

Abstract
The endogenous cannabinoid, anandamide, has been reported to induce an 'endothelium-derived hyperpolarizing factor (EDHF)-like' relaxation in vitro. We therefore investigated the effects of cannabinoid CB1 receptor agonists; HU 210, Δ9-tetrahydrocannabinol (Δ9-THC) and anandamide, and a CB1 antagonist/inverse agonist, SR 141716A, on nitric oxide (NO) and EDHF-mediated relaxation in precontracted rings of porcine coronary, rabbit carotid and mesenteric arteries.
In rings of mesenteric artery HU 210 and Δ9-THC induced endothelium- and cyclo-oxygenase-independent relaxations which were sensitive to SR 141716A. Anandamide (0.03–30 μM) induced a slowly developing, endothelium-independent relaxation which was abolished by diclofenac and was therefore mediated by cyclo-oxygenase product(s). None of the CB1 agonists tested affected the tone of precontracted rings of rabbit carotid or porcine coronary artery.
In endothelium-intact segments, HU 210, Δ9-THC and anandamide did not affect NO-mediated responses but under conditions of continuous NO synthase/cyclo-oxygenase blockade, significantly inhibited acetylcholine and bradykinin-induced relaxations which are attributed to the production of EDHF. The effects of HU 210 and Δ9-THC were not observed when experiments were performed in the presence of SR 141716A suggesting the involvement of the CB1 receptor.
In a patch clamp bioassay of EDHF production, HU 210 decreased the EDHF-mediated hyperpolarization of detector smooth muscle cells when applied to the donor segment but was without effect on the membrane potential of detector cells. The inhibition of EDHF production was unrelated to alterations in Ca2+-signalling or cytochrome P450 activity.
These results suggest that the activation of endothelial CB1 receptors appears to be negatively coupled to the production of EDHF.
Keywords: Endothelium-derived hyperpolarizing factor (EDHF), nitric oxide (NO), cannabinoid CB1 receptor, electrophysiology
Introduction
Δ9-Tetrahydrocannabinol (Δ9-THC) has long been known to exert distinct effects on the cardiovascular system inducing a maintained decrease in blood pressure (Graham & Li, 1973; Estrada et al., 1987). Δ9-THC- and the endogenously generated cannabinoid, anandamide, induce hypotension and bradycardia which are generally attributed to a centrally mediated reduction in sympathetic tone (Berdyshev et al., 1996; Varga et al., 1995). However, in anaesthetized rats the cannabinoid CB1 receptor agonist, HU 210, induces a long-lasting hypotension and bradycardia which cannot be attributed to sympathetic withdrawal (Vidrio et al., 1996). Indeed, cannabinoid receptors have recently been reported in the peripheral organs (Galiegue et al., 1995; Munro et al., 1993) as well as in endothelial cells (Deutsch et al., 1997). Thus, the activation of the peripheral CB1 receptor on endothelial cells may well influence vascular tone and the generation of endothelium-derived autacoids. While some of the effects of anandamide may be attributed to its metabolism to arachidonic acid and subsequently to cyclo-oxygenase or cytochrome P450 (CYP) products via a receptor-independent mechanism (Ellis et al., 1995; Pratt et al., 1998), others are clearly mediated by CB1 receptor activation (Hunter & Burstein, 1997). The recent discovery of cannabinoid receptors and some of their endogenous ligands has in turn focused attention on the possible signalling pathways that may be activated by these seven transmembrane, G protein-coupled receptors. Evidence for at least three, apparently different pathways has been proposed and depending on the cell type under investigation, cannabinoids are able to stimulate eicosanoid synthesis to inhibit the activation of adenylyl cyclase and to affect Ca2+ signalling (Pertwee, 1997). CB1 receptors are also involved in G protein receptor cross talk and anandamide is, for example, able to inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurones (Fan, 1995).
Endothelial cells generate and release anandamide (Deutsch et al., 1997) as well as a second endogenous cannabimimetic molecule, 2-arachidonoylglycerol (Sugiura et al., 1998), and as such may also be involved in the local regulation of vascular responsiveness. Indeed, just such a role for anandamide was recently proposed when it was suggested that a CB1 agonist produced by endothelial cells could account for the, as yet still elusive, endothelium-derived hyperpolarizing factor (EDHF) (Randall et al., 1996). The aim of the present study was to determine whether a crosstalk occurs between CB1 receptor activation or inactivation and endothelial autacoid production. To this end we studied the effects of CB1 receptor agonists (HU 210, Δ9-THC and anandamide) and the selective antagonist/inverse agonist SR 141716A (Bouaboula et al., 1997) on the vascular tone of three different arteries all of which synthesize both nitric oxide (NO) and an EDHF which is characterized as a CYP-dependent metabolite of arachidonic acid (Bauersachs et al., 1994; Popp et al., 1996; 1998).
Methods
Materials
Bradykinin was from Bachem Biochemica GmbH (Heidelberg, Germany), N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) and NGnitro-L-arginine (L-NA) from Serva (Heidelberg, Germany). Diclofenac (Voltaren injection solution) was from Ciba-Geigy, (Wehr, Germany), M-199 medium from GIBCO (Berlin, Germany), U46619 (9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α) was provided by Upjohn (Ann Arbor, MI, U.S.A.) and SR 141716A was provided by Sanofi Research (Montpellier, France). Rp-cyclic AMPS was from Alexis Biochemicals (Grünberg, Germany), 3-(1,1-dimethylheptyl)-(−)-11-hydroxy-Δ8-tetrahydrocannabinol (HU 210) was purchased from Tokris Cookson (Langford, England). Arachidoylethanolamide (anandamide), Δ9-tetrahydrocannabinol (Δ9-THC), acetylcholine (ACh), isobutyl-methyl-xanthine (IBMX), sodium nitroprusside and all other chemicals were purchased from Sigma (Deisenhofen, Germany).
Vessel preparation
Porcine coronary arteries: Porcine hearts were obtained from a local slaughterhouse, placed immediately into ice-cold Krebs-Henseleit solution and transported to the laboratory. The coronary arteries were dissected, cleaned of adventitial adipose and connective tissue and cut into rings (4 mm) for organ bath studies. Coronary segments (40 mm length) were used for the bioassay of EDHF.
Rabbit carotid and mesenteric arteries: New Zealand White rabbits of either sex (1.5–2.5 kg body weight) were anaesthetized with pentobarbitone sodium (90 mg kg−1 i.v.) and exsanguinated by cutting through both the abdominal aorta and vena cava. The carotid arteries and the main mesenteric artery were dissected, cleaned of adventitial adipose and connective tissue, and cut into rings (4 mm). In a number of experiments the endothelium was removed mechanically using blunt forceps.
Vascular reactivity studies
Porcine coronary artery, rabbit carotid or mesenteric artery rings were mounted between force transducers (Scaime, France) and a rigid support for measurement of isometric force and incubated in organ baths containing warmed (37°C), oxygenated (95% O2, 5% CO2) Krebs-Henseleit solution (pH 7.4) of the following composition (mM): NaCl, 119; NaHCO3, 25.0; KCl, 4.7; CaCl2, 1.6; KH2PO4, 1.2; MgSO4, 1.2; glucose 12.
Porcine coronary arteries
Passive tension was adjusted over a 60 min equilibration period to 5 g; thereafter the segments were exposed to a Krebs-Henseleit solution rich in KCl (80 mM) until stable contractions were obtained. After washing, arterial rings were exposed to U46619 (0.1–0.3 μM) to achieve a stable contraction which was approximately 80% of the maximal KCl-induced contraction. Thereafter, the integrity of the endothelium was assessed by the application of bradykinin (1 μM) and vessels exhibiting less than 90% relaxation were discarded.
Rabbit carotid and mesenteric arteries
Passive tension was adjusted over a 60 min equilibration period (2 g for carotid arteries and 2.5 g for mesenteric arteries), thereafter the segments were exposed twice to K+-rich Krebs solution. After washing and a further equilibration period of 40–60 min the arterial rings were contracted to 80% of the maximal KCl response using phenylephrine (0.1–1 μM). The integrity of the endothelium was assessed using ACh (1 μM) and vessels exhibiting less than 90% relaxation were discarded. As some of the relaxant responses observed were transient, the concentration-response curves presented represent the peak response observed to cumulative applications of the agonists.
Cell culture
Human umbilical vein endothelial cells were seeded on fibronectin-coated culture dishes containing medium M-199 and 20% heat-inactivated foetal calf serum (Greiner) supplemented with penicillin (50 u ml−1), streptomycin (50 μg ml−1) and L-glutamine (1 mM). CYP activity, cyclic AMP levels and [Ca2+]i were estimated in confluent cells.
Rat aortic smooth muscle cells were isolated and cultured as described (Popp et al., 1996). Experiments were performed using confluent smooth muscle cells grown on glass coverslips between passages 6 and 16.
Electrophysiological measurements
The membrane potential of cultured rat aortic smooth muscle cells was recorded using the slow whole-cell configuration of the patch-clamp technique. The patch pipettes used had an input resistance of 8–10 MΩ when filled with standard KCl solution (mM: KCl, 140; MgCl2, 1; CaCl2, 1.3; HEPES, 10; D-glucose, 5; pH 7.4). GΩ seals (approximately 10 GΩ) were established by gentle suction. The slow whole-cell configuration was obtained using nystatin (100 μg ml−1) in the pipette. This nystatin concentration provides a low resistance access to the cytosol to measure intracellular potentials under current clamp conditions. The electrical contact with the cytosol was established within 1–2 min of seal formation, during this time the input resistance decreased to approximately 1 GΩ. The cell membrane potential, measured in the current clamp mode, was recorded continuously and only detector smooth muscle cells which had a stable resting membrane potential for more than 2 min and exhibited no further change in the input resistance were used.
Detection of EDHF release
The release of EDHF was detected by recording changes in membrane potential of cultured rat aortic smooth muscle cells exposed to the effluate of a perfused porcine coronary artery. The transit time between the coronary artery and the detector smooth muscle cells was approximately 2 s. Briefly, a segment of porcine coronary artery was cannulated at both ends, mounted in an organ chamber and perfused intraluminally (1 ml min−1) with HEPES-modified Tyrode's solution of the following composition (in mM): NaCl, 132; KCl, 4; CaCl2, 1; MgCl2, 0.5; HEPES, 9.5 and glucose, 5 (pH 7.4, 37°C) containing the NO synthase inhibitor L-NA (100 μM) and diclofenac (10 μM). The perfusate from a bradykinin-stimulated, L-NA/diclofenac-treated porcine coronary artery was directed over cultured rat aortic smooth muscle cells, the membrane potential of which was recorded using the slow whole-cell configuration of the patch clamp technique as described above. The resting membrane potential as determined in the whole cell configuration was approximately–48.2 mV (n=30) in cultured rat aortic smooth muscle cells. Continuous superfusion with HEPES-Tyrode (1 ml min−1) did not alter the resting membrane potential. We have previously reported that freshly isolated porcine coronary smooth muscle cells and cultured rat aortic smooth muscle cells respond in a similar manner to the luminal incubate from endothelium-intact porcine coronary arteries (Popp et al., 1998).
Cell culture
Human umbilical vein endothelial cells (HUVEC), isolated from umbilical cords as described previously (Hopkins & Gorman, 1981) were seeded on coverslips in culture dishes containing medium M–199 and 20% heat-inactivated foetal calf serum supplemented with penicillin (50 u ml−1), streptomycin (50 μg ml−1), L-glutamine (1 mM), glutathione (5 mg ml−1) and L(+)ascorbic acid (5 mg ml−1) (GIBCO) and cultured until confluent.
Measurement of [Ca2+]i
For the measurement of [Ca2+]i, HUVEC were loaded with the fluorescent Ca2+-sensitive dye fura 2 by incubation with 3 μM fura 2-AM and 0.025% (w/v) Pluronic F-127 at 37°C for 60 min. Thereafter the coverslips were washed in HEPES-Tyrode solution and [Ca2+]i was determined fluorometrically in thermostatically controlled cuvette as described previously (Fleming et al., 1996).
Measurement of cytochrome P450 (CYP) activity
CYP-dependent metabolic activity was assayed as the dealkylation of 7-ethoxyresorufin (Yatomi et al., 1992) in confluent HUVEC in which P450 activity had been induced by incubation with β-naphthoflavone (3 μM, 48 h). Briefly, cells were washed twice with HEPES-Tyrode and thereafter incubated with 7-ethoxyresorufin (3.4 μM) for 10 min. The supernatant was then removed and the resorufin formed was measured at an excitation of 522 nm and emission at 586 nm using a fluorescence spectrophotometer (Deltascan, Photon-Technology, Wedel, Germany). Cell-mediated conversion of ethoxyresorufin to resorufin was linear over at least 30 min and was abolished by the P450-inhibitor clotrimazole (3 μM).
Determination of cyclic AMP concentration
In order to assess the effects of CB1 agonists on the isoprenaline-induced increase in HUVEC cyclic AMP, cells were pretreated with the phosphodiesterase inhibitor IBMX (0.1 mM) in the absence and presence of either HU 210 (30 μM) or Δ9-THC (50 μM). Cells were then incubated with isoprenaline (0.3 μM, 37°C), and after 5 min the incubation was stopped by the aspiration of the buffer and addition of 600 μl of ice cold 6% (v/v) trichloroacetic acid. Samples were left on ice for 30 min, cells and supernatants were then harvested by scraping and the precipitates centrifuged at 10,000×g (15 min, 4°C). Thereafter, the supernatants were extracted four times with 5 volumes of water-saturated diethyl ether and the concentration of cyclic AMP was determined by a specific radioimmunoassay. Cyclic AMP content is expressed as pmol per mg protein.
Statistical analysis
Unless otherwise indicated data are expressed as mean±s.e.mean. Statistical evaluation was performed using Student's t-test for unpaired data, one-way analysis of variance (ANOVA) followed by a Bonferroni t-test, or ANOVA for repeated measures where appropriate. Values of P<0.05 were considered statistically significant.
Results
Effects of HU 210, Δ9-THC and anandamide on vascular tone
In phenylephrine-precontracted rabbit mesenteric artery rings, the selective non-hydrolysable CB1 agonist, HU 210, induced an endothelium-independent decrease in mesenteric artery tone which was slightly, but not significantly, attenuated by diclofenac (Figure 1A) and was abolished by the CB1 antagonist SR 141716A (data not shown). Δ9-THC also induced an endothelium-independent relaxation which was mostly insensitive to diclofenac (Figure 1B). The endocannabinoid, anandamide, elicited a slowly developing relaxation, with a threshold of 0.1 μM (Figure 1C). Although the removal of the endothelium induced a distinct rightward shift in the concentration-response curve to anandamide (EC50 −6.0±0.05 and −5.39±0.02 M, in endothelium-intact and endothelium denuded rings respectively, P<0.05), the maximal relaxation (Rmax) of vessels precontracted to 80% of the maximal KCl-induced contraction was comparable in both the presence and absence of the endothelium. Inclusion of diclofenac (10 μM) in the organ bath abolished the anandamide-induced relaxation of both endothelium-intact and -denuded mesenteric artery rings (Figure 1C).
Figure 1.
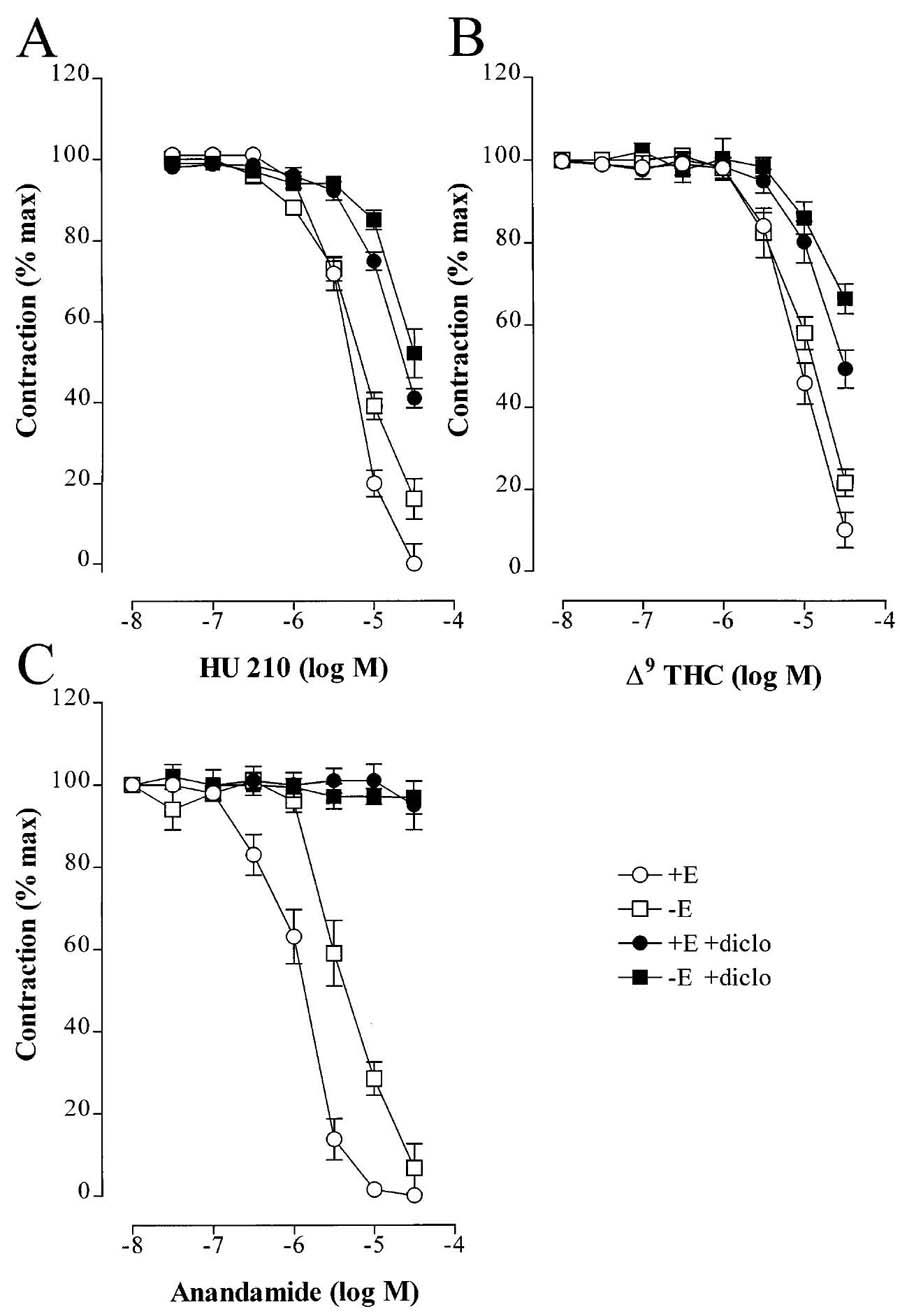
Effect of CB1 agonists of the tone of phenylephrine-precontracted mesenteric artery rings. Concentration-response curves showing the relaxant effect of (A) HU 210, (B) Δ9-THC and (C) anandamide on rings of rabbit mesenteric artery precontracted to 80% of the maximal KCl-induced response with phenylephrine. Experiments were performed using endothelium-intact (+E) and -denuded (−E) arteries in the absence and presence of diclofenac (diclo, 10 μM). Results are presented as the mean±s.e.mean of 12 separate experiments.
Neither HU 210, Δ9-THC nor anandamide relaxed rabbit carotid or porcine coronary artery rings (data not shown).
Effect of SR 141716A, HU 210, Δ9-THC and anandamide on EDHF-mediated relaxations
In endothelium-intact phenylephrine-contracted rabbit mesenteric artery rings under conditions of combined NO synthase/cyclo-oxygenase blockade (L-NA 100 μM; diclofenac 10 μM), ACh induced a concentration-dependent relaxation which was attributed to the synthesis and release of EDHF (Figure 2). The CB1 antagonist, SR 141716A (30 μM) did not alter the basal tension of mesenteric arteries or significantly affect tone in preconstricted arteries. However, SR 141716 (30 μM) attenuated the ACh-induced EDHF-mediated relaxation (Figure 2, Table 1).
Figure 2.
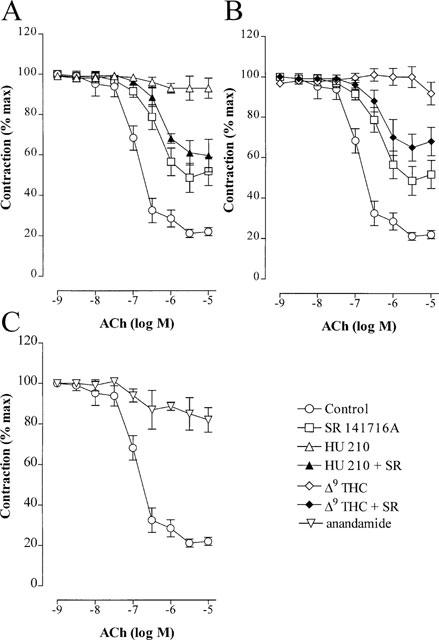
Effect of CB1 agonists on the EDHF-mediated relaxation of the rabbit mesenteric artery. Concentration-response curves to acetylcholine (ACh) were obtained using phenylephrine precontracted mesenteric artery rings in the presence of either solvent (0.06% ethanol), SR 141716A (30 μM), HU 210 (30 μM; A), Δ9-tetrahydrocannabinol (Δ9-THC, 30 μM; B), anandamide (30 μM; C) or combinations of CB1 agonist and SR 141716A. All experiments were performed in the continuous presence of NGnitro-L-arginine (100 μM) and diclofenac (10 μM) and the results are presented as the mean±s.e.mean of nine separate experiments.
Table 1.
Effect of CB1 agonists and a CB1 antagonist on the ACh-induced, EDHF-mediated relaxation of the rabbit mesenteric and carotid arteries
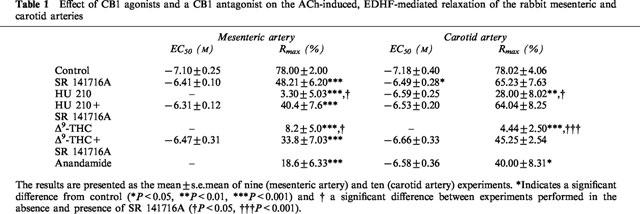
The ACh-induced, EDHF-mediated relaxations of mesenteric arteries was concentration-dependently inhibited by HU 210 (1–30 μM) and was abolished at the highest concentration studied. This effect was not observed in experiments performed in the presence of SR 141716A (Figure 2A, Table 1). The sensitivity of arterial rings to an exogenous NO donor (sodium nitroprusside, 1 μM) was unaffected by HU 210 (Rmax was 103±6% and 98±10% in vessels with solvent and HU 210 respectively, n=9). Similarly, Δ9-THC (30 μM; Figure 2B) also resulted in the SR 141716A-sensitive inhibition of EDHF-mediated relaxation, while the anandamide-induced inhibition (30 μM; Figure 2C) was not sensitive to SR 141716A (not shown).
In the rabbit carotid artery, which did not relax in response to either HU 210, Δ9-THC or anandamide, all of these agents inhibited EDHF-mediated relaxation to a similar extent as in the rabbit mesenteric artery (Figure 3, Table 1). Similar results were obtained using the porcine coronary artery.
Figure 3.
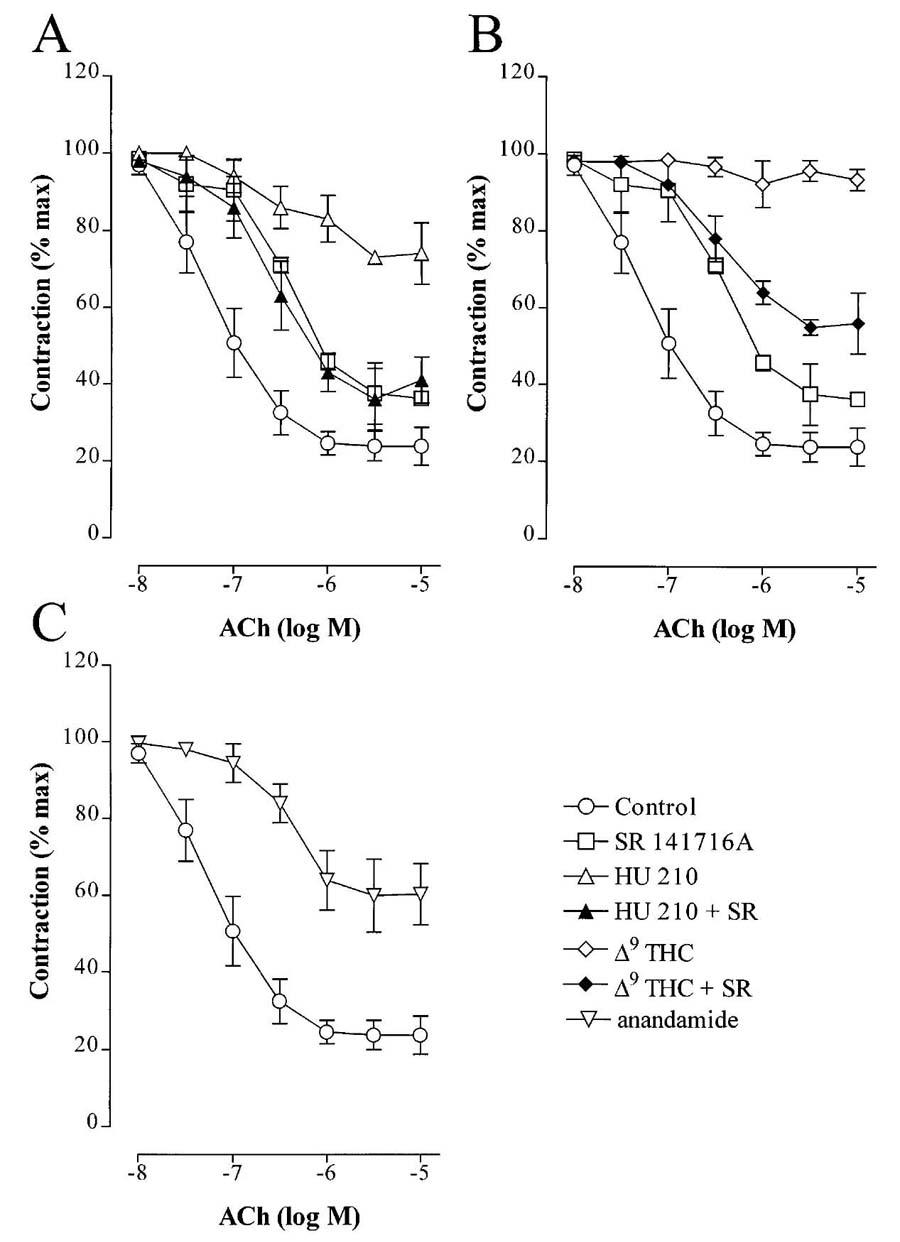
Effect of CB1 agonists on the EDHF-mediated relaxation of the rabbit carotid artery. Concentration-response curves to acetylcholine (ACh) were obtained using phenylephrine precontracted carotid artery rings in the presence of either solvent (0.06% ethanol), SR 141716A (30 μM), HU 210 (30 μM; A), Δ9-tetrahydrocannabinol (Δ9-THC, 30 μM; B), anandamide (30 μM; C) or combinations of CB1 agonist and SR 141716A. All experiments were performed in the continuous presence of NGnitro-L-arginine (100 μM) and diclofenac (10 μM) and the results are presented as the mean±s.e.mean of ten separate experiments.
To determine whether the effect of the CB1 agonists on EDHF-mediated relaxations was specific, the NO-mediated relaxation to ACh was determined in the absence and presence of HU 210 (30 μM, Figure 4). The CB1 agonist failed to affect the ACh-induced, NO-mediated relaxation of vessels precontracted to 80% of the maximal KCl-induced contraction, demonstrating that these compounds selectively affect EDHF production. The subsequent addition of L-NA completely abolished ACh-induced relaxation (not shown). Although the second concentration response curve to ACh was shifted slightly to the right with respect to the first (Figure 4), this was the case in vessels treated with either solvent or HU 210. Similar results were obtained using the rabbit carotid artery (data not shown).
Figure 4.
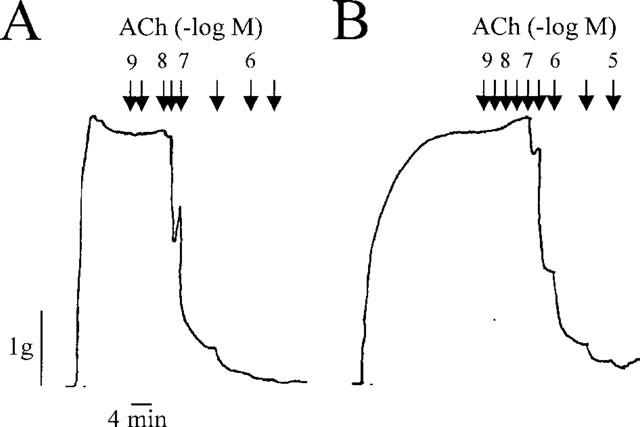
Effect of HU 210 on the acetylcholine-induced, NO-mediated relaxation of precontracted rabbit mesenteric arteries. Concentration-response curves to acetylcholine (1 nM to 3 μM) were obtained in rings of rabbit mesenteric artery precontracted to 80% of the maximal KCl-induced tone with phenylephrine. Experiments were performed in (A) the absence (0.06% ethanol) and (B) the presence of HU 210 (30 μM). Diclofenac (10 μM) was present throughout. The tracings shown are representative of results obtained in four separate experiments.
Effect of HU 210 on the production and activity of EDHF
A patch clamp bioassay for EDHF production was used to assess the effects of the CB1 agonists on the production and actions of EDHF. In this model the perfusate from a bradykinin-stimulated, L-NA/diclofenac-treated porcine coronary artery is directed over cultured rat aortic smooth muscle cells, the membrane potential of which is continuously monitored using the patch clamp technique (Popp et al., 1996; Bauersachs et al., 1996).
The effects of ‘authentic EDHF', anandamide and HU 210 on smooth muscle membrane potential were determined. The EDHF present in the perfusate of bradykinin-stimulated arteries induced an immediate biphasic change in smooth muscle membrane potential which consisted of an initial hyperpolarization of −11.4±1.0 mV, followed by a depolarization of 4.7±1.0 mV, as previously described (Popp et al., 1996). Anandamide used at a concentration (30 μM) which induced complete relaxation of rabbit mesenteric artery rings induced a small and slowly developing smooth muscle cell hyperpolarization which reached a maximum of −3.04±0.5 mV approximately 3 min after its application (Figure 5A). Neither the resting membrane potential nor the magnitude of the EDHF-induced hyperpolarization were markedly altered when detector cells were pre-treated with HU 210 (30 μM) or the CYP inhibitor miconazole (3 μM, Figure 5B). However, incubation of donor segments with either HU 210 or miconazole attenuated the production of EDHF, as demonstrated by a decrease in the hyperpolarization of detector smooth muscle cells induced by the perfusate from HU 210 or miconazole-treated, bradykinin-stimulated porcine coronary artery segments (Figure 5B).
Figure 5.
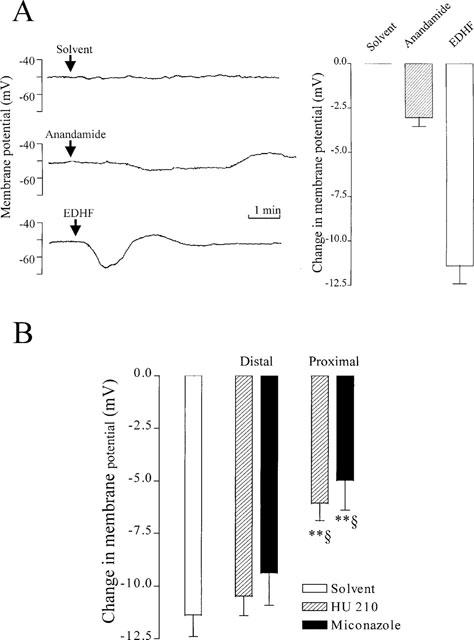
Effect of anandamide and HU 210 on smooth muscle membrane potential and the generation of EDHF from porcine coronary arteries. (A) Comparison of the effects on anandamide and ‘authentic EDHF' on the membrane potential of cultured rat aortic smooth muscle cells. Original tracings (left panel) and statistical summary (right panel) illustrating the hyperpolarization of rat aortic smooth muscle cells induced by either the direct application of solvent (0.06% ethanol), anandamide (30 μM) or the luminal perfusate removed from bradykinin-stimulated porcine coronary artery segments in the continuous presence of NGnitro-L-arginine (100 μM) and diclofenac (10 μM). Results are expressed as the mean±s.e.mean of four separate experiments. (B) Effect of HU 210 and miconazole on the generation of EDHF by bradykinin-stimulated porcine coronary arteries. The membrane potential of cultured vascular smooth muscle cells superfused with the effluate from bradykinin-stimulated porcine coronary arteries was monitored (open columns). Experiments were performed in the absence (0.06% ethanol) and presence of HU 210 (30 μM) and miconazole (3 μM) added to the perfusate either proximal or distal to the donor segment (incubation time 15 min). Results are presented as the mean±s.e.mean of six separate experiments, **P<0.01 vs control and §P<0.05 vs response when the substance was applied distal to the donor artery.
Effects of CB1 agonists on endothelial [Ca2+]i, CYP activity and cyclic AMP levels
Since HU 210 decreased the synthesis and/or release of EDHF we determined its effect on endothelial [Ca2+]i and CYP activity. These responses were assessed using human umbilical vein endothelial cells which produce an EDHF which we have previously characterized as a CYP-dependent metabolite of arachidonic acid (Popp et al., 1996). In fura 2-loaded human umbilical vein endothelial cells, bradykinin (10 nM) induced a biphasic increase in [Ca2+]i as previously described (Fleming et al., 1995). Neither basal [Ca2+]i nor the bradykinin-induced Ca2+-response were altered by HU 210 (50 μM, Figure 6A) irrespective of whether experiments were performed in the absence or presence of L-NA and diclofenac (data not shown).
Figure 6.
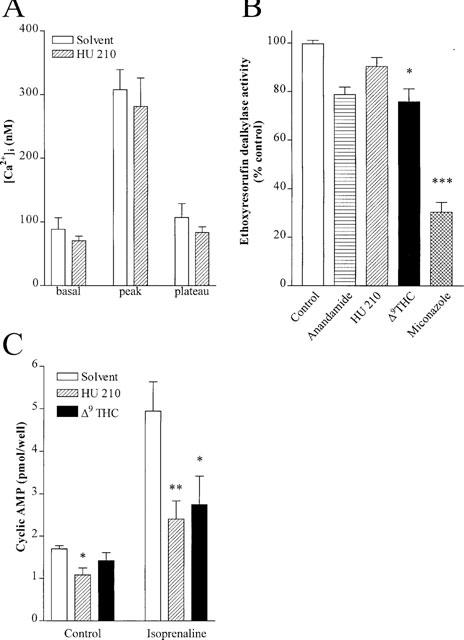
Effect on CB1 agonists on endothelial [Ca2+]i, P450 activity and cyclic AMP levels. (A) Basal [Ca2+]i and the bradykinin (10 nM)-induced Ca2+ response in fura 2-labelled human endothelial cells. Experiments were performed in the absence (0.06% ethanol) and presence of HU 210 (30 μM). Results are presented as the mean±s.e.mean of eight separate experiments. (B) Ethoxyresorufin dealkylase activity was assessed in endothelial cells pre-incubated with either solvent, anandamide (30 μM), HU 210 (30 μM), Δ9-THC (30 μM) or miconazole (3 μM). Results are presented as the mean±s.e.mean of five separate experiments. (C) Intracellular cyclic AMP levels in endothelial cells under basal and isoprenaline (0.3 μM)-stimulated conditions. Experiments were performed in the absence and presence of HU 210 (30 μM) or Δ9THC (30 μM). Results are presented as the mean±s.e.mean of six separate experiments *P<0.05, **P<0.01, ***P<0.001 vs the appropriate solvent.
As the production of EDHF by all of the arteries tested as well as that produced by cultured endothelial cells stimulated with bradykinin (Bauersachs et al., 1994; 1996; Popp et al., 1996), could be attenuated by inhibitors of CYP we determined whether the CB1 agonists affected P450 activity in cultured human umbilical vein endothelial cells. CYP activity, assayed as the dealkylation of 7-ethoxyresorufin, was slightly attenuated by anandamide and Δ9-THC. HU 210 (30 μM) did not affect ethoxyresorufin dealkylase activity in intact endothelial cells, whereas miconazole inhibited the formation of resorufin by approximately 70% (Figure 6B). None of the CB1 agonists tested inhibited CYP activity in purified endothelial cell microsome preparations (data not shown).
As CB1 receptors are negatively coupled to adenylyl cyclase, we determined the effect of the CB1 agonists on the isoprenaline-induced increase in endothelial cyclic AMP. In confluent human umbilical vein endothelial cells HU 210 markedly reduced cyclic AMP levels under basal conditions and following stimulation with isoprenaline. Δ9-THC induced a similar, though less pronounced effect on cyclic AMP levels (Figure 6C).
In order to determine whether an increase in cellular cyclic AMP is a prerequisite for the production of EDHF we performed experiments using a specific protein kinase A inhibitor. Rp-cyclic AMPs (25 μM) was without effect on the EDHF-mediated relaxation of the phenylephrine-precontracted rabbit mesenteric artery (data not shown).
Discussion
The results of the present study demonstrate that in rabbit mesenteric and carotid arteries as well as porcine coronary arteries, the CB1 agonists HU 210, Δ9-THC and anandamide inhibit the production of EDHF without affecting the synthesis of NO. The inhibitory effect of these agonists on EDHF production was not related to changes in endothelial [Ca2+]i or CYP activity and could be reversed by the inclusion of the CB1 receptor antagonist, SR 141716A. Thus, the activation of endothelial CB1 receptors appears to be negatively coupled to the production of EDHF.
Anandamide has been reported to dilate rat mesenteric and coronary vascular beds via an EDHF-like (i.e. NO/PGI2-independent) mechanism (Randall et al., 1996; Randall & Kendall, 1997). The observation that the selective CB1 antagonist SR 141716A inhibited EDHF-mediated relaxations in these preparations led to the suggestion that EDHF may be an endocannabinoid. This hypothesis is however a matter of debate, as other investigators have since reported that anandamide-induced relaxations are not sensitive to SR 141617A and appear not to be mediated by the CB1 receptor or Ca2+-dependent K+ channel activation (Plane et al., 1997; Chataigneau et al., 1998). Although anandamide does hyperpolarize vascular smooth muscle cells, the response obtained in the present study bore characteristics markedly different to that induced by EDHF, being delayed in onset and exhibiting a different kinetic. Anandamide also relaxed rabbit mesenteric arteries but as this effect was sensitive to diclofenac the reduction in vascular tone was more likely to be mediated by a cyclo-oxygenase product. Indeed, the cyclo-oxygenase-dependent metabolism of anandamide underlies its vasodilator action in cerebral arterioles (Facci et al., 1995) and bovine coronary arteries (Pratt et al., 1998; Ellis et al., 1995).
While the discussion relating to the role played by anandamide in EDHF-mediated relaxations seems to be concluded, endocannabinoids do appear to play a significant role in the regulation of vascular tone in certain vascular beds. In the rat kidney, which has previously been reported to express both anandamide amidase and the CB1 receptor (Deutsch & Chin, 1993; Shire et al., 1995), anandamide vasodilates juxtamedullary afferent arterioles by a process involving the generation of endothelium-derived NO (Deutsch et al., 1997). This anandamide-induced production of NO inhibited the release of norepinephrine from sympathetic nerve endings in the renal artery (Deutsch et al., 1997) demonstrating that anandamide possesses both vasorelaxant and neuromodulatory properties. Similarly, anandamide is also reported to stimulate the production of NO from the vascular endothelium of the rat portal plexus (Prevot et al., 1998). The effects of CB1 agonists appear to vary with the vascular bed under investigation since none of the CB1 agonists used affected tone in rabbit carotid or porcine coronary arteries. Only rings from the rabbit mesenteric artery relaxed in response to CB1 agonists. HU 210 and Δ9-THC relaxed both endothelium-intact and denuded mesenteric arterial rings to a similar extent, thus excluding a role for endothelium-derived NO. The inclusion of diclofenac shifted the concentration-response curve of both agonists to the right suggesting that both cyclo-oxygenase-dependent and -independent processes were involved in mediating the response observed. However, the fact that the relaxation induced by HU 210 and Δ9-THC was resistant to L-NA and diclofenac does not suggest that these agents represent an EDHF-like substance as neither compound hyperpolarized vascular smooth muscle cells.
The data in Figures 2 and 3 clearly show that SR 141716A alleviated (between 70 and 100%) the inhibition of EDHF-mediated responses by HU 210 and Δ9-THC suggesting that this effect may be mediated by the CB1 receptor. However, since high concentrations of the inverse agonist (i.e., a compound which exhibits biological activity by blocking the signal transduction mediated by constitutively activated receptors) were required to observed any effect, we cannot rule out the possibility that the effects observed may be indirect. Indeed, high concentrations of SR 141716A have recently been reported to block the agonist-induced release of arachidonic acid (Pratt et al., 1998) and to hyperpolarize guinea-pig carotid arteries and thus attenuate the hyperpolarization observed in response to ACh (Chataigneau et al., 1998).
The patch clamp bioassay, in which HU 210 attenuated EDHF-mediated hyperpolarizations only when applied to the donor artery, provided the most convincing evidence that CB1 agonists interfere with the generation of EDHF by endothelial cells rather that its effect on smooth muscle membrane potential. Exactly how the CB1 agonists interfere with the production of EDHF is at the moment unclear but cannabinoids are able to intercalate into cell membranes and have been reported to alter the properties of cellular proteins including G protein-coupled receptors. Such effects are however unlikely to account for the results observed in the present study as neither anandamide nor HU 210 markedly altered basal [Ca2+]i or the bradykinin-induced Ca2+-response in cultured human umbilical vein endothelial cells, which express mRNA for the CB1 receptor (data not shown), and neither Δ9-THC nor HU 210 affected ACh or bradykinin-induced, NO-mediated relaxations.
Given that the exact chemical identity of EDHF remains to be elucidated, it is difficult to investigate the signal transduction cascade which results in the activation of the ‘EDHF synthase'. Current opinion is that the synthesis of EDHF is secondary to the activation of phospholipase A2 (PLA2), and the liberation of arachidonic acid (Quilley et al., 1997). Indeed, the synthesis of EDHF is thought to be Ca2+-dependent by virtue of the reported Ca2+-dependency of PLA2. Thereafter, the signalling pathway is obscure and although EDHF may be generated from arachidonic acid by CYP enzymes in some vascular beds this is almost certainly not the case in others (Mombouli & Vanhoutte, 1997; Quilley et al., 1997). Since the EDHF produced in all of the arteries under investigation has been pharmacologically characterized as a CYP-dependent metabolite of arachidonic acid, it seemed plausible to suggest that the CB1 agonists attenuated EDHF production by decreasing the activity of the ‘EDHF synthase'. However, although cannabidiol has been reported to inactivate several CYP enzymes (Bornheim et al., 1993), and anandamide and Δ9-THC did attenuate ethoxyresorufin dealkylase activity in intact endothelial cells, HU 210 was without effect. Thus CYP inhibition seems unlikely to be the major mechanism by which the CB1 agonists attenuate the production of EDHF. It is nonetheless impossible to rule out the possibility that HU 210, Δ9-THC and anandamide affect the activity of CYP enzymes which do not metabolise 7-ethoxyresorufin.
The binding of anandamide to cannabinoid receptors leads to the inhibition of adenylyl cyclase and anandamide has been shown to inhibit forskolin-stimulated cyclic AMP accumulation in a number of cell systems expressing the CB1 receptor (Felder et al., 1993; Vogel et al., 1993; Childers et al., 1994). One previous study showed that isoprenaline induced an endothelium-dependent relaxation in rat aortic rings which appeared to be mediated by the release of a CYP-dependent metabolite of arachidonic acid (Satake et al., 1997). Although HU 210 and Δ9-THC attenuated both basal cyclic AMP levels as well as the isoprenaline-stimulated increase in cyclic AMP in cultured endothelial cells, a selective inhibitor of protein kinase A did not affect EDHF-mediated dilation.
Novel effects of CB1 receptor activation are continuously being described, for example it was recently reported that coupling exists between CB1 receptors and receptor tyrosine kinase-activated transduction pathways (Bouaboula et al., 1997). Since SR 141716A has been shown to interfere with the activation of mitogen-activated protein (MAP) kinases following receptor tyrosine kinase activation by insulin and IGF1 (Bouaboula et al., 1997), it is feasible that CB1 agonists affect signalling pathways which may be involved in the generation of EDHF. Although it is tempting to suggest that CB1 agonists interfere with the signal transduction pathway initiated by ACh and bradykinin, such a general effect cannot account for the selective inhibition of EDHF production. The only way that signal crosstalk could influence the production of EDHF selectively is if the pathways for EDHF production are regulated independently of that resulting in an increase in NO production. Such an idea would contradict the general concept that EDHF synthesis is activated simply in response to an increase in [Ca2+]i.
Assuming that an increase in [Ca2+]i is the essential signal to initiate the generation of EDHF, one might speculate that the CB1 agonists interfere with EDHF-mediated responses by uncoupling cell-cell communication. The possibility that EDHF is a substance which is capable of passing through gap junctions is not necessarily at odds with the observations made using bioassay models, as it is conceivable that this factor may spill over into the extracellular fluid. Anandamide has been reported to inhibit gap junction conductance and dye permeability in astrocytes in a pertussis toxin-sensitive manner (Venance et al., 1995; Schmilinsky-Fluri et al., 1997). Moreover, cell-cell communication in connexin 43-expressing cells is rapidly and transiently disturbed by G protein-coupled receptors (Postma et al., 1998) via a process involving the activation of p60Src. Other indications that CB1 agonists interfere with gap junction conductance can be indirectly inferred from observations that MAP kinases are involved in the regulation of connexin 43-based cell-cell communication (Kanemitsu & Lau, 1993) and that anandamide (Wartmann et al., 1995) and HU 210 (authors unpublished observations) can activate MAP kinase in cells expressing the CB1 receptor. Given that peptides homologous to extracellular loop motifs of connexin 43 attenuate EDHF-mediated relaxations (Chaytor et al., 1997), an inhibitory effect of CB1 agonists on myo-endothelial and/or inter-endothelial gap junctions is an attractive hypothesis, currently under investigation, which may explain the abolition of EDHF-mediated relaxations by HU 210 and Δ9-THC.
Acknowledgments
The authors are indebted to Isabel Winter for expert technical assistance. This study was supported by the Deutsche Forschungsgemeinschaft (SFB 553, B1) and the Commission of the European Communities (BMH4-CT96-0979).
Abbreviations
- ACh
acetylcholine
- CB1
cannabinoid CB1 receptor
- CYP
cytochrome P450
- Δ9-THC
Δ9-tetrahydrocannabinol
- EDHF
endothelium-derived hyperpolarizing factor
- HUVEC
human umbilical vein endothelial cells
- L-NA
NGnitro-L-arginine
- NO
nitric oxide
References
- BAUERSACHS J., HECKER M., BUSSE R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid metabolite in the coronary microcirculation. Br. J. Pharmacol. 1994;113:1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUERSACHS J., POPP R., HECKER M., SAUER E., FLEMING I., BUSSE R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- BERDYSHEV E.V., BOICHOT E., LAGENTE V. Anandamide – A new look on fatty acid ethanolamides. J. Lipid Mediators. Cell Signal. 1996;15:49–67. doi: 10.1016/s0929-7855(96)00548-2. [DOI] [PubMed] [Google Scholar]
- BORNHEIM L.M., KIM K.Y., CHEN B., CORREIA M.A. The effect of cannabidiol on mouse hepatic microsomal cytochrome P450-dependent anandamide metabolism. Biochem. Biophys. Res. Commun. 1993;197:740–746. doi: 10.1006/bbrc.1993.2541. [DOI] [PubMed] [Google Scholar]
- BOUABOULA M., PERRACHON S., MILLIGAN L., CANAT X., RINALDI-CARMONA M., PORTIER M., BARTH F., CALANDRA B., PECCEU F., LUPKER J., MAFFRAND J.-P., LE FUR G., CASELLAS P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FELETOU M., THOLLON C., VILLENEUVE N., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Cannabinoid CB1 receptor and endothelium-dependent hyperpolarization in guinea-pig carotid, rat mesenteric and porcine coronary arteries. Br. J. Pharmacol. 1998;123:968–974. doi: 10.1038/sj.bjp.0701690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Peptides homologous to the extracellular loop motifs of connexin 43 reversibly abolish rhythmic contractile activity in rabbit arteries. J. Physiol. (Lond.) 1997;503:99–110. doi: 10.1111/j.1469-7793.1997.099bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHILDERS S.R., SEXTON T., ROY M.B. Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem. Pharmacol. 1994;47:711–715. doi: 10.1016/0006-2952(94)90134-1. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., GOLIGORSKY M.S., SCHMID P.C., KREBSBACH R.J., SCHMID H.H., DAS S.K., DEY S.K., ARREAZA G., THORUP C., STEFANO G., MOORE L.C. Production and physiological actions of anandamide in the vasculature of the rat kidney. J. Clin. Invest. 1997;100:1538–1546. doi: 10.1172/JCI119677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLIS E.F., MOORE S., WILLOUGHBY K.A. Anandamide and D9-THC dilation of cerebral arterioles is blocked by indomethacin. Am. J. Physiol. 1995;269:H1859–H1864. doi: 10.1152/ajpheart.1995.269.6.H1859. [DOI] [PubMed] [Google Scholar]
- ESTRADA U., BRASE D.A., MARTIN B.R., DEWEY W.L. Cardiovascular effects of Δ9- and Δ9(11)-tetrahydrocannabinol and their interaction with norepinephrine. Life Sci. 1987;41:79–87. doi: 10.1016/0024-3205(87)90559-5. [DOI] [PubMed] [Google Scholar]
- FACCI L., DAL TOSO R., ROMANELLO S., BURIANI A., SKAPER S.D., LEON A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAN P. Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurones. J. Neurophysiol. 1995;736:907–910. doi: 10.1152/jn.1995.73.2.907. [DOI] [PubMed] [Google Scholar]
- FELDER C.C., BRILEY E.M., AXELROD J., SIMPSON J.T., MACKIE K., DEVANE W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEMING I., FISSLTHALER B., BUSSE R. Calcium signaling in endothelial cells involves activation of tyrosine kinases and leads to activation of MAP kinase. Circ. Res. 1995;76:522–529. doi: 10.1161/01.res.76.4.522. [DOI] [PubMed] [Google Scholar]
- FLEMING I., FISSLTHALER B., BUSSE R. Interdependence of calcium signaling and protein tyrosine phosphorylation in human endothelial cells. J. Biol. Chem. 1996;271:11009–11015. doi: 10.1074/jbc.271.18.11009. [DOI] [PubMed] [Google Scholar]
- GALIEGUE S., MARY S., MARCHAND J., DUSSOSSOY D., CARRIERE D., CARAYON P., BOUABOULA M., SHIRE D., LE FUR G., CASELLAS P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- GRAHAM J.D.P., LI D.M.F. Cardiovascular and respiratory effects of cannabis in cat and rat. Br. J. Pharmacol. 1973;49:1–10. [PMC free article] [PubMed] [Google Scholar]
- HOPKINS N.K., GORMAN R.R. Regulation of endothelial cell cyclic nucleotide metabolism by prostacyclin. J. Clin. Invest. 1981;67:540–546. doi: 10.1172/JCI110064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNTER S.A., BURSTEIN S.H. Receptor mediation in cannabinoid stimulated arachidonic acid mobilization and anandamide synthesis. Life Sci. 1997;60:1563–1573. doi: 10.1016/s0024-3205(97)00122-7. [DOI] [PubMed] [Google Scholar]
- KANEMITSU M.Y., LAU A.F. Epidermal growth factor stimulates the disruption of gap junction communication and connexin43 phosphorylation independent of 12-O-tetradeacnoyl-phorbol 13-acetate-sensitive protein kinase C: the possible involvement of mitogen-activated protein kinase. Mol. Biol. Cell. 1993;4:827–848. doi: 10.1091/mbc.4.8.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOMBOULI J.-V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MUNRO S., THOMAS K.L., ABU-SHAAR M. Molecular characterization of a peripheral receptor for cannabinoid. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- PLANE F., HOLLAND M., WALDRON G.J., GARLAND C.J., BOYLE J.P. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br. J. Pharmacol. 1997;121:1509–1512. doi: 10.1038/sj.bjp.0701361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesised by native and cultured porcine coronary endothelial cells. J. Physiol. (Lond.) 1996;497.3:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., FLEMING I., BUSSE R. Pulsatile stretch elicits the release of the endothelium-derived hyperpolarizing factor from isolated coronary arteries: a modulator of arterial compliance. Circ. Res. 1998;81:696–703. doi: 10.1161/01.res.82.6.696. [DOI] [PubMed] [Google Scholar]
- POSTMA F.R., HENGEVELD T., ALBLAS J., GIEPMANS B.N., ZONDAG G.C., JALINK K., MOOLENAAR W.H. Acute loss of cell-cell communication caused by G protein-coupled receptors: A critical role for c-Src. J. Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRATT P.F., HILLARD C.J., EDGEMOND W.S., CAMPBELL W.B. N-arachidonylethanolamide relaxation of bovine coronary artery is not mediated by CB1 cannabinoid receptor. Am. J. Physiol. 1998;43:H375–H381. doi: 10.1152/ajpheart.1998.274.1.H375. [DOI] [PubMed] [Google Scholar]
- PREVOT V., RIALAS C.M., CROIX D., SALZET M., DUPOUY J.P., POULAIN P., BEAUVILLAIN J.C., STEFANO G.B. Morphine and anandamide coupling to nitric oxide stimulates GnRH and CRF release from rat median eminence: neurovascular regulation. Brain Res. 1998;790:236–244. doi: 10.1016/s0006-8993(98)00066-3. [DOI] [PubMed] [Google Scholar]
- QUILLEY J., FULTON D., MCGIFF J.C. Hyperpolarizing factors. Biochem. Pharmacol. 1997;54:1059–1070. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., ALEXANDER S.P.H., BENNETT T., BOYD E.A., FRY J.R., GARDINER S.M., KEMP P.A., MCCULLOCH A.I., KENDALL D.A. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem. Biophys. Res. Commun. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur. J. Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- SATAKE N., SHIBATA M., SHIBATA S. Endothelium- and cytochrome P-450-dependent relaxation induced by isoproterenol in rat aortic rings. Eur. J. Pharmacol. 1997;319:37–41. doi: 10.1016/s0014-2999(96)00822-9. [DOI] [PubMed] [Google Scholar]
- SCHMILINSKY-FLURI G., VALIUNAS V., WILLI M., WEINGART R. Modulation of cardiac gap junctions: the mode of action of arachidonic acid. J. Mol. Cell Cardiol. 1997;29:1703–1713. doi: 10.1006/jmcc.1997.0409. [DOI] [PubMed] [Google Scholar]
- SHIRE D., CARILLON C., KAGHAD M., CALANDRA B., RINALDI-CARMONA M., LE FUR G., CAPUT D., FERRARA P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J. Biol. Chem. 1995;270:3726–3731. doi: 10.1074/jbc.270.8.3726. [DOI] [PubMed] [Google Scholar]
- SUGIURA T., KODAKA T., NAKANE S., KISHIMOTO S., KONDO S., WAKU K. Detection of an endogenous cannabimimetic molecule, 2- arachidonoylglycerol, and cannabinoid CB1 receptor mRNA in human vascular cells: Is 2-arachidonoylglycerol a possible vasomodulator. Biochem. Biophys. Res Commun. 1998;243:838–843. doi: 10.1006/bbrc.1998.8187. [DOI] [PubMed] [Google Scholar]
- VARGA K., LAKE K., MARTIN B.R., KUNOS G. Novel antagonist implicates the CB1 cannabinoid receptor in the hypotensive action of anandamide. Eur. J. Pharmacol. 1995;278:279–283. doi: 10.1016/0014-2999(95)00181-j. [DOI] [PubMed] [Google Scholar]
- VENANCE L., PIOMELLI D., GLOWINSKI J., GIAUME C. Inhibition by anandamide of gap junctions and intercellular calcium signaling in striatal astrocytes. Nature. 1995;376:590–594. doi: 10.1038/376590a0. [DOI] [PubMed] [Google Scholar]
- VIDRIO H., SÁNCHEZ-SALVATORI M.A., MEDINA M. Cardiovascular effects of (−)-11-OH-delta8-tetrahydrocannabinol-dimethylheptyl in rats. J. Cardiovasc. Pharmacol. 1996;28:332–336. doi: 10.1097/00005344-199608000-00022. [DOI] [PubMed] [Google Scholar]
- VOGEL Z., BARG J., LEVY R., SAYA D., HELDMAN E., MECHOULAM R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 1993;61:352–355. doi: 10.1111/j.1471-4159.1993.tb03576.x. [DOI] [PubMed] [Google Scholar]
- WARTMANN M., CAMPBELL D., SUBRAMANIAN A., BURSTEIN S.H., DAVIS R.J. The MAP kinase signal transduction pathway is activated by the endogenous cannabinoid anandamide. FEBS Lett. 1995;359:133–136. doi: 10.1016/0014-5793(95)00027-7. [DOI] [PubMed] [Google Scholar]
- YATOMI Y., OZAKI Y., KUME S. Synthesis of phosphatidylinositol 3,4-bisphosphate but not phosphatidylinositol 1,3,5-trisphosphate is closely correlated with protein tyrosine phosphorylation in thrombin-activated human platelets. Biochem. Biophys. Res. Commun. 1992;186:1480–1486. doi: 10.1016/s0006-291x(05)81573-6. [DOI] [PubMed] [Google Scholar]