

Abstract
Objective:
To examine the mechanism by which androstenediol improves cardiac function following trauma-hemorrhage (T-H).
Summary Background Data:
Androstenediol administration improves cardiovascular function and attenuates proinflammatory cytokine production following T-H. Activation of the peroxisome proliferator-activated receptor-gamma (PPAR-γ) has been shown to be protective following ischemic conditions. We hypothesized that PPAR-γ activation plays a role in the androstenediol-mediated salutary effects on cardiac function following T-H.
Methods:
Male rats underwent laparotomy and hemorrhagic shock (40 mm Hg for 90 minutes), followed by resuscitation with 4 times the shed blood volume in the form of Ringer's lactate. Androstenediol (1 mg/kg body weight, i.v.) was administrated at the end of resuscitation. In a separate group of animals, a PPAR-γ antagonist (GW9662) was administered simultaneously with androstenediol and animals were killed at 5 hours thereafter.
Results:
A decrease in cardiac function and an increase in IL-6 and iNOS gene expression were observed following T-H. Androstenediol treatment normalized cardiac function, increased PPAR-γ DNA binding activity, attenuated IL-6 and iNOS gene expressions, and reduced plasma IL-6. Plasma 15-deoxy-Δ12, 14-prostaglandin J2 (PGJ2, an endogenous PPAR-γ agonist) levels were also increased in androstenediol-treated T-H rats, but these levels were lower than those observed in shams. Coadministration of PPAR-γ antagonist along with androstenediol, however, prevented the androstenediol-mediated reduction in cardiac iNOS and IL-6 expressions and abolished the improvements in cardiac function.
Conclusion:
The androstenediol-mediated salutary effects on cardiac function following T-H appear to be mediated at least in part via PPAR-γ activation, which down-regulates IL-6 and iNOS gene expression in the heart.
Although androstenediol (Adiol, a metabolite of dehydroepiandrosterone) improves cardiovascular function and attenuates proinflammatory cytokine production following trauma-hemorrhage, its mechanism of action remains unknown. The present study indicates that the Adiol-mediated salutary effects on cardiac function following trauma-hemorrhage are mediated via PPAR-γ-dependent down-regulation of cardiac IL-6 and iNOS mRNA expression.
Hemorrhagic shock results in a rapid decrease in cardiac output and organ blood flow.1 The depressed organ perfusion and excessive production of pro-inflammatory mediators play an important role in the development of multiple organ dysfunction following hemorrhagic shock.2,3 It has been reported that systemic IL-6 levels increase following trauma-hemorrhage (T-H), and a sustained elevation in plasma IL-6 levels is correlated with the evolving organ dysfunction.4–6 Clinical data also confirmed that the increased IL-6 production is associated with poor outcome in patients with trauma or hemorrhagic shock.7 IL-6 has been described as a multifunctional cytokine, produced during the acute phase by macrophages, T and B cells, and nonimmune cells such as myocytes or endothelial cells.8–10
Studies have demonstrated that the IL-6-mediated elevation in nitric oxide (NO) production is likely to be one of the detrimental effects induced by the IL-6 in the heart.11,12 Yu et al demonstrated that IL-6 decreases cardiac contractility and enhances the synthesis of the inducible nitric oxide synthase (iNOS) resulting in an excess of NO production.11 In line with these findings, previous studies from our laboratory have shown that cardiac dysfunction was associated with elevated plasma IL-6 levels.6,13–15 Furthermore, our recent study suggested that the depressed cardiac function following T-H is associated with an increase in cardiomyocyte IL-6 production.16 The causal role of IL-6 in producing organ dysfunction has been further substantiated by studies which showed that administration of anti-IL-6 antibodies results in improved organ function following T-H.17
The peroxisome proliferator-activated receptor-gamma (PPAR-γ), a member of the nuclear hormone receptor superfamily, was originally reported to have a role in adipocyte differentiation and glucose homeostasis. However, subsequent studies revealed that PPAR-γ is a potent regulator of genes implicated in inflammatory responses.18 PPAR-γ activators can exert anti-inflammatory activity in a wide variety of cell types such as monocyte/macrophages, endothelial cells, epithelial cells, and smooth muscle cells.19 In agreement with these results, recent studies have reported that PPAR-γ agonists can reduce organ injury in hemorrhagic shock20,21 and attenuate the inflammatory response in sepsis.22 Additional studies have demonstrated that ligands of the PPAR-γ also have protective effects on heart against ischemia/reperfusion injury via reduction of pro-inflammatory factors as TNF-α, MCP-1, or iNOS.23,24
Androstenediol (adiol or 5-androstene-3β,17β-diol), which is one of the metabolites of dehydroepiandrosterone (DHEA), was shown to have greater protective capacity than DHEA in lethal bacterial infections and endotoxin shock.25 Following T-H shock, androstenediol administration also proved to be effective in reducing plasma IL-6 production and improving cardiovascular function and splanchnic perfusion.26 Since androstenediol has been reported to influence the activity of a peroxisome proliferator,27 we hypothesized that the salutary effects of androstenediol on cardiac functions following T-H are mediated via PPAR-γ activation. To test the hypothesis, we first determined the effect of androstenediol administration on expression and PPAR-γ DNA binding activity and measured plasma level of its endogenous agonist, 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2) following T-H. In a second set of experiments, we used the PPAR-γ antagonist GW9662 along with androstenediol to determine the effects of PPAR-γ blockade on androstenediol-induced effects on cardiac functions following T-H.
METHODS
Animals
Adult male (275–325 g) Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were used in this study. All experiments were performed in adherence to the National Institutes of Health guidelines for the use of experimental animals and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Experimental Procedures
A nonheparinized model of T-H as previously described, was used in this study.6 Briefly, male Sprague-Dawley rats were fasted overnight before the experiment, but allowed water ad libitum. Animals were anesthetized using isoflurane (Attane, Minrad Inc., Bethlehem, PA) inhalation following, which a 5-cm midline laparotomy was performed to induce soft-tissue trauma. The abdomen was closed in layers and catheters were placed in both femoral arteries and the right femoral vein (polyethylene (PE-50) tubing; Becton-Dickinson, Sparks, MD). The animals were restrained in a supine position and the wounds bathed with 1% lidocaine (Elkins-Sinn, Cherry Hill, NJ) throughout the surgical procedure to minimize postoperative pain. The rats were then allowed to awaken following which they were bled rapidly to a mean arterial pressure (MAP) of 35 to 40 mm Hg within 10 minutes. The time at which the animals could no longer maintain a MAP of 35 to 40 mm Hg without infusing some fluid defined as maximum bleed-out volume. The rats were maintained at this MAP until 40% of the shed blood was returned in the form of Ringer's lactate. The animals were then resuscitated with 4 times the shed blood volume with Ringer's lactate over 60 minutes, the catheters were removed, the vessels ligated and the skin incisions closed with sutures. Sham-operated animals underwent the same surgical procedure, which included the ligation of the femoral arteries and a femoral vein; however, neither hemorrhage nor resuscitation was carried out. The animals were returned to their cages, allowed food and water ad libitum, and killed at 5 hours after the end of resuscitation to obtain blood and tissue samples.
Experimental Groups
Androstenediol (1 mg/kg BW, Steraloids, Inc., Newport, RI) was administered intravenously at the end of the resuscitation. The same volume of vehicle (Intralipid, 1 mL/kg BW, Sigma, St. Louis, MO) was administered in the nontreated group rats. To block PPAR-γ activity, a separate group of animals was treated with PPAR-γ antagonist, GW9662 (1 mg/kg BW, Sigma) intraperitoneally at the beginning of resuscitation with or without androstenediol administration. The dose of GW9622 used was according to the previously published studies.21,22
Measurement of Heart Performance and Cardiac Output
Cardiac output (CO) and heart performance were evaluated at 5 hours after resuscitation or sham operation. CO was determined by using an indocyanine green (ICG) dilution technique as previously described.6 Briefly, the right jugular vein was isolated and cannulated by using polyethylene-50 tubing under pentobarbital sodium (∼30 mg/kg BW). A 2.4-Fr fiberoptic catheter (Hospex Fiberoptics, Chestnut Hill, MA) was inserted into the right carotid artery and placed at the level of the aortic arch for continuous measurement of ICG concentration with an in vivo hemoreflectometer (Schwarzer-Picker International, Munich, Germany). ICG (50 μL, 1 mg/mL) was injected via the right jugular vein catheter. The area under the curve was determined, and CO calculated according to the principle of dye dilution. To measure left ventricular heart performance, a PE-50 catheter was inserted into the left ventricle via right carotid artery. The maximal rates of left ventricular pressure increase (+dp/dtmax) and decrease (−dp/dtmax) were determined with a heart performance analyzer (Digi-Med).
Measurement of Plasma Levels of PGJ2 and IL-6
Five hours after either the completion of resuscitation or sham operation, blood samples were obtained from cardiac puncture and placed in microcentrifuge tubes, and plasma was separated by centrifugation, immediately frozen and stored at −80°C until assayed. Plasma levels of PGJ2 (Assay Designs Inc., Ann Arbor, MI) and IL-6 (Pharmingen, San Diego, CA) were measured using a commercially available ELISA kit according to the manufacturers′ instructions.
Gene Expression in the Heart
Gene expressions of IL-6, iNOS, and PPAR-γ in the heart were determined by quantitative real-time PCR as previously described.16 Total RNA was isolated from left ventricular samples using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The cDNA was generated from the total RNA samples by using TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA). Each real-time PCR reaction was performed in a mix of 10 μL reaction mixture containing 20 ng of cDNA, 2× PCR Master Mix (Applied Biosystems), and each probe and primer set. TaqMan Gene Expression Assays (Applied Biosystems) for IL-6, iNOS, and PPAR-γ were purchased as probe and primer sets. The reaction mixture was denatured for 1 cycle of 2 minutes at 50°C, 10 minutes at 95°C and incubated for 40 cycles (denaturing for 15 seconds at 95°C and annealing and extending for 1 minute at 60°C) using ABI Prism 7900HT (Applied Biosystems). All samples were tested in triplicate, and average values were used for quantification. 18S rRNA was used as an endogenous control. The comparative CT method (ΔΔCT, cycle threshold) was used for quantification of gene expression. Analysis was performed using SDS v2.2 software (Applied Biosystems) according to the manufacturer's instruction.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts in whole heart samples were prepared as previously described.28 DNA binding activity of PPAR-γ was determined using EMSA technique. Oligonucleotide probes corresponding to PPAR-responsive elements (PPRE) consensus sequence (5′-GATGCTGTACAGGATGTTCTAGCTAGA-3′) were labeled with γ-32P-ATP (≥6000 Ci/mmoL, Amersham, Piscataway, NJ) using T4 polynucleotide kinase (NewEngland Biolab, Beverly, MA) according to the manufacturer's instructions. The labeled oligonucleotides were purified in Bio-Spin chromatography columns (Amersham). Each 20-μL assay contained 20 ng of nuclear extracts, ≥20,000 cpm of radiolabeled double-stranded target oligonucleotide, poly(dI-dC) and incubation buffer (Promega, Madison, WI). For the supershift assay, antibodies specific to PPAR-γ were added to the reaction mixture and incubated for an additional 20 minutes. After a 30-minute incubation period at room temperature, each of the samples was loaded onto a 6% DNA retardation gel (Novex, Carlsbad, CA). The gels were run at 10 to 15 mA for 90 minutes at 4°C in Tris-borate-EDTA buffer. Following electrophoresis, band intensities were quantified using autoradiography. Signal densities were evaluated by ChemiImager 5500 software (Alpha Innotech, San Leandro, CA).
Western Blot Analysis
Western blot analysis was performed to determine PPAR-γ protein levels of the above-mentioned nuclear extracts.29 Electrophoresis was performed on precasted gels (Nupage, Bis-Tris; Invitrogen). Proteins from the gel were transferred to nitrocellulose membranes at 30 V for 60 minutes. Primary anti-PPAR-γ antibody (1:1000, Santa Cruz, CA) and the horseradish peroxidase-conjugated secondary antibody (1:1500, Stressgen, Victoria, BC, Canada) were incubated overnight at 4°C or for 1 hour at room temperature, respectively. Blots were immersed for 5 minutes in SuperSignal West Pico (Pierce Biotechnologies, Rockford, IL) detection reagent and then exposed to film. Membranes were then stripped and reblotted with anti-GAPDH antibody (1:5000, Abcam Inc., Cambridge, MA) to confirm equal protein loading in each lane. Protein levels were determined by densitometric evaluation and were normalized according to the corresponding GAPDH densities. Signals were quantified using ChemiImager 5500 software (Alpha Innotech).
Statistical Analysis
Data are presented as mean ± SEM. Statistical differences between groups were calculated by one-way analysis of variance, followed by Student-Newman Keuls method as a post hoc test. Differences were considered significant if P < 0.05.
RESULTS
Effect of Androstenediol Administration on Sham and Trauma-Hemorrhaged Animals
Effect of Androstenediol Administration on Cardiac Function
In sham-operated animals, no significant difference in cardiac function was observed between animals receiving vehicle and those receiving androstenediol. A decrease in cardiac output (Fig. 1A) as well as in the absolute dp/dtmax values (Fig. 1B, C) was observed in the vehicle-treated animals at 5 hours after T-H. Androstenediol administration normalized cardiac output (Fig. 1A), attenuated the decrease in +dp/dtmax (Fig. 1B), and induced a small but insignificant increase in −dp/dtmax values at 5 hours following T-H (Fig. 1C).
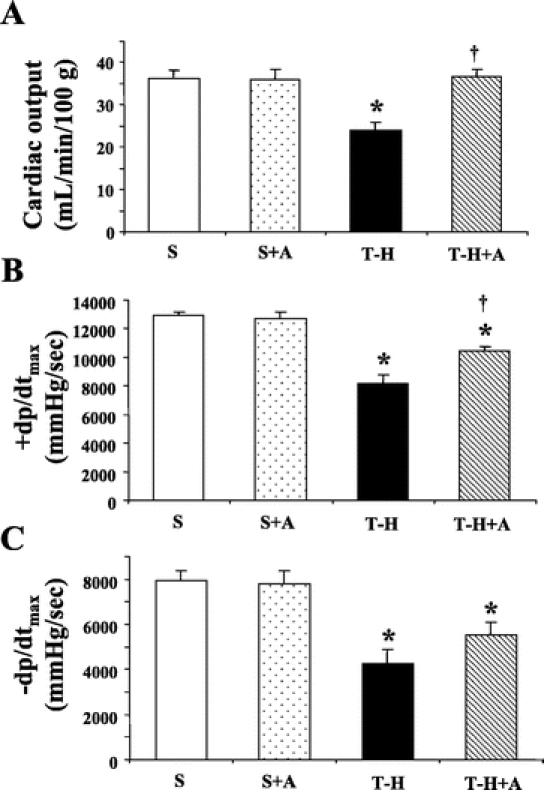
FIGURE 1. Effects of androstenediol on cardiac function at 5 hours after sham operation or trauma-hemorrhage (T-H). A, Cardiac output. B, +dp/dtmax. C, −dp/dtmax. Data are mean ± SEM (n = 6 animals/group). *P < 0.05 versus corresponding sham group. †P < 0.05 versus T-H. S, sham; A, androstenediol.
Effects of Androstenediol Administration on PPAR-γ Gene Expression and Protein Level in the Heart
The PPAR-γ gene expression value showed a small but insignificant increase in the hearts of androstenediol-treated sham animals. Similarly, there was no significant increase in PPAR-γ gene expression in heart at 5 hours following T-H in vehicle-treated animals (Fig. 2A). Androstenediol administration following resuscitation, however, significantly increased PPAR-γ gene expression compared with all other groups. In agreement with the gene expression data, PPAR-γ protein levels in the nuclear extracts prepared from heart samples were significantly increased in trauma-hemorrhaged animals treated with androstenediol compared with all other groups (Fig. 2B, C).
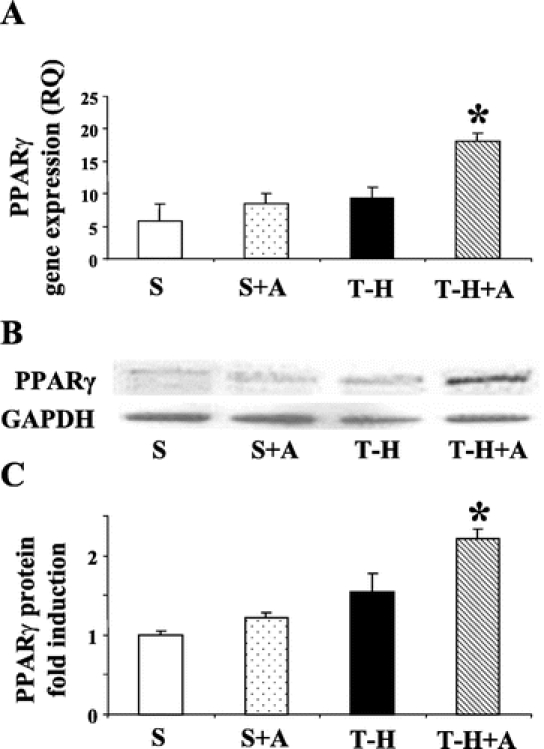
FIGURE 2. Effects of androstenediol on PPAR-γ gene and protein expression in heart at 5 hours after sham operation or trauma-hemorrhage (T-H). A, PPAR-γ gene expression in heart. B, PPAR-γ protein expression in cardiac nuclear extract. C, Folds induction of PPAR-γ protein expression in cardiac nuclear extract compared with sham. Data are mean ± SEM (n = 3–5 animals/group). *P < 0.05 versus the other groups. S, sham; A, androstenediol; RQ, relative quantity.
Effects of Androstenediol Administration on DNA Binding Activity of PPAR-γ in the Heart
To determine PPAR-γ activation, DNA binding activity to PPAR response elements (PPRE) in cardiac nuclear extract was evaluated. As shown in Figure 3, PPAR-γ DNA binding activity to PPRE was not influenced by T-H (lanes 4 and 5) compared with sham (lane 1) at 5 hours following resuscitation. Androstenediol administration, however, significantly increased DNA binding activity to PPRE in both sham (lane 2) and trauma-hemorrhaged animals (lanes 6 and 7) compared with vehicle-treated groups (Fig. 3). In addition, we performed competitive and supershift assays to determine the specificity of this binding activity. As shown in lane 8 of Figure 3A, DNA-binding activity was completely abolished in the presence of specific oligonucleotide cold probe (lane 8). The incubation with anti-PPAR-γ specific antibody also abolished PPAR/PPRE complex (lane 9) (Fig. 3A).
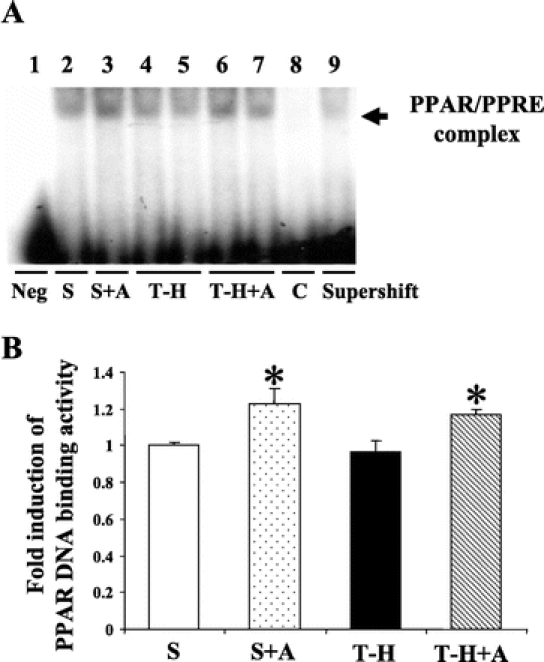
FIGURE 3. Effects of androstenediol on PPAR-γ DNA binding activity in heart at 5 hours after sham operation or trauma-hemorrhage (T-H). A, PPAR-γ DNA binding activity to PPAR response elements (PPRE). Lane 1, negative (Neg., probe only). Lane 2, sham. Lane 3, sham + androstenediol (S+A). Lanes 4 and 5, T-H. Lanes 6 and 7, T-H + androstenediol (T-H+A). Lane 8, competitive assay (C) using 100 times higher concentration of excessive probe. Lane 9, supershift assay of PPAR-γ. B, The index of PPAR-γ DNA binding activity to PPRE. Data are mean ± SEM (n = 3–5 animals/group): *P < 0.05 versus sham and T-H. S, sham; A, androstenediol.
Effects of Androstenediol Administration on Plasma PGJ2 Levels
As shown in Figure 4, androstenediol administration did not influence plasma levels of PGJ2 in sham groups. T-H markedly reduced plasma PGJ2 in vehicle-treated T-H animals at 5 hours after resuscitation. The decrease in plasma PGJ2 was significantly attenuated by androstenediol administration; nevertheless, plasma PGJ2 level remained significantly lower compared with shams.
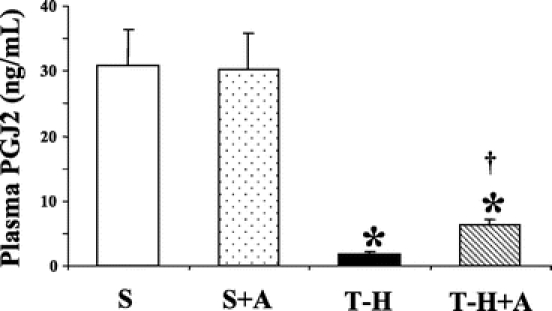
FIGURE 4. Effects of androstenediol on 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2) at 5 hours after sham operation or trauma-hemorrhage (T-H). Data are mean ± SEM (n = 6 animals/group). *P < 0.05 versus sham. †P < 0.05 versus T-H. S, sham; A, androstenediol.
Effects of PPAR-γ Inhibition on Androstenediol-Mediated Effects
Effects of PPAR-γ Inhibition on Cardiac Performance
To evaluate whether the salutary effects of androstenediol on cardiac function mediated via PPAR-γ, a separate group of sham and T-H animals were administered with PPAR-γ inhibitor GW9662 along with androstenediol. Cardiac performance was determined 5 hours after resuscitation. The results indicate that the salutary effects of androstenediol administration following resuscitation on cardiac output (Fig. 5A), +dp/dtmax (Fig. 5B), and −dp/dtmax (Fig. 5C) were significantly prevented by coadministration of PPAR-γ antagonist GW9662 following T-H.
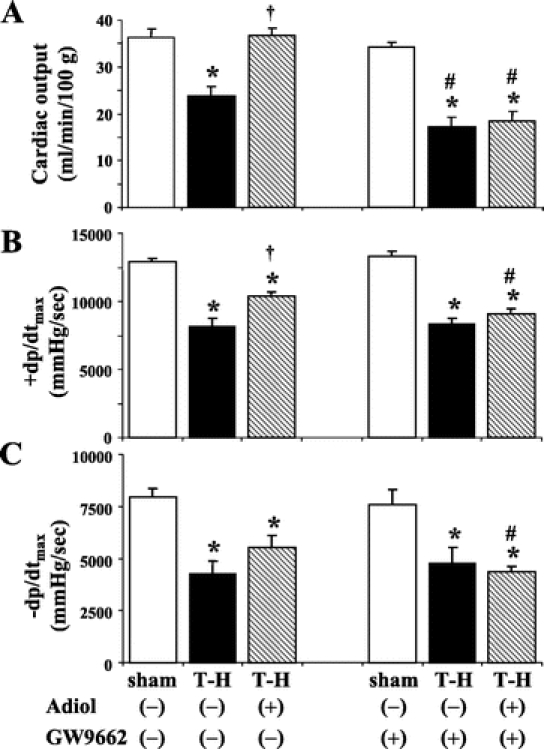
FIGURE 5. Effects of PPAR-γ antagonist on androstenediol-mediated restoration of cardiac function at 5 hours after sham operation or trauma-hemorrhage (T-H). A, Cardiac output. B, +dp/dtmax. C, −dp/dtmax. Data are mean ± SEM (n = 6 animals/group). *P < 0.05 versus corresponding sham group. †P < 0.05 versus corresponding T-H. #P < 0.05 versus corresponding group without GW9662. Adiol, androstenediol; GW9662, PPAR-γ antagonist.
Effects of PPAR-γ Inhibition on Cardiac Gene Expression
Following T-H, IL-6 and iNOS gene expressions were increased in the heart. Androstenediol administration following resuscitation decreased these pro-inflammatory gene expressions, but IL-6 mRNA level remained higher than shams. Administration of GW9662 along with androstenediol prevented the androstenediol-induced attenuation of IL-6 and iNOS gene expressions (Fig. 6).
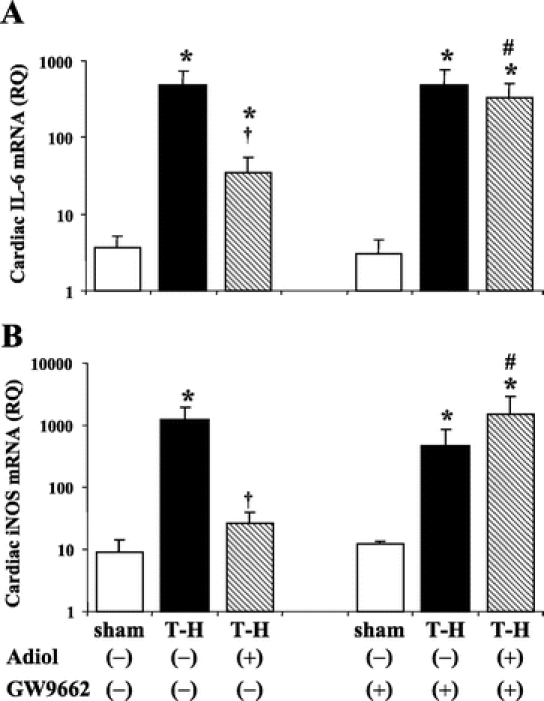
FIGURE 6. Effects of PPAR-γ antagonist on androstenediol-mediated alterations in cardiac gene expression of IL-6 (A) and iNOS (B) at 5 hours after sham operation or trauma-hemorrhage (T-H). Data are mean ± SEM (n = 3–5 animals/group). *P < 0.05 versus corresponding sham group. #P < 0.05 versus corresponding group without GW9662. †P < 0.05 versus vehicle-treated T-H. Adiol, androstenediol; GW9662, PPAR-γ antagonist; RQ, relative quantity.
Effects of PPAR-γ Inhibition on Plasma IL-6 and PGJ2
T-H induced a significant elevation in plasma IL-6 (Fig. 7A) and a decrease in PGJ2 (Fig. 7B) levels. Administration of androstenediol following resuscitation completely blocked the increase in plasma IL-6. PGJ2 levels also improved in T-H animals treated with androstenediol; however, these levels remained significantly lower than shams. Coadministration of PPAR-γ inhibitor GW9662 prevented the androstenediol-induced attenuation of plasma IL-6 elevation (Fig. 7A). Furthermore, GW9662 did not influence PGJ2 levels in androstenediol-treated trauma-hemorrhaged rats (Fig. 7B).
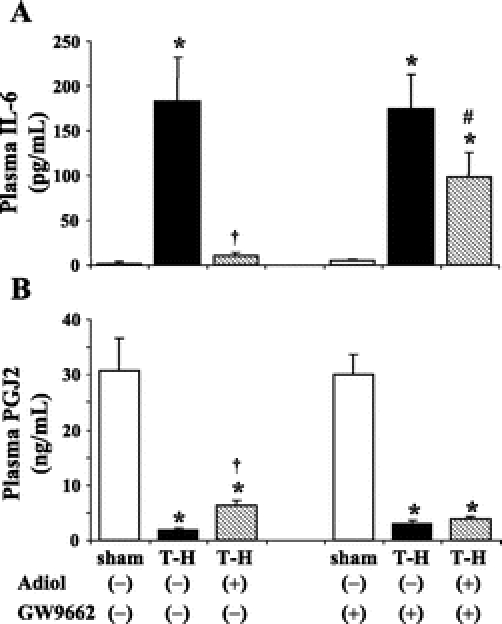
FIGURE 7. Effects of PPAR-γ antagonist on androstenediol-mediated alterations in plasma IL-6 (A) and PGJ2 (B) at 5 hours after sham operation or trauma-hemorrhage (T-H). Data are mean ± SEM (n = 6 animals/group). *P < 0.05 versus corresponding sham group. †P < 0.05 versus vehicle-treated T-H. #P < 0.05 versus corresponding group without GW9662. Adiol, androstenediol; GW9662, PPAR-γ antagonist.
DISCUSSION
The present study demonstrates that androstenediol administration following T-H and resuscitation increases cardiac PPAR-γ DNA binding activity. This is accompanied by improvement in left ventricular performance and decrease in cardiac IL-6 and iNOS gene expressions following T-H. Blockade of PPAR-γ activation by cotreating animals with PPAR-γ inhibitor GW9662, however, prevented the androstenediol-induced improvement in cardiac function and the pro-inflammatory gene expression in the heart. These findings therefore suggest that the salutary effects of androstenediol on cardiac function following T-H are mediated at least in part via an enhanced PPAR-γ activity in the heart.
Although our previous studies have shown that androstenediol administration following T-H decreased hepatic iNOS protein expression and plasma IL-6 levels,26,29 the precise mechanisms responsible for these anti-inflammatory effects of androstenediol remains unknown. PPAR-γ, which is a member of the nuclear hormone receptor superfamily, was originally reported to play a role in adipocyte differentiation and glucose homeostasis.18 However, later subsequent studies showed that PPAR-γ also plays a role in inflammatory responses.19 PPAR-γ have been reported to control the expression of genes implicated in the inflammatory response since it negatively regulates the different inflammatory pathways, such as nuclear factor kappa B (NFκB) and activator protein 1 (AP-1).18,22 A number of studies have shown that PPAR-γ ligands, such as troglitazone or PGJ2, have protective effects against organ injury and inflammatory response following either hemorrhagic or septic shock.18,20,22,30 Zingarelli et al demonstrated that administration of PPAR-γ ligands improves pro-inflammatory conditions in a cecal ligation and puncture model of sepsis.22 They also showed that PPAR-γ ligands, PGJ2, treatment reduced NFκB and AP-1 DNA binding activity in the lung and decreased plasma IL-6 levels during sepsis.22 Cuzzocrea et al reported that pretreatment of rats with PPAR-γ ligand significantly attenuated iNOS activity and consequent NO production induced by carrageenan administration.31 Another study showed that PPAR-γ activators, PGJ2 and antidiabetic thiazolidinediones, inhibited iNOS expression induced by LPS and IFN-γ in rat glial cells.32 Uchimura et al have also shown that PPAR-γ activation interferes with NFκB and AP-1 transcriptional activity and suppresses LPS-induced NO and TNF-α production in Kupffer cells.33 Wayman et al demonstrated that PPAR-γ ligands reduce tissue necrosis associated with acute myocardial infarction. They also showed that regional myocardial ischemia and reperfusion result in a significant increase in iNOS expression, which could be prevented by PGJ2 pretreatment.23 Furthermore, Liu et al reported a PPAR-γ agonist-induced protection from ischemic/reperfusion-induced myocardial apoptosis.34 In another study, Yue et al have shown that PPAR-γ activation reduced myocardial infarction and improved contractile function caused by ischemia/reperfusion injury.24 These studies collectively suggest that an increase in PPAR-γ activity can reduce pro-inflammatory responses and protect myocardium under various stressful conditions. Consistent with these studies, we observed that PPAR-γ activation induced by androstenediol administration following T-H and resuscitation plays an important role in protecting cardiac function under those conditions. Furthermore, our results support the suggestion that inhibition in cardiac IL-6 and iNOS gene expressions is likely the mechanism by which PPAR-γ mediates its protective effects.
Several studies have shown that injury including hemorrhagic shock induces a cascade of pro-inflammatory cytokines that are associated with immunosuppression, hemodynamic depression, and organ dysfunction.35,36 Since elevated plasma IL-6 levels have been associated with cardiac dysfunction following burn, pancreatitis,37,38 or T-H,6,13–15,26 attenuation of IL-6 production could be one of the downstream mechanism responsible for the androstenediol-mediated salutary effects in preventing cardiac depression following T-H. Our previous studies have also shown that progesterone,6 tyrosine kinase inhibitor,14 metoclopramide,13 or estrogen15 mediate their salutary effects on cardiac function following T-H by modulating plasma IL-6 levels under those conditions. Furthermore, our recent findings indicate that the depressed cardiac functions following T-H are associated with an increase in IL-6 production locally in cardiomyocytes.16 Consistent with these findings, the present results suggest that the improvement in cardiac functions by androstenediol treatment is due to the reduction in cardiac IL-6 gene expression and plasma IL-6 through PPAR-γ following T-H.
Studies have demonstrated that IL-6 decreases cardiac contractility of adult ventricular myocytes and this was associated with an enhanced synthesis of iNOS and increased NO production.11 The induction of iNOS and the release of NO have been show to have detrimental effects on myocardial contractile functions.39,40 Finkel et al demonstrated that the direct negative inotropic effects of IL-6 on the heart are mediated by nitric oxide.12 Another study reported that NO overproduction in cardiac myocytes lowers energy consumption and myocardial contractility by impairing mitochondrial function.40 Ungureanu-Longrois et al also showed that the induction of iNOS by inflammatory mediators, including cytokines, causes a marked depression of myocyte contractile responsiveness.41 In this study, we observed a significant increase in cardiac iNOS gene expression following T-H. It is likely that the observed down-regulation of iNOS gene expression in the heart may contribute to the androstenediol-mediated salutary effects on cardiac function following T-H.
Although our results collectively suggest that androstenediol-mediated PPAR-γ activation may regulate cardiac function at multiple levels following T-H, the precise mechanism by which androstenediol regulates PPAR-γ remains unknown. In our study, we measured PGJ2, which is an endogenous PPAR-γ ligand and found that plasma PGJ2 levels decreased significantly after T-H. Although androstenediol administration significantly increased PGJ2 levels in T-H animals, these levels remained significantly lower compared with those observed in sham rats. While such an increase in PGJ2 levels may contribute to PPAR-γ activation, additional studies are needed to determine the mechanism by which androstenediol activates PPAR-γ following T-H. These studies are important especially in view of findings suggesting that PGJ2 can also reduce inflammatory responses by directly inhibiting IkB kinase, a parameter that is independent of PPAR-γ activation.42,43 Modulation of mitogen activated protein kinase (MAPK) is another potential mechanism by which androstenediol may mediate its effect on PPAR-γ. Our recent study has shown that administration of PD 98059, an inhibitor of extracellular signal-regulated kinases (ERKs), reduces plasma IL-6 levels and hepatocellular damage following T-H.44 Studies have also reported that the phosphorylation of ERKs or JNK decreases the activity of PPAR-γ, but the activators of phosphokinase A (PKA) stimulate PPAR-γ activity even in the absence of exogenous ligands.45 Although the influence of androstenediol on MAPK activity is not investigated in this study, a reduction in MAPK activation following DHEA administration has been reported.46 Nonetheless, further studies are needed to determine whether androstenediol has any effect on MAPK activity following T-H and resuscitation.
CONCLUSION
The salutary effects of androstenediol administration on cardiac function following trauma-hemorrhage appear to be mediated at least in part via PPAR-γ activation, which involves the down-regulation of cardiac IL-6 and iNOS gene expressions. We propose that a better understanding of the androstenediol-mediated downstream mechanisms will enable us to develop novel adjunct therapeutic modalities for the treatment of trauma-hemorrhagic shock.
ACKNOWLEDGMENTS
The authors thank Mr. Zheng F. Ba and Mr. Philip Sohn for their superb technical assistance during this study.
Footnotes
Supported by NIH Grant No. R37 GM39519.
Reprints: Irshad H. Chaudry, PhD, Center for Surgical Research, University of Alabama at Birmingham, 1670 University Boulevard, Volker Hall, Room G094, Birmingham, AL 35294-0019. E-mail: Irshad.Chaudry@ccc.uab.edu.
REFERENCES
- 1.Ba ZF, Wang P, Koo DJ, et al. Alterations in tissue oxygen consumption and extraction after trauma and hemorrhagic shock. Crit Care Med. 2000;28:2837–2842. [DOI] [PubMed] [Google Scholar]
- 2.Ayala A, Ertel W, Chaudry IH. Trauma-induced suppression of antigen presentation and expression of major histocompatibility class II antigen complex in leukocytes. Shock. 1996;5:79–90. [DOI] [PubMed] [Google Scholar]
- 3.Jarrar D, Chaudry IH, Wang P. Organ dysfunction following hemorrhage and sepsis: mechanisms and therapeutic approaches [Review]. Int J Mol Med. 1999;4:575–583. [DOI] [PubMed] [Google Scholar]
- 4.Diodato MD, Knoferl MW, Schwacha MG, et al. Gender differences in the inflammatory response and survival following haemorrhage and subsequent sepsis. Cytokine. 2001;14:162–169. [DOI] [PubMed] [Google Scholar]
- 5.Angele MK, Schwacha MG, Ayala A, et al. Effect of gender and sex hormones on immune responses following shock. Shock. 2000;14:81–90. [DOI] [PubMed] [Google Scholar]
- 6.Kuebler JF, Jarrar D, Bland KI, et al. Progesterone administration after trauma and hemorrhagic shock improves cardiovascular responses. Crit Care Med. 2003;31:1786–1793. [DOI] [PubMed] [Google Scholar]
- 7.Roumen RM, Hendriks T, van der Ven-Jongekrijg J, et al. Cytokine patterns in patients after major vascular surgery, hemorrhagic shock, and severe blunt trauma: relation with subsequent adult respiratory distress syndrome and multiple organ failure. Ann Surg. 1993;218:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akira S, Hirano T, Taga T, et al. Biology of multifunctional cytokines: IL 6 and related molecules (IL 1 and TNF). FASEB J. 1990;4:2860–2867. [PubMed] [Google Scholar]
- 9.Simpson RJ, Hammacher A, Smith DK, et al. Interleukin-6: structure-function relationships. Protein Sci. 1997;6:929–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ancey C, Corbi P, Froger J, et al. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine. 2002;18:199–205. [DOI] [PubMed] [Google Scholar]
- 11.Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem. 2003;278:16304–16309. [DOI] [PubMed] [Google Scholar]
- 12.Finkel MS, Oddis CV, Jacob TD, et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. [DOI] [PubMed] [Google Scholar]
- 13.Jarrar D, Wang P, Song GY, et al. Metoclopramide: a novel adjunct for improving cardiac and hepatocellular functions after trauma-hemorrhage. Am J Physiol Endocrinol Metab. 2000;278:E90–E95. [DOI] [PubMed] [Google Scholar]
- 14.Jarrar D, Wang P, Song GY, et al. Inhibition of tyrosine kinase signaling after trauma-hemorrhage: a novel approach for improving organ function and decreasing susceptibility to subsequent sepsis. Ann Surg. 2000;231:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima Y, Wang P, Jarrar D, et al. Estradiol administration after trauma-hemorrhage improves cardiovascular and hepatocellular functions in male animals. Ann Surg. 2000;232:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S, Zheng R, Hu S, et al. Mechanism of cardiac depression after trauma-hemorrhage: increased cardiomyocyte IL-6 and effect of sex steroids on IL-6 regulation and cardiac function. Am J Physiol Heart Circ Physiol. 2004;287:H2183–H2191. [DOI] [PubMed] [Google Scholar]
- 17.Toth B, Yokoyama Y, Schwacha MG, et al. Insights into the role of interleukin-6 in the induction of hepatic injury after trauma-hemorrhagic shock. J Appl Physiol. 2004;97:2184–2189. [DOI] [PubMed] [Google Scholar]
- 18.Blanquart C, Barbier O, Fruchart JC, et al. Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol. 2003;85:267–273. [DOI] [PubMed] [Google Scholar]
- 19.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. [DOI] [PubMed] [Google Scholar]
- 20.Abdelrahman M, Collin M, Thiemermann C. The peroxisome proliferator-activated receptor-gamma ligand 15-deoxyDelta12,14 prostaglandin J2 reduces the organ injury in hemorrhagic shock. Shock. 2004;22:555–561. [DOI] [PubMed] [Google Scholar]
- 21.Collin M, Abdelrahman M, Thiemermann C. Endogenous ligands of PPAR-gamma reduce the liver injury in haemorrhagic shock. Eur J Pharmacol. 2004;486:233–235. [DOI] [PubMed] [Google Scholar]
- 22.Zingarelli B, Cook JA. Peroxisome proliferator-activated receptor-gamma is a new therapeutic target in sepsis and inflammation. Shock. 2005;23:393–399. [DOI] [PubMed] [Google Scholar]
- 23.Wayman NS, Hattori Y, McDonald MC, et al. Ligands of the peroxisome proliferator activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002;16:1027–1040. [DOI] [PubMed] [Google Scholar]
- 24.Yue Tl TL, Chen J, Bao W, et al. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2001;104:2588–2594. [DOI] [PubMed] [Google Scholar]
- 25.Ben Nathan D, Padgett DA, Loria RM. Androstenediol and dehydroepiandrosterone protect mice against lethal bacterial infections and lipopolysaccharide toxicity. J Med Microbiol. 1999;48:425–431. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu T, Choudhry MA, Szalay L, et al. Salutary effects of androstenediol on cardiac function and splanchnic perfusion after trauma-hemorrhage. Am J Physiol Regul Integr Comp Physiol. 2004;287:R386–R390. [DOI] [PubMed] [Google Scholar]
- 27.Waxman DJ. Role of metabolism in the activation of dehydroepiandrosterone as a peroxisome proliferator. J Endocrinol. 1996;150(suppl):129–147. [PubMed] [Google Scholar]
- 28.Schreiber E, Matthias P, Muller MM, et al. Rapid detection of octamer binding proteins with ‘mini-extracts,’ prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu T, Szalay L, Choudhry MA, et al. Mechanism of salutary effects of androstenediol on hepatic function after trauma-hemorrhage: role of endothelial and inducible nitric oxide synthase. Am J Physiol Gastrointest Liver Physiol. 2005;288:G244–G250. [DOI] [PubMed] [Google Scholar]
- 30.Collin M, Patel NS, Dugo L, et al. Role of peroxisome proliferator-activated receptor-gamma in the protection afforded by 15-deoxydelta12,14 prostaglandin J2 against the multiple organ failure caused by endotoxin. Crit Care Med. 2004;32:826–831. [DOI] [PubMed] [Google Scholar]
- 31.Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces acute inflammation. Eur J Pharmacol. 2004;483:79–93. [DOI] [PubMed] [Google Scholar]
- 32.Kitamura Y, Kakimura J, Matsuoka Y, et al. Activators of peroxisome proliferator activated receptor-gamma (PPARgamma) inhibit inducible nitric oxide synthase expression but increase heme oxygenase-1 expression in rat glial cells. Neurosci Lett. 1999;262:129–132. [DOI] [PubMed] [Google Scholar]
- 33.Uchimura K, Nakamuta M, Enjoji M, et al. Activation of retinoic X receptor and peroxisome proliferator-activated receptor-gamma inhibits nitric oxide and tumor necrosis factor-alpha production in rat Kupffer cells. Hepatology. 2001;33:91–99. [DOI] [PubMed] [Google Scholar]
- 34.Liu HR, Tao L, Gao E, et al. Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc Res. 2004;62:135–144. [DOI] [PubMed] [Google Scholar]
- 35.Hierholzer C, Billiar TR. Molecular mechanisms in the early phase of hemorrhagic shock. Langenbecks Arch Surg. 2001;386:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy KD, Lee JO, Herndon DN. Current pharmacotherapy for the treatment of severe burns. Expert Opin Pharmacother. 2003;4:369–384. [DOI] [PubMed] [Google Scholar]
- 37.Jambrik Z, Gyongyosi M, Hegyi P, et al. Plasma levels of IL-6 correlate with hemodynamic abnormalities in acute pancreatitis in rabbits. Intensive Care Med. 2002;28:1810–1818. [DOI] [PubMed] [Google Scholar]
- 38.Maass DL, White J, Horton JW. IL-1beta and IL-6 act synergistically with TNF-alpha to alter cardiac contractile function after burn trauma. Shock. 2002;18:360–366. [DOI] [PubMed] [Google Scholar]
- 39.Shah AM, Prendergast BD, Grocott-Mason R, et al. The influence of endothelium-derived nitric oxide on myocardial contractile function. Int J Cardiol. 1995;50:225–231. [DOI] [PubMed] [Google Scholar]
- 40.Tatsumi T, Matoba S, Kawahara A, et al. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs contractile function in rat cardiac myocytes. J Am Coll Cardiol. 2000;35:1338–1346. [DOI] [PubMed] [Google Scholar]
- 41.Ungureanu-Longrois D, Balligand JL, Kelly RA, et al. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: role of a cytokine-inducible nitric oxide synthase in cardiac myocytes. J Mol Cell Cardiol. 1995;27:155–167. [DOI] [PubMed] [Google Scholar]
- 42.Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. [DOI] [PubMed] [Google Scholar]
- 43.Forman BM, Tontonoz P, Chen J, et al. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. [DOI] [PubMed] [Google Scholar]
- 44.Jarrar D, Song GY, Kuebler JF, et al. The effect of inhibition of a major cell signaling pathway following trauma hemorrhage on hepatic injury and interleukin 6 levels. Arch Surg. 2004;139:896–901. [DOI] [PubMed] [Google Scholar]
- 45.Lazennec G, Canaple L, Saugy D, et al. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol Endocrinol. 2000;14:1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshimata T, Yoneyama A, Jin-no Y, et al. Effects of dehydroepiandrosterone on mitogen-activated protein kinase in human aortic smooth muscle cells. Life Sci. 1999;65:431–440. [DOI] [PubMed] [Google Scholar]