Results are expressed as fold of induction compared with control group.
NaB and TGF-β Synergistically Inhibit Akt-Mediated Transformation
Since NaB enhances TGF-β signaling and TGF-β is an important tumor suppressor in the gut, we next examined whether NaB enhanced the tumor suppressor activities of TGF-β. The tumor suppressor function of TGF-β has been attributed to its ability to inhibit cell cycle progression and induce apoptosis. However, we were not able to detect any synergistic effects of NaB and TGF-β on cell proliferation and apoptosis in RIE-1 cells (data not shown). One of the key phenotype during carcinogenesis is the ability of tumors cells to grow in the absence of anchorage, and this has been linked to the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt oncogenic pathway. Recently, we have shown that TGF-β inhibits Akt-induced transformation of intestinal epithelial cells (unpublished data). Therefore, we examined whether NaB enhanced the inhibitory effects of TGF-β on Akt-mediated transformation. RIE-1 cells overexpressing myristylated Akt (mAkt), a constitutively active form of Akt, were treated with NaB and TGF-β and plated in soft agar. Anchorage-independent growth was determined by quantifying the number of colonies formed after 14 days of growth. As expected, RIE-1/mAkt cells formed colonies on an average of 11 colonies per microscopic field. Treatment with low dosages of TGF-β or NaB alone did not inhibit colony formation (Fig. 7). However, when RIE-1/mAkt cells were treated with both NaB and TGF-β, a significant inhibition (>85%) of colony formation was observed (Fig. 7). These results showed that NaB and TGF-β synergistically inhibited anchorage-independent growth of mAkt-transformed RIE-1 cells.
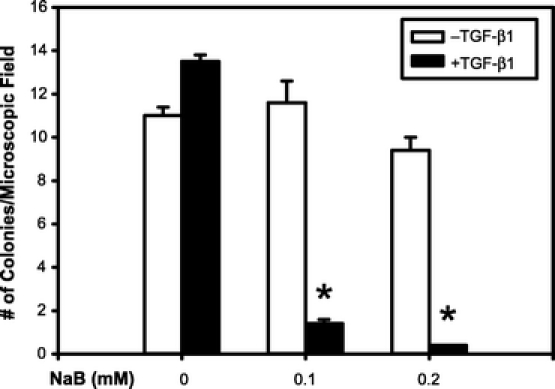
FIGURE 7. NaB and TGF-β synergistically inhibited anchorage-independent growth. RIE-1 cells overexpressed constitutively active, myristylated Akt (RIE-1/mAkt) were treated with TGF-β1 (20 pmol/L), NaB (0.1 or 0.2 mmol/L), and in combinations as indicated. Cells were grown in soft agar for 14 days and colonies formed were quantified under a light microscope at 100× magnification. Results are expressed as mean ± SEM *P < 0.05 compared with the control group that did not receive TGF-β1 or NaB.
DISCUSSION
There appears to be a positive association between high fiber intake and a lower risk of colon tumors and cancers.4–6 The mechanisms by which dietary fiber decreases the risk of colon cancer are poorly characterized. One possible mechanism is through SCFAs produced by bacterial fermentation of dietary fiber. NaB, a major SCFA, induces cell cycle arrest, differentiation, and apoptosis in various colon cancer cell lines.30–32 As evidence indicating the anticancer effects of NaB accumulates, research aiming at defining the mechanisms of NaB action at the cellular level takes a priority. Since NaB has similar biologic activities as TGF-β in regulating gut epithelial cells, we designed studies to answer the question of whether NaB enhances the TGF-β signaling pathway and its tumor suppressor activities. The findings from our study may contribute significantly to the understanding of the relationship between dietary fiber and colon cancer risks. We found that NaB selectively induced the expression of Smad3 (Fig. 1) and enhanced TGF-β-induced activation of Smad3 (Fig. 5) in RIE-1 cells. We also found that NaB induced Smad3 in 4 of the 6 human colon cancer cell lines examined (Fig. 4). Interestingly, NaB did not induce Smad2 expression, which is also a key mediator of TGF-β signal transduction. Previously, we have shown that Smad2 and Smad3 mediate different biologic effects of TGF-β, with Smad3 being essential for TGF-β-induced apoptosis.11 Therefore, our results suggest that only a subset of TGF-β signal is enhanced by NaB in the gut.
TGF-β signaling serves as an important tumor suppressor pathway during colon carcinogenesis. We examined whether NaB enhanced the tumor suppressor activities of TGF-β in gut epithelial cells. NaB did not enhance TGF-β-induced cell cycle arrest or apoptosis in RIE-1 cells (data not shown). One of the key steps during carcinogenesis is the ability of tumors cells to grow in the absence of anchorage and this has been linked to the activation of the PI3K/Akt pathway. In RIE-1 cells transformed by the overexpression of mAkt, low doses of NaB and TGF-β synergistically inhibited anchorage-independent growth (Fig. 7). These results provide the first evidence that NaB may enhance the tumor suppressor function of TGF-β.
Although we have shown that NaB enhances Smad3 expression and TGF-β signaling, the molecular mechanisms by which NaB and TGF-β synergistically induce PAI-1 expression and inhibit anchorage-independent growth are not known. NaB is a direct inhibitor of HDACs leading to increased histone acetylation, modification of chromatin structure, changes in accessibility of transcription factors to DNA and the interaction of transcription factors and their cofactors. These alterations lead to changes in the transcription of many genes and may contribute to the effects of NaB on TGF-β-mediated gene expression. To determine whether the induction of Smad3 by NaB may be due to its ability to inhibit HDACs, we treated RIE-1 cells with another HDAC inhibitor, all-trans retinoic acid (RA). RA induced Smad3 at early time points and to a greater extent than NaB (data not shown), suggesting that the induction of Smad3 in RIE-1 cells may be the result of inhibiting HDACs.
TGF-β has paradoxical effects during colon carcinogenesis, serving both as a tumor suppressor and a tumor promoter.33 TGF-β is overexpressed in colon cancers,34 and high level expression of TGF-β in the primary tumor is associated with advanced stages,35 tumor recurrence,36 and decreased survival.35 TGF-β induces COX-2 expression,28 and COX-2 appears to play an important role during colon carcinogenesis.37 Although NaB potentiated TGF-β-induced PAI-1 gene expression (Fig. 6A), similar effects were not observed with COX-2 (Fig. 6B). Surprisingly, NaB inhibited TGF-β-induced COX-2 expression at the 4-hour time point (Fig. 6B). These results suggest that NaB does not have a broad effect on TGF-β signaling and function but may selectively target a subset of TGF-β signaling and biologic activities such as those related to its tumor suppressor function.
CONCLUSION
Our results demonstrate that NaB selectively induces Smad3 and potentiates TGF-β signaling in gut epithelial cells. Furthermore, low levels of NaB are sufficient to enhance the tumor suppressor function of TGF-β. Our data reveal a novel molecular mechanism that may explain in part the beneficial effects of dietary fiber, especially poorly fermentable fiber such as those from wheat bran, in decreasing the risk of colon cancers.
ACKNOWLEDGMENTS
The authors thank Chunyan Deng, Dr. Weili Zhang, and Dr. Junmei Zhao for technical assistance, and Steve Schuenke and Karen Martin for manuscript preparation.
Discussions
Dr. John B. Hanks (Charlottesville, Virginia): Dr. Ko’s group from Galveston has provided a scholarly and reasoned approach to an exciting possibility, that being a possible mechanism that might suggest a dietary influence on the suppression of colorectal cancer. The mechanism proposed is complex, but this investigation is an elegant set of experiments that takes advantage of the Galveston group’s expertise in signaling pathways that has been led so long and brilliantly by Drs. Courtney Townsend and Jim Thompson. In this study, the group proposes that dietary fiber intake results in increased intraluminal levels of sodium butyrate, which in turn sets off a cascade of events whereby malignant transformation of colonic mucosa is suppressed in cell preparations. I have a couple of questions for the group.
Sodium butyrate enhanced Smad3 but not Smad2 expression. Do you have any thoughts or could you say a few words about the importance of that observation?
The second question is: the TGF beta has important tumor suppression function by producing apoptosis and cell cycle arrest, as you point out. Does the sodium butyrate enhance those effects?
Third, your studies assume that dietary fiber might affect the suppression of tumor growth by the release of sodium butyrate. Do you have any direct evidence that dietary fiber enhances Smad3 production in colonic cell preparations? Said another way, what is the next step in correlating these findings to the clinical circumstances?
Dr. Tien C. Ko (Galveston, Texas): First, you ask about the biological significance of the differential effects of sodium butyrate on Smad3 and Smad2 expression. Both Smad2 and Smad3 are signaling proteins that are phosphorylated and activated by TGF-β receptors after ligand binding and they have been shown to mediate distinct biological effects of TGF-β. For example, we have shown that only Smad3 appears to mediate the apoptotic effects of TGF-β in intestinal epithelial cells. The specific activation of Smad3 by butyrate suggests that only a subset of TGF-β-mediated target genes are induced by butyrate and may explain why butyrate only synergistically enhances TGF-β-induced PAI-1 expression and has no effect on TGF-β-induced COX-2 level.
In response to whether sodium butyrate affects other tumor suppressor activities of TGF-β, we have examined the effects of butyrate on TGF-β-induced cell cycle arrest and apoptosis in RIE-1 cells. Our studies did not demonstrate any enhancement of these biological effects of TGF-β by butyrate, supporting the concept that butyrate only enhances a subset of TGF-β signaling and biological activities.
Lastly, you asked whether we have any direct evidence that dietary fiber enhances Smad3 in the colon and what is our next step? We do not have any direct evidence that dietary fiber enhances Smad3 expression in vivo, but this is our next step. We have begun in vivo studies in mice to examine the effects of high-fiber diets on the expression of Smads and TGF-β-regulated genes in the colon.
Dr. David H. Berger (Houston, Texas): Over the past few decades, there has been a controversy over the role of dietary fiber in the prevention of human colorectal cancer. As Dr. Ko mentioned, three large epidemiologic studies published within the last 3 years would seem to put this controversy to rest. These studies demonstrate a 25% to 40% decreased risk of both polyps and colon cancer in patients who consume high-fiber diets. Dr. Ko and his colleagues are to be commended for their attempt to investigate the molecular events responsible for this important clinical observation.
As Dr. Ko mentioned, highly fermentable fiber sources, such as fruits and vegetables, produce high amounts of short-chain fatty acids, of which butyrate is the most important. Dr. Ko and his colleagues have demonstrated that butyrate interacts with the TGF-β signaling pathway, one of the most important pathways in maintaining normal gut homeostasis. Specifically, Dr. Ko and his colleagues have demonstrated that butyrate upregulates Smad3 and potentiates TGF-β signaling and its tumor suppressor activity in gut epithelial cells.
I have several questions for Dr. Ko.
As you mentioned, butyrate is a potent inhibitor of histone deacetylases, suggesting that its effects are on transcription; however, based on the time course of Smad3 mRNA and protein expression you report in your paper, it appears the effect of butyrate on Smad3 expression is post-transcriptional. Can you tell us the mechanism by which butyrate regulates Smad3?
You demonstrated that butyrate acts synergistically with TGF-β to upregulate PAI-1 in RIE-1 cells. As you know, elevated PAI-1 has been demonstrated to be a poor prognostic indicator in colorectal cancer. How do you explain the effect you saw on PAI-1 with the potential preventive effects of butyrate?
You demonstrated very nicely that sodium butyrate in combination with TGF-β suppresses tumorigenesis induced by the Akt pathway. Does this combination affect other molecular mechanisms of tumor formation through other signaling pathways?
As you know, Dr. Ko, as colorectal cancers progress there is a change in the response of TGF-β. Early on, TGF-β is a tumor suppressor, as you mentioned. However, eventually TGF-β can act as a tumor promoter. Is the synergism between butyrate and TGF-β only seen in the tumor suppressor activity of TGF-β or is it possible in advanced cancer that butyrate potentiates the tumor promoting effects of TGF-β? If butyrate indeed potentiates the harmful effects of TGF-β in advanced cancer, should we be placing those patients on low fiber diets?
Dr. Tien C. Ko (Galveston, Texas): First, you ask about the mechanism by which butyrate regulates Smad3. Although our experiments do not address the mechanism by which butyrate regulates Smad3, our time-course studies on Smad3 mRNA and protein results do suggest that butyrate regulation of Smad3 is complex. Sodium butyrate is a well-known inhibitor of histone deacetylases and has been shown to regulate the transcription of many genes, some as early as 30 minutes after sodium butyrate treatment. One possible explanation for the upregulation of Smad3 protein expression is due to an increase to protein stability secondary to the downregulation of the protein degradation pathway by butyrate. Other mechanisms may involve the regulation of Smad3 gene transcription by butyrate.
Your second question regarding the induction of PAI-1 and its implication on the chemo-preventive effects of butyrate. PAI-1 regulates the plasmin/plasminogen system by inhibiting urokinase plasminogen activator. Although PAI-1 elevation has been associated with worse prognosis in colon cancer, the molecular mechanism by which PAI-1 enhances carcinogenesis is much more complicated. It now appears that carcinogenesis is linked to the ratio of activators and inhibitors of the plasmin/plasminogen system and not just simply the level of PAI-1. Therefore, we are in the process of examining the effects of butyrate on urokinase plasminogen activator and tissue plasminogen activator levels in gut epithelial cells. It may very well be that the ratio of activators and inhibitors of plasmin/plasminogen system is altered by butyrate to favor the prevention of tumor development.
You asked whether we have examined the effects of butyrate and TGF-β on other signaling pathways. The Ras oncogenic pathway has been shown to be important in colorectal carcinogenesis. We are in the process of determining whether butyrate and TGF-β also synergistically inhibit Ras-mediated transformation in RIE-1 cells.
Finally, you asked whether the synergism between butyrate and TGF-β is also seen in the tumor-promoting effects of TGF-β. Our study has focused primarily on the tumor suppressor activities of TGF-β; however, we did examine the effects of butyrate on TGF-β-induced COX-2 expression, which is linked to the tumor promoting effects of TGF-β. In our study, butyrate did not enhance, butrather inhibited, TGF-β-induced COX-2 expression. Therefore, it appears that butyrate selectively enhances the tumor suppressor function of TGF-β and may inhibit the tumor-promoting effect of TGF-β. From these results, our study supports putting patients with colon cancer on low-fiber diets.
Footnotes
Supported by the National Institutes of Health (grant nos. R01 DK60105 and P01 DK35608).
Reprints: Tien C. Ko, MD, Department of Surgery, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555-0737. E-mail: tko@utmb.edu.
REFERENCES
- 1.Greenlee RT, Murray T, Bolden S, et al. Cancer statistics. CA Cancer J Clin. 2000;50:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Seifeldin R, Hantsch JJ. The economic burden associated with colon cancer in the United States. Clin Ther. 1999;21:1370–1379. [DOI] [PubMed] [Google Scholar]
- 3.Burkitt DP. Related disease–related cause? Lancet. 1969;2:1229–1231. [DOI] [PubMed] [Google Scholar]
- 4.Peters U, Sinha R, Chatterjee N, et al. Dietary fibre and colorectal adenoma in a colorectal cancer early detection programme. Lancet. 2003;361:1491–1495. [DOI] [PubMed] [Google Scholar]
- 5.Bingham SA, Day NE, Luben R, et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361:1496–1501. [DOI] [PubMed] [Google Scholar]
- 6.Larsson SC, Giovannucci E, Bergkvist L, et al. Whole grain consumption and risk of colorectal cancer: a population-based cohort of 60,000 women. Br J Cancer. 2005;92:1803–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Argenio G, Cosenza V, Delle CM, et al. Butyrate enemas in experimental colitis and protection against large bowel cancer in a rat model. Gastroenterology. 1996;110:1727–1734. [DOI] [PubMed] [Google Scholar]
- 8.Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol Cell Biochem. 1982;42:65–82. [DOI] [PubMed] [Google Scholar]
- 9.Augeron C, Laboisse CL. Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res. 1984;44:3961–3969. [PubMed] [Google Scholar]
- 10.Ko TC, Sheng HM, Reisman D, et al. Transforming growth factor-beta 1 inhibits cyclin D1 expression in intestinal epithelial cells. Oncogene. 1995;10:177–184. [PubMed] [Google Scholar]
- 11.Conery AR, Cao Y, Thompson EA, et al. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–372. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Han W, Zborowska E, et al. Reduced expression of transforming growth factor beta type I receptor contributes to the malignancy of human colon carcinoma cells. J Biol Chem. 1996;271:17366–17371. [DOI] [PubMed] [Google Scholar]
- 13.Matsushita M, Matsuzaki K, Date M, et al. Down-regulation of TGF-beta receptors in human colorectal cancer: implications for cancer development. Br J Cancer. 1999;80:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riggins GJ, Thiagalingam S, Rozenblum E, et al. Mad-related genes in the human. Nat Genet. 1996;13:347–349. [DOI] [PubMed] [Google Scholar]
- 15.Eppert K, Scherer SW, Ozcelik H, et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. [DOI] [PubMed] [Google Scholar]
- 16.Thiagalingam S, Lengauer C, Leach FS, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–346. [DOI] [PubMed] [Google Scholar]
- 17.Takagi Y, Kohmura H, Futamura M, et al. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996;111:1369–1372. [DOI] [PubMed] [Google Scholar]
- 18.MacGrogan D, Pegram M, Slamon D, et al. Comparative mutational analysis of DPC4 (Smad4) in prostatic and colorectal carcinomas. Oncogene. 1997;15:1111–1114. [DOI] [PubMed] [Google Scholar]
- 19.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 1999;9:274–279. [DOI] [PubMed] [Google Scholar]
- 21.Quaroni A, Wands J, Trelstad RL, et al. Epithelioid cell cultures from rat small intestine: characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heid CA, Stevens J, Livak KJ, et al. Real time quantitative PCR. Genome Res. 1996;6:986–994. [DOI] [PubMed] [Google Scholar]
- 23.Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol. 2002;76:6213–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheng H, Shao J, DuBois RN. Akt/PKB activity is required for Ha-Ras-mediated transformation of intestinal epithelial cells. J Biol Chem. 2001;276:14498–14504. [DOI] [PubMed] [Google Scholar]
- 25.McBain JA, Eastman A, Nobel CS, et al. Apoptotic death in adenocarcinoma cell lines induced by butyrate and other histone deacetylase inhibitors. Biochem Pharmacol. 1997;53:1357–1368. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Friedman EA. Short-chain fatty acids induce cell cycle inhibitors in colonocytes. Gastroenterology. 1998;114:940–946. [DOI] [PubMed] [Google Scholar]
- 27.Slivka SR, Loskutoff DJ. Platelets stimulate endothelial cells to synthesize type 1 plasminogen activator inhibitor: evaluation of the role of transforming growth factor beta. Blood. 1991;77:1013–1019. [PubMed] [Google Scholar]
- 28.Sheng H, Shao J, Hooton EB, et al. Cyclooxygenase-2 induction and transforming growth factor beta growth inhibition in rat intestinal epithelial cells. Cell Growth Differ. 1997;8:463–470. [PubMed] [Google Scholar]
- 29.Berger DH. Plasmin/plasminogen system in colorectal cancer. World J Surg. 2002;26:767–771. [DOI] [PubMed] [Google Scholar]
- 30.Hague A, Manning AM, Hanlon KA, et al. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer. 1993;55:498–505. [DOI] [PubMed] [Google Scholar]
- 31.Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288–3293. [PubMed] [Google Scholar]
- 32.Litvak DA, Evers BM, Hwang KO, et al. Butyrate-induced differentiation of Caco-2 cells is associated with apoptosis and early induction of p21Waf1/Cip1 and p27Kip1. Surgery. 1998;124:161–169. [PubMed] [Google Scholar]
- 33.Roman C, Saha D, Beauchamp RD. TGF-beta and colorectal carcinogenesis. Microsc Res Tech. 2001;52:450–457. [DOI] [PubMed] [Google Scholar]
- 34.Derynck R, Goeddel DV, Ullrich A, et al. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987;47:707–712. [PubMed] [Google Scholar]
- 35.Robson H, Anderson E, James RD, et al. Transforming growth factor beta 1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. Br J Cancer. 1996;74:753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman E, Gold LI, Klimstra D, et al. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol Biomarkers Prev. 1995;4:549–554. [PubMed] [Google Scholar]
- 37.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. [DOI] [PubMed] [Google Scholar]