

Abstract
The aim of this study was to assess the inhibitory activities of phosphodiesterase type 4 (PDE4) inhibitors on tumour necrosis factor-α (TNF-α) and leukotriene B4 (LTB4) production in a novel human whole blood assay.
Lipopolysaccharide (LPS) stimulation of human whole blood caused a time dependent increase in TNF-α and prostaglandin E2 (PGE2) plasma levels. Inhibition of LPS-induced TNF-α by the selective PDE4 inhibitor RP73401 was proportionally enhanced with endogenous PGE2 (maximal after 24 h). In contrast, blocking endogenous PGE2 production with indomethacin in blood stimulated with LPS for 24 h decreased the potency of RP73401 to that observed with a 4 h LPS incubation.
Non-selective and selective PDE4 inhibitors showed greater inhibition of LPS-induced TNF-α after 24 h compared to 4 h. Stereoselectivity was only achieved in the 24 h method.
LPS-stimulation of whole blood for either 30 min or 24 h followed by N-formyl-Met-Leu-Phe (fMLP) activation resulted in low plasma LTB4 levels. Combination of both treatments resulted in a greater than 7 fold increase in plasma LTB4 levels. Inhibition of the double LPS and fMLP-activated LTB4 production was observed with non-selective and PDE4-selective inhibitors. Their LTB4 inhibitory potencies were similar to that observed in the 24 h LPS-induced TNF-α assay. Thus, stimulation of human whole blood with two LPS stimulations followed by fMLP gives rise to both TNF-α and LTB4 and their inhibition by various compounds can be assessed in the same blood sample.
Calcium ionophore (A23187) stimulation of whole blood resulted in plasma LTB4 levels similar to the double LPS and fMLP method. Inhibition of A23187-induced LTB4 biosynthesis was also achieved by PDE4-selective inhibitors as well as the direct 5-lipoxygenase (5-LO) inhibitor L-739,010.
These results confirm the anti-inflammatory properties of PDE4 inhibitors. Thus, this novel human whole blood can be used to assess the biochemical efficacy of PDE4 inhibitors in human subjects.
Keywords: Phosphodiesterase, tumour necrosis factor α, leukotriene B4, whole blood
Introduction
Phosphodiesterase 4 (PDE4), a member of the phosphodiesterase family of at least nine known distinct types (PDE1 to PDE9), is a cyclic adenosine monophosphate (cyclic AMP)-specific enzyme and is responsible for the hydrolysis of intracellular cyclic AMP to 5′-AMP. Inhibition of this enzyme leads to increases in intracellular cyclic AMP which can subsequently activate protein kinase A (PKA) (Torphy & Undem, 1991; Seldon et al., 1995). It has been shown with various PDE4-specific inhibitors that an increase in intracellular cyclic AMP can inhibit the expression and release of several inflammatory mediators, prevent superoxide anion formation in neutrophils, inhibit eosinophil chemotaxis and degranulation as well as lymphocyte proliferation, and causes relaxation of bronchial smooth muscles resulting in bronchodilatation (Nielson et al., 1990; Hartman et al., 1993; Underwood et al., 1994; Schudt et al., 1995; Essayan et al., 1997). PDE4 is predominantly expressed in monocytes, neutrophils and eosinophils (Schudt et al., 1995) and its inhibition could be beneficial in treating the underlying inflammation associated with asthma.
Tumour Necrosis Factor-α (TNF-α) is a very powerful proinflammatory mediator produced by activated macrophages, blood monocytes and mast cells (Gordon & Galli, 1991). In addition to its anti-tumour properties (Jia & Kleinerman, 1991), it plays a central role in many inflammatory processes (Warren et al., 1988). It is a critical cytokine in the host defence of bacterial infections (Beutler & Cerami, 1989; Nakane et al., 1992) but it is also associated with shock and tissue injury during sepsis leading to organ failure (Tracey et al., 1986). It has many biological activities on a wide variety of cell types (endothelial cells, lymphocytes, osteoclasts) and can upregulate or induce expression of receptors (e.g. interferon (IFN)-gamma, TNF-α p55 receptors), cytokines, growth factors, inflammatory mediators and adhesion molecules as well as activate transcription factors (e.g. NF-κB) (Dinarello et al., 1986; Scheurich et al., 1987; Doerfler et al., 1989; Valyi-Nagy et al., 1992; Krakauer & Oppenheim, 1993; Trefzer et al., 1993; Mackay et al., 1993; Arias-Negrete et al., 1995; Schutze et al., 1995; Batten et al., 1996). This cytokine has been implicated in several inflammatory diseases (rheumatoid arthritis and Crohn's disease) and its inhibition has been the focus of many studies (Eigler et al., 1997Eigler et al., 1997).
Leukotriene B4 (LTB4) is a product of arachidonic acid metabolism of the 5-lipoxygenase (5-LO) pathway (Borgeat & Samuelsson, 1979). Leukotrienes are also proinflammatory mediators and are synthesized by many cells such as macrophages, monocytes, eosinophils, mast cells and neutrophils. LTB4 is a major product of activated neutrophils and a potent chemoattractant. It can exert its effects on cells and tissues by increasing aggregation and release of lysosomal enzymes, stimulating superoxide anion production in neutrophils, increasing interleukin-2 (IL-2) release and receptors in lymphocytes and enhancing vascular permeability in tissues (Ford-Hutchinson, 1990). Psoriasis, inflammatory bowel disease (IBD), rheumatoid arthritis and mostly asthma have all been associated with the potent inflammatory properties of LTB4 (Davidson et al., 1983; Brain et al., 1984; Grabbe et al., 1984; Sharon & Stenson, 1984). Inhibitors of 5-LO and 5-LO activating protein (FLAP) have been developed as therapeutic agents for the treatment of asthma and their effects on psoriasis and IBD have also been studied (Larsen & Jackson, 1996).
TNF-α has been shown to be inhibited at the transcriptional level by increases in cyclic AMP due to PDE4 inhibition or activation of adenylyl cyclase (Han et al., 1990). A synergistic effect with both PDE4 inhibitors and activators of adenylyl cyclase (e.g. PGE2) has been shown to greatly inhibit TNF-α (Schade & Schudt, 1993). The effects of PDE4 inhibitors on LTB4 have not been studied as much as TNF-α but it has been shown that inhibition of LTB4 in polymorphonuclear cells (PMNs) can be achieved by the non-selective PDE inhibitors IBMX (3-isobutyl-1-methylxanthine) and theophylline, the PDE4 selective inhibitor rolipram and the β-agonist isoproteronol. Studies have shown that arachidonic acid metabolites such as platelet activating factor (PAF) and LTB4 are inhibited by increases in cyclic AMP via the regulation of phospho-lipase A2 (PLA2) activity and the modulation of intracellular calcium concentrations (Fonteh et al., 1993; Villagrasa et al., 1996). For the reason that TNF-α and LTB4 are key mediators in inflammation and are both sensitive to intracellular cyclic AMP levels, they represent good surrogate markers of PDE4 activity.
Activation of isolated blood leukocytes is the most common in vitro method used to examine the intrinsic potency of PDE4 inhibitors, however these assays usually use low serum concentrations, hence a low protein environment, and cannot predict activity in whole blood. Furthermore, the effects of PDE4 inhibition have only been studied on one surrogate marker at a time per cell type using various stimulations such as LPS-induced TNF-α in monocytes or fMLP activated PMNs. In this study, we have developed a human whole blood assay which can be used to examine the effects of non-selective and selective PDE inhibitors as well as activators of adenylyl cyclase on both TNF-α and LTB4 simultaneously in a protein-rich environment.
Methods
LPS-induced TNF-α
Fresh blood was collected in heparinized tubes by venipuncture from both male and female volunteers with consent. The subjects had no apparent inflammatory conditions and had not taken any nonsteroidal anti-inflammatory drugs (NSAIDs) for at least 4 days prior to blood collection. Five hundred μl aliquots of blood were pre-incubated with either 2 μl vehicle or test compound at 37°C for 15 min. This was followed by incubation of the blood with 10 μl lipopolysaccharide (LPS) (Sigma, St Louis, MO, U.S.A., from E. coli serotype 0111 : B4, 1 μg ml−1 final concentration, diluted in 0.1% bovine serum albumin (BSA) (Sigma, St Louis, MO, U.S.A., fraction V, diluted in phosphate buffered saline (PBS))) for 0.5–24 h at 37°C. Appropriate PBS controls (no LPS) were used as blanks. After the desired incubation period, the samples were centrifuged at 1500×g at 4°C for 10 min. Plasma TNF-α was quantified ELISA (Cistron Biotechnology, Pine Brook, NJ, U.S.A.). A 50 μl aliquot of plasma was mixed with 200 μl methanol for protein precipitation and centrifuged at 1500×g at 4°C for 10 min. The supernatant was obtained and PGE2 was determined by radioimmunoassay (Amersham, Oakville, Ont, Canada) after conversion of PGE2 to its methyl oximate derivative according to manufacturer's procedure.
fMLP-stimulation of LTB4
Heparinized human whole blood was pre-incubated with either vehicle or test compound as described above and then incubated with 10 μl of LPS (1 μg ml−1 final concentration, diluted in 0.1% BSA) for either 30 min or 24 h at 37°C. This was followed by incubation of 10 μl of the chemotactic peptide n-formyl-Met-Leu-Phe (fMLP) (Sigma, St Louis, MO, U.S.A., diluted in 1% BSA, 1 μM final concentration), or PBS for blank controls, for 15 min at 37°C. For some experiments, the blood was incubated with fMLP immediately after drug pre-incubation without pre-stimulation with LPS. At the end of the fMLP incubation, the blood was immediately centrifuged in a pre-cooled centrifuge at 4°C for 10 min. The samples were left on ice during plasma transfer. The plasma was extracted in methanol as described above and the supernatant was assayed for LTB4 by enzyme immunoassay (Cayman Chemicals, Ann Arbor, MI, U.S.A.) according to manufacturer's instructions.
Double LPS-induced and fMLP-activated TNF-α and LTB4 assay
Five hundred μl aliquots of blood were pre-incubated with either vehicle or test compound at 37°C for 15 min. This was followed by incubation of the blood with 10 μl LPS (1 μg ml−1 final concentration, diluted in 0.1% BSA) at 37°C in a humidified incubator (5% CO2). Appropriate PBS control (no LPS) were used as blanks. After the 24 h incubation, another 10 μl of LPS (1 μg ml−1 final concentration, diluted in 0.1% BSA) was added to blood (PBS for blanks) and incubated for 30 min at 37°C. The samples were then challenged for 15 min at 37°C with either 10 μl fMLP (diluted in 1% BSA, 1 μM final concentration) or PBS for blanks. The blood was centrifuged as above and the plasma obtained was quantified for both TNF-α and LTB4 content as described above.
A23187-stimulation of LTB4
Heparinized human whole blood was pre-incubated with either vehicle or test compound as described above and then incubated with 2 μl of the calcium ionophore A23187 (Sigma, St Louis, MO, U.S.A., diluted in DMSO, 25 μM final concentration) or 2 μl of dimethyl sulphoxide (DMSO) as blank controls for 30 min at 37°C. The following procedure was identical to the fMLP method for measurement of LTB4.
Statistics
Results are expressed as mean and s.e.mean. Differences between controls and treatment groups were tested using the Student's t-test. A P-value of less than 0.05 was considered statistically significant.
Materials
RS14203 (Wilhelm & Fatheree, 1994), RP73401 (Souness et al., 1995), T-440 (Kaminuma et al., 1996), R- and S-rolipram, SB207499 (Christensen et al., 1998), CDP840 (Hughes et al., 1996), CT1731 (Hughes et al., 1996), Trequinsin, L-739,010 (Hamel et al., 1997), MF-tricyclic (selective cyclo-oxygenase-2 (COX-2) inhibitor) (Oshima et al., 1996) were synthesized by the Medicinal Chemistry departments at Merck Frosst Centre for Therapeutic Research and Celltech Therapeutics, U.K. Other compounds and their sources were: Indomethacin (Merck & Co., Rahway, NJ, U.S.A.), PGE2 (Cayman Chemicals, Ann Arbor, MI, U.S.A.), amrinone, dibutyryl-cyclic AMP, isoproteronol, 3-isobutyl-1-methylxanthine (IBMX), pentoxyfylline, theophylline, zaprinast (Sigma, St Louis, MO, U.S.A.), dipyridamole, milrinone, quazinone, forskolin (Calbiochem, La Jolla, CA, U.S.A.) and cholera toxin (Biomol, Plymouth Meeting, PA, U.S.A.).
Results
Time course
Figure 1 shows the time course of TNF-α and PGE2 production after LPS-stimulation of whole blood. During the incubation period, a sharp rise in TNF-α levels was observed from 2 h (2.70±0.76 ng ml−1) to 4 h (15.59±3.61 ng ml−1) and peaked at 8 h (22.92±4.77 ng ml−1). The levels obtained at 4, 6, 8 and 24 h were not significantly different from each other. Out of the three subjects used for this experiment, two had maximal TNF-α levels at 8 h whereas the other reached its peak at 4 h. In all cases the levels were lower after 24 h (17.33±2.52 ng ml−1) compared to levels at 8 h. PGE2 levels did not increase significantly over baseline until 4 h (0.88±0.15 nM) and rose steadily from 8 h (1.96±0.16 nM) to 24 h (16.94±2.79 nM) due to increased COX-2 expression as previously shown (Brideau et al., 1996). Addition of 10 μM RP73401 inhibited TNF-α production at 4, 6, 8 and 24 h by 48, 67, 75 and 78% respectively. Interestingly, this inhibition increased with rising levels of PGE2 and was most significant at 24 h.
Figure 1.
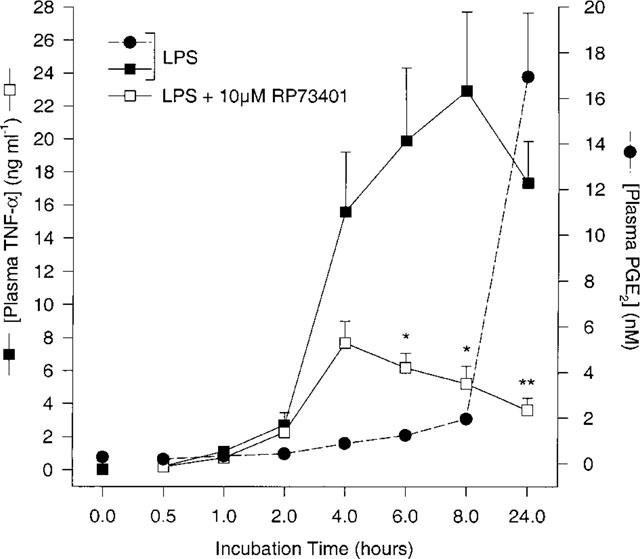
Time course of TNF-α and PGE2 production in human whole blood following LPS (1 μg ml−1) incubation in the presence or absence of a PDE4 inhibitor (10 μM RP73401). n=3 donors, *P<0.05 and **P<0.01 vs LPS-induced TNF-α alone.
TNF-α production: Effect of PGE2 and LPS incubation time
Most of the published methods for TNF-α induction and inhibition by various agents in human whole blood use a 4 h LPS incubation period (Hartman et al., 1993; 1995). To determine whether 4 h is optimal for characterizing PDE4 inhibitors, we examined the effects of LPS incubation time and PGE2 on TNF-α inhibition. The concentration of PGE2 required to inhibit half of the TNF-α produced (or IC50) in the 4 h LPS-stimulation assay was 15 nM in whole blood (Figure 2a). In the same assay, the IC50 for RP73401 was 9.5 μM and the maximum inhibition observed was 68% at 10 μM. The addition of 1 nM or 10 nM PGE2 resulted in a dose dependent increase in TNF-α inhibition by RP73401 (Figure 2b). The maximal response observed with 10 μM RP73401 rose to 97% in the presence of 10 nM PGE2.
Figure 2.
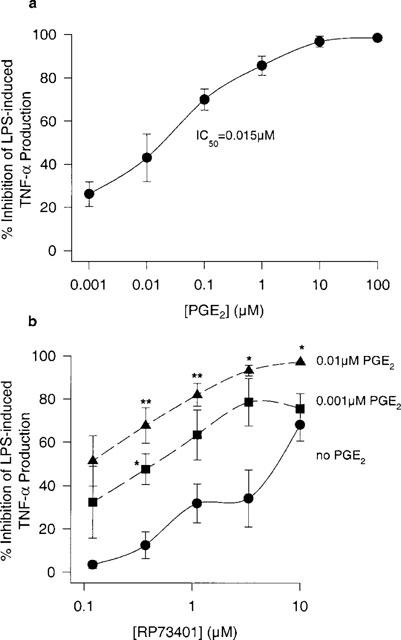
The potentiation of the inhibitory effect of a PDE4 inhibitor (RP73401) by exogenous PGE2 on LPS-induced TNF-α production (4 h incubation). (a) Effect of exogenous PGE2 alone. (b) Effect of RP73401 in the absence or presence of PGE2. n=3 donors. *P<0.05 and **P<0.01 vs RP73401 alone.
In many experiments performed in our laboratory using PDE4-selective inhibitors, maximal inhibitions of greater than 70% could not be achieved in the 4 h LPS-induced TNF-α assay (data not shown). Moreover, we showed greater inhibition of LPS-induced TNF-α with RP73401 after 8 h of incubation which coincided with increased PGE2 levels (Figure 1). Therefore, we compared the potency of RP73401 after 4 and 24 h LPS incubations. As represented in Figure 3a, the mean plasma TNF-α levels increased at least 6 fold after 24 h LPS incubation compared to 4 h. In Figure 3b, the inhibitory effect of RP73401 on LPS-induced TNF-α was greater after 24 h. To establish the importance of PGE2 in the 24 h assay, indomethacin was incubated simultaneously with LPS for 24 h to inhibit the synthesis of PGE2. Indomethacin completely blocked the 24 h LPS-induced PGE2 production and had no effect on TNF-α levels (Figure 3a). The potency of RP73401 on LPS-induced TNF-α was significantly reduced in the presence of indomethacin and was similar to that observed with the 4 h LPS incubation (Figure 3b).
Figure 3.
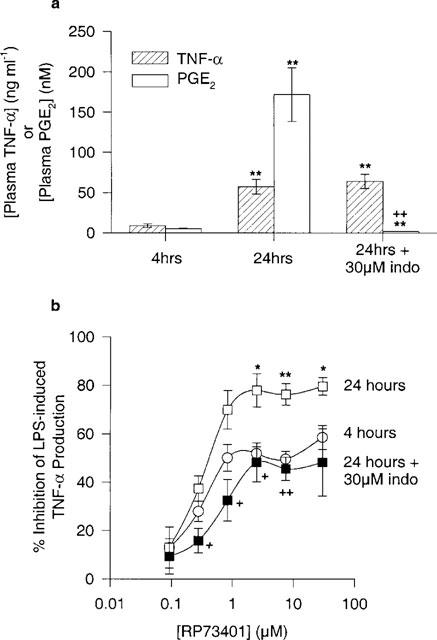
Comparison between 4 and 24 h incubation of human whole blood with 1 μg ml−1 LPS in addition to a 24 h LPS incubation in the presence of 30 μM indomethacin. (a) plasma TNF-α and PGE2 levels and (b) effect of RP73401 on LPS-induced TNF-α. Experiments performed in same blood sample. n=3 donors. *P<0.05 and **P<0.01 vs 4 h incubation and +P<0.05 and ++P<0.01 vs 24 h LPS only.
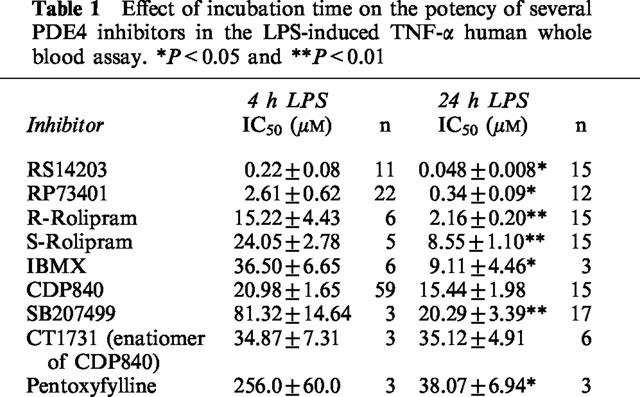
Table 1 summarizes the IC50 values of several PDE4 inhibitors in both the 4 and 24 h assays. All inhibitors, except CT1731 and CDP840, showed significantly higher potency in the 24 h assay. The maximum inhibitions observed in the 24 h assay were greater than 80% for all compounds. Significant (P<0.01) differences between enantiomeric pairs such as R-rolipram and S-rolipram as well as CDP840 and CT1731 were only achieved in the 24 h LPS incubation method.
Table 1.
Effect of incubation time on the potency of several PDE4 inhibitors in the LPS-induced TNF-α human whole blood assay. *P<0.05 and **P<0.01
LTB4 production: Effects of LPS and fMLP at different incubation times
We compared the effects of LPS stimulation, fMLP activation and incubation time on LTB4 production in human whole blood. Figure 4 demonstrates the results of four different methods based on published procedures (Surette et al., 1993) as well as procedures modified in our laboratory. Incubation of blood with 1 μM fMLP for 15 min failed to increase LTB4 production compared to baseline and the negative PBS control (method A). Priming blood with LPS for 30 min prior to fMLP addition resulted in a significant increase in LTB4 levels (3.44±1.11 ng ml−1) over baseline whereas a 30 min LPS incubation without fMLP had no effect (method B, Surette et al., 1993). When LPS priming time was increased to 24 h as shown in method C, we obtained the similar results as in method B. More than a 6 fold increase (25.20±7.92 ng ml−1) in LTB4 levels was observed when whole blood was pre-stimulated with LPS for 24 h and then again for 30 min followed by fMLP stimulation (method D). Thus, combining the LPS priming from methods B and C had a much greater effect on LTB4 synthesis in human whole blood. We compared the effects of two PDE4 inhibitors on LTB4 synthesis in whole blood using methods B and D. Figure 5 demonstrates the inhibition curves for RS14203 (panel a) and RP73401 (panel b) in both methods. A greater maximal inhibition was observed for both compounds when the blood was pre-incubated with LPS for 24 h (method D). RS14203 exhibited a flat titration curve without the LPS pre-incubation and a plateau of approximately 66% inhibition was observed in the highest doses (method B) while 90% inhibition was achieved with the 24 h LPS pre-incubation (method D).
Figure 4.
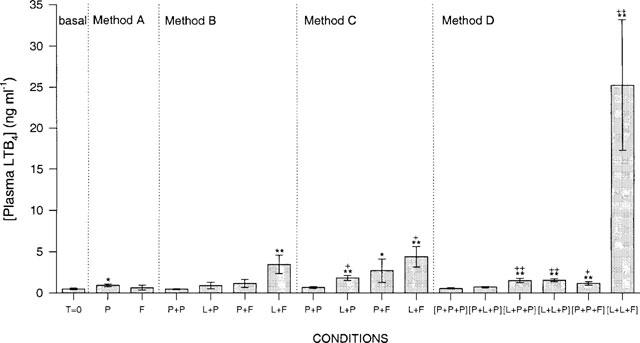
Plasma LTB4 levels from different conditions. Basal represents unstimulated blood plasma at time 0. P=PBS (blanks), L=LPS (1 μg ml−1) and F=fMLP (1 μM). Method A: human whole blood stimulated with fMLP for 15 min. Method B: pre-stimulation with either PBS or LPS (first letter on graph) for 30 min followed by incubation with either PBS or fMLP for 15 min (second letter on graph). Method C: pre-stimulation with either PBS or LPS for 24 h (first letter on graph) followed by incubation with either PBS or fMLP for 15 min (second letter on graph). Method D: pre-stimulation with either PBS or LPS (first letter on graph) for 24 h, then a 30 min stimulation with either PBS or LPS (second letter on graph) followed by incubation with either PBS or fMLP (third letter on graph) for 15 min. n=3–6 donor. *P<0.05 and **P<0.01 vs time 0 and +P<0.05 and ++P<0.01 vs PBS blank for each condition.
Figure 5.
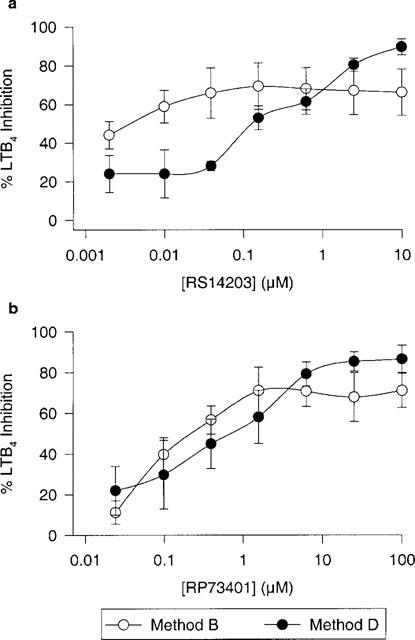
Comparison between two methods of fMLP-stimulated LTB4 human whole blood assays using two different PDE4 inhibitors (RS14203 and RP73401). Method B: whole blood pre-stimulated with LPS (1 μg ml−1) for 30 min followed by 15 min incubation with fMLP (1 μM) and Method D: whole blood pre-stimulated with LPS (1 μg ml−1) for 24 h followed by 30 min stimulation with LPS (1 μg ml−1) then a 15 min incubation with fMLP (1 μM). n=3 donors. Both methods were performed in the same donor sample.
Effect of double LPS pre-incubations and fMLP stimulation on TNF-α
As demonstrated above, TNF-α inhibition by PDE inhibitors was much more effective when whole blood was incubated with LPS for 24 h because of the presence of PGE2. We examined whether an additional 30 min incubation with LPS and a 15 min fMLP-stimulation would alter the levels of TNF-α and the efficacy of PDE inhibitors. Figure 6 shows a very good correlation between the simple 24 h assay (TNF-α method) and the elaborated method used for LTB4 (method D). TNF-α levels in the absence of PDE4 inhibitors were approximately 25% lower in method D but the inhibition responses of PDE inhibitors in the same blood sample were almost identical. Thus, method D can be used to study the effects of PDE4 inhibitors on both TNF-α and LTB4.
Figure 6.
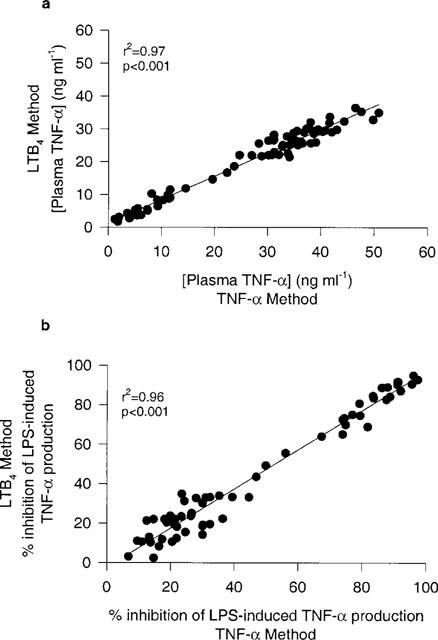
Linear correlation of results obtained from two methods of LPS-induced TNF-α in human whole blood. TNF-α Method: whole blood samples incubated with LPS (1 μg ml−1) for 24 h. LTB4 Method D: whole blood samples incubated with LPS (1 μg ml−1) for 24 h followed by 30 min incubation of LPS (1 μg ml−1) then a 15 min incubation with fMLP (1 μM). (a) represents plasma TNF-α levels from an experiment where both methods were done in the same blood sample in the presence and absence of varying concentrations of 10 different PDE4 inhibitors. n=71 data points. (b) represents the percentage inhibition of TNF-α of the same data points (except data points from positive no drug controls from which the percentage inhibition was calculated). n=61 data points. All data represented were blood samples from one donor.
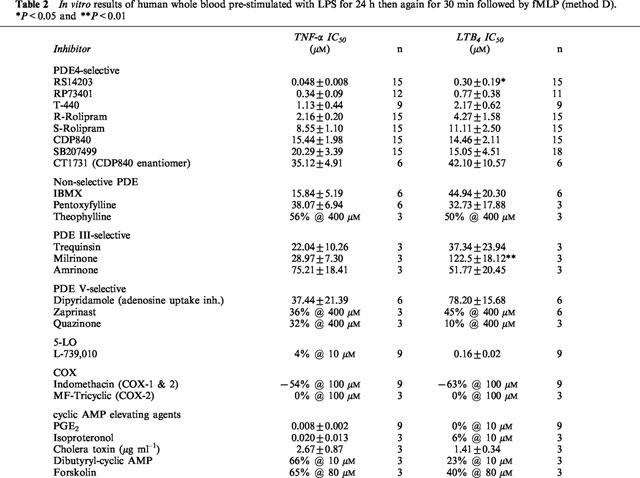
Table 2 summarizes the IC50 values of several PDE inhibitors, cyclic AMP elevating agents, inhibitors of cyclo-oxygenase (COX-1/COX-2) and a 5-LO inhibitor on inhibition of either TNF-α or LTB4 from the same blood sample. All of the PDE4-selective inhibitors were able to inhibit both TNF-α and LTB4 with similar potency except RS14203 and milrinone which were significantly more potent against TNF-α. Agents which elevate intracellular cyclic AMP by activating adenylyl cyclase are more potent inhibitors of TNF-α than LTB4 except for cholera toxin which had similar potency. The non-selective PDE inhibitors and the PDE III-selective inhibitors were not potent against either mediator and the PDE V-selective inhibitors were not active. The activity observed with dipyridamole was probably due to its inhibition on adenosine uptake by red blood cells rather than its inhibition of PDE V (Krump et al., 1996; Eigler et al., 1997Eigler et al., 1997). The 5-LO inhibitor L-739,010 had no effect on TNF-α but was very potent against LTB4. The selective inhibition of COX-2 by MF-tricyclic had no effect on either TNF-α or LTB4 whereas an increase in TNF-α and LTB4 production was observed with the non-selective COX inhibitor indomethacin.
Table 2.
In vitro results of human whole blood pre-stimulated with LPS for 24 h then again for 30 min followed by fMLP (method D). *P<0.05 and **P<0.01
Effects of PDE inhibitors on A23187-stimulated LTB4
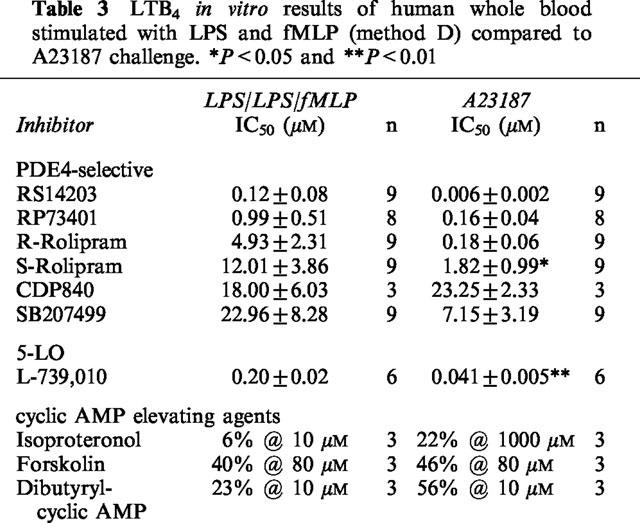
To confirm the LTB4 inhibitory effects of PDE4 inhibitors, a non-receptor mediated whole blood assay was performed using the calcium ionophore, A23187, without LPS pre-stimulation. PDE4-selective inhibitors, cyclic AMP elevating agents and a 5-LO inhibitor were examined in whole blood challenged with 25 μM A23187 for 30 min. The plasma LTB4 levels obtained by A23187 were very similar to the double LPS-fMLP-stimulated whole blood method (method D), 19.30±3.76 ng ml−1 and 20.87±4.41 ng ml−1 respectively (Figure 7a). Inhibition of LTB4 by RS14203 was significantly greater in blood challenged with A23187 (Figure 7b). Several compounds were tested in both assays using the same blood sample and their results are summarized in Table 3. Although these compounds were more potent in A23187-challenged blood, with the exception of CDP840, none were found to be significantly different aside from S-rolipram and L-739,010.
Figure 7.
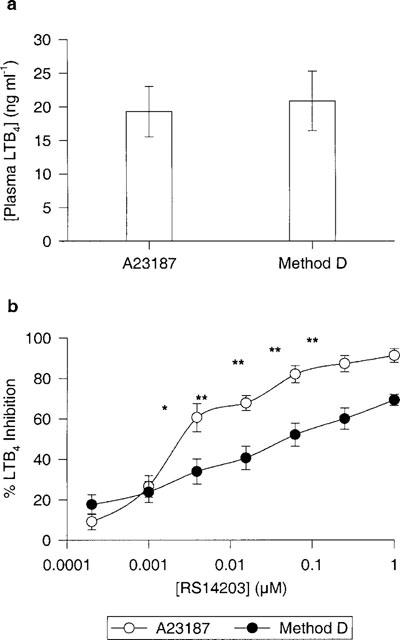
Comparison between two methods of LTB4 human whole blood assays. (a) represents plasma LTB4 levels for both assays and (b) represents the titration curve for RS14203 in both assays. A23187 method: whole blood challenged with 25 μM A23187 for 30 min. Method D: whole blood pre-stimulated with LPS (1 μg ml−1) for 24 h followed by 30 min stimulation with LPS (1 μg ml−1) then a 15 min incubation with fMLP (1 μM). n=3 donors. Both methods were performed in the same donor sample. *P<0.05 and **P<0.01.
Table 3.
LTB4 in vitro results of human whole blood stimulated with LPS and fMLP (method D) compared to A23187 challenge. *P<0.05 and **P<0.01
Discussion
We have developed a novel method of assessing the effects of PDE4 inhibitors on both TNF-α and LTB4 in the same human whole blood sample. Using this assay, it was possible to study PDE4 activity by measuring the levels of two surrogate markers in distinct cell types such as monocytes for TNF-α and neutrophils for LTB4.
Inhibition of PDE4 using selective inhibitors was not sufficient to completely block the synthesis of TNF-α in LPS-stimulated blood in the absence of PGE2. However, PGE2 can completely block TNF-α synthesis albeit at concentrations greater than 500 fold the IC50. As we did not use a direct method of establishing PDE4 inhibition in human blood cells such as measuring intracellular cyclic AMP or PKA activity, it is possible that complete inhibition of PDE4 may not raise intracellular cyclic AMP levels high enough for full inhibition of TNF-α synthesis. COX-2 enzyme expression and activity is upregulated in LPS-treated human monocytes (Patrignani et al., 1994; Brideau et al., 1996) and increases endogenous synthesis of PGE2. PGE2, an activator of adenylyl cyclase through the prostanoid receptor subtypes EP2 or EP4, leads to elevation of intracellular cyclic AMP (Coleman et al., 1994), which, in combination with PDE4 inhibition, enhances inhibition of TNF-α synthesis. Complete TNF-α inhibition by PDE4 inhibitors was only observed in the 24 h LPS-stimulated whole blood assay. Using this assay, the best dose response curves for PDE4 inhibitors were achieved and thus more accurate IC50 values were calculated. This was particularly important when comparing the activity of enantiomeric pairs of PDE4 inhibitors. The activity of R-rolipram in whole blood was 4 fold more potent than its enantiomer, S-rolipram, which is consistent with previously published data on LPS-stimulated TNF-α in human monocytes (Souness et al., 1996). In the absence of PGE2 (4 h LPS incubation), the difference in activity between R-rolipram and S-rolipram and was not significant. As demonstrated in the indomethacin experiment, PGE2 is important for complete inhibition of TNF-α by PDE4 inhibitors since in its absence there was approximately 30% of residual TNF-α which could not be inhibited with higher doses of inhibitor. PGE2 may be responsible for further activation of PKA through the direct activation of adenylyl cyclase. It has been shown that PKA is involved in the inhibition of TNF-α transcription by inhibiting NF-κB-mediated transcription (Ollivier et al., 1996), however, the mechanism is still unclear as there are other transcription factors involved in the regulation of TNF-α such as the p38 and p42 MAPK cascades (Foey et al., 1998). It is possible that a 24 h incubation is longer than necessary and that 8 h may be sufficient for complete TNF-α inhibition by PDE4 inhibitors. The results from the time course experiment showed little difference in TNF-α inhibition by RP73401 at 8 and 24 h. However, the 24 h incubation method was considered more pragmatic taking into account the higher donor variability in expressing COX-2 and the subsequent release of PGE2 in an 8 h method.
It has been shown that priming human whole blood with LPS for 30 min is necessary for the stimulation of LTB4 production by fMLP (method B) and that exogenous TNF-α has an additive effect with LPS (Surette et al., 1993). We confirmed this by obtaining similar results when whole blood was incubated with LPS for 24 h (method C) to increase both TNF-α and PGE2 levels before the subsequent fMLP-stimulated release of LTB4. The LTB4 levels in either assay were approximately 7 fold higher compared to their blank controls. A greater than 50 fold window was obtained when whole blood was primed twice with LPS before fMLP-stimulation (method D). This substantial increase in LTB4 may be due to further activation of cytosolic PLA2 by the additional LPS incubation releasing a large pool of free arachidonate to be readily utilized by activated 5-LO. LPS-stimulated LTB4 production was not TNF-α dependent since 10 μM PGE2 totally abolished TNF-α but had no effect on LTB4. As observed in the TNF-α experiments, the presence of PGE2 enhanced the inhibitory activity of PDE4 inhibitors on LTB4 production. Compared to method B, LTB4 inhibition was enhanced using method D (24 h LPS) even though LTB4 levels were 7 fold higher. Furthermore, the potency of the PDE4-selective inhibitor, RP73401, was reduced in the presence of indomethacin using the method D assay (data not shown).
Of the non-selective PDE inhibitors, IBMX was most potent followed by pentoxyfylline yet theophylline displayed marginal activity. For both TNF-α and LTB4, the PDE V-selective compounds and the COX-2 selective inhibitor, MF-tricyclic, showed no effect. The dual COX-1 and COX-2 inhibitor, indomethacin, potentiated TNF-α and LTB4 production equally. In contrast, the 5-LO inhibitor, L-739,010, was most potent against LTB4 and as expected was inactive against TNF-α.
PDE4 is most pre-dominant in monocytes and neutrophils however PDE III is also present in monocytes (Souness et al., 1996). PDE III-selective inhibitors have shown inhibitory activity on monocyte-derived TNF-α production and on neutrophil-derived leukotriene production (Banner et al., 1996; Schudt et al., 1991). In our assay, the PDE III-selective inhibitors were also active but to a lesser extent than the PDE4 inhibitors. The activity observed may also be due to the loss of selectivity of these inhibitors at high concentrations (IC50s: >20 μM).
While TNF-α levels were significantly decreased by 25% after an additional LPS priming and fMLP-stimulation following the initial 24 h LPS incubation, the effects of PDE4 inhibitors remained the same. Thus, this assay was used to determine the inhibitory effects of various agents on TNF-α and LTB4. Most of the PDE inhibitors were equipotent against TNF-α and LTB4. Among all of the agents tested in this assay, the PDE4-selective inhibitor, RS14203, was most potent against both TNF-α and LTB4 despite the weaker activity seen on LTB4. Of the PDE4 selective compounds tested, CDP840 and SB207499, both being clinical development compounds, exhibited the least potency in whole blood. It is well recognized that synthetic small molecule inhibitors bind extensively to plasma protein. Experiments using isolated human mononuclear cells (PBMC) treated with LPS for 20 h in the presence of 1% human serum were performed to assess protein shifts in terms of TNF-α inhibition. It was observed in our laboratory that CDP840 activity decreased by more than 40 fold in the whole blood assay compared to the PBMC assay (data not shown). However, SB207499 demonstrated lower intrinsic potency in the PBMC assay than CDP840 and was shifted approximately 26 fold in whole blood. Thus, high intrinsic potency does not always translate in whole blood assays.
CDP840 and SB207499 are the most interesting PDE4-selective inhibitors in that both were investigated in clinic. Of particular interest is CDP840 for it has been reported to be 20 fold more potent on the native PDE4 than (±)-rolipram (Hughes et al., 1996) and our TNF-α assay results show a 7 fold difference when compared to R-rolipram. Using isolated human monocytes in the presence of 1% human serum such as described by Sounness et al., 1996, we found that CDP840 and R-rolipram had equivalent activity on the inhibition of LPS-stimulated TNF-α, 365 nM and 330 nM respectively (data not shown). Thus, it appears CDP840 is highly shifted (42 fold) in human whole blood compared to R-rolipram (7 fold).
SB207499 has been reported to be approximately 10 fold more selective on the D isoform of PDE4 than A, B or C isoforms (Torphy et al., 1997). PDE4 inhibitors with greater selectivity for each of the four isoforms (at least >100 fold) will be useful to investigate the expression and regulation of PDE4 isoenzymes and to establish which subtype is most involved in inflammation and the negative side effects of PDE4 inhibition such as emesis. Previously reported studies have already shown that PDE4 A, B and D are expressed in human monocytes (C was not detected) and that prolonged exposure of these cells to elevated cyclic AMP upregulated PDE4 A and B expression and activity (Souness et al., 1996; Manning et al., 1996).
Using the whole blood assay, it appears that monocytes are preferentially sensitive to PGE2 and isoproteronol compared to neutrophils since both agents were very potent inhibitors of TNF-α but had no effect on LTB4. Conversely, cholera toxin, a stimulator of Gαs protein, had the same activity on both TNF-α and LTB4 whereas forskolin and dibutyryl cyclic AMP showed weak activity. The different inhibitory activities of PGE2 and isoproteronol in comparison to cholera toxin involve receptor binding and activation. The prostanoid receptor subtype (EP1, 2, 3 or 4) distribution on human leukocytes is still unknown. Further studies are necessary to elucidate the mechanisms involved in neutrophil-derived LTB4 modulation by cyclic AMP-elevating agents and PDE4 inhibition. Nonetheless, it is known that increases in cyclic AMP lead to a reduction in cytosolic free calcium (Torphy et al., 1991) and it was shown that rolipram was an effective inhibitor of both LTB4 synthesis and intracellular calcium elevation in human PMNs (Cortijo et al., 1996; Villagrasa et al., 1996). Other studies have shown that increases in cyclic AMP can regulate PLA2 activity and inhibit the ability of activated cells to mobilize arachidonic acid (Fonteh et al., 1993).
The non-receptor mediated A23187-stimulation of whole blood (without LPS priming) resulted in the same LTB4 levels as the double LPS and fMLP-stimulation assay (method D) yet the potency of PDE4 inhibitors were enhanced in this assay. This increased inhibitory activity by PDE4 inhibitors may be due to lower levels of active PDE4 in comparison to a long exposure of blood cells to LPS. It has been shown that PDE4 expression and activity are upregulated during long exposures to β-adrenoceptor stimulation in the presence of rolipram (Manning et al., 1996). Other studies have shown that PDE4 activity could be augmented by 8-bromo-AMP alone in U937 cells and that this increase was due to new protein synthesis (Torphy et al., 1992).
L-739,010 was significantly more potent in the A23187-stimulated LTB4 assay which may be due to different sequestrations of intracellular calcium. Interestingly, isoproteronol, forskolin and dibutyryl cyclic AMP exhibited very little effect on LTB4 in both the LPS-fMLP and A23187 assays.
In conclusion, we have established a method to assess the biochemical efficacy of PDE4-selective inhibitors in human whole blood. We have shown that PDE4-selective inhibitors are potent inhibitors of TNF-α and LTB4 biosynthesis and thus should be effective anti-inflammatory agents. Since TNF-α and LTB4 are implicated in asthma, this assay shall be useful in clinical settings where ex vivo experiments can be performed on human subjects.
Acknowledgments
We wish to thank Cairine Brideau, R.N., Linda Godin, R.N. and Yvonne Baise, R.N. for their expert technical assistance.
Abbreviations
- 5′-AMP
adenosine 5′-monophosphate
- 5-LO
5-lipoxygenase
- A23187
calcium ionophore
- BSA
bovine serum albumin
- cyclic AMP
adenosine 3′ : 5′-cyclic monophosphate
- COX
cyclo-oxygenase
- DMSO
dimethyl sulphoxide
- ELISA
enzyme-linked immunosorbent assay
- EP
PGE2 receptor
- FLAP
5-lipoxygenase activating protein
- fMLP
n-formyl-Met-Leu-Phe
- IBD
inflammatory bowel disease
- IBMX
3-isobutyl-1-methylxanthine
- IFN
interferon
- IL-2
interleukin-2
- LTB4
leukotriene B4
- LPS
lipopolysaccharide
- NSAIDS
non-steroidal anti-inflammatory drugs
- PAF
platelet activating factor
- PBS
phosphate buffered saline
- PDE
phosphodiesterase
- PGE2
prostaglandin E2
- PKA
protein kianse A
- PLA2
phospholipase A2
- PBMC
peripheral blood mononuclear cell
- PMN
polymorphonuclear cell
- TNF-α
tumour necrosis factor-alpha
References
- ARIAS-NEGRETE S., KELLER K., CHADEE K. Proinflammatory cytokines regulate cyclooxygenase-2 mRNA expression in human macrophages. Biochem. Biophys. Res. Commun. 1995;208:582–589. doi: 10.1006/bbrc.1995.1378. [DOI] [PubMed] [Google Scholar]
- BANNER K.H., MORIGGI E., DA ROS B., SCHIOPPACASSI G., SEMERARO C., PAGE C.P. The effect of selective phosphodiesterase 3 and 4 isoenzyme inhibitors and established anti-asthma drugs on inflammatory cell activation. Br. J. Pharmacol. 1996;119:1255–1261. doi: 10.1111/j.1476-5381.1996.tb16030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATTEN P., YACOUB M.H., ROSE M.L. Effect of human cytokines (IFN-gamma, TNF-alpha, IL-1 beta, IL-4) on porcine endothelial cells: induction of MHC and adhesion molecules and functional significance of these changes. Immunology. 1996;87:127–133. [PMC free article] [PubMed] [Google Scholar]
- BEUTLER B., CERAMI A. The biology of cachectin/TNF-a primary mediator of the host response. Annu. Rev. Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- BORGEAT P., SAMUELSSON B. Arachidonic acid metabolism in polymorphonuclear leukocytes: effects of ionophore A23187. Proc. Natl. Acad. Sci. U.S.A. 1979;76:2148–2152. doi: 10.1073/pnas.76.5.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRAIN S., CAMP R., DOWD P., BLACK A.K., GREAVES M. The release of leukotriene B4-like material in biologically active amounts from the lesional skin of patients with psoriasis. J. Invest. Dermatol. 1984;83:70–73. doi: 10.1111/1523-1747.ep12261712. [DOI] [PubMed] [Google Scholar]
- BRIDEAU C., KARGMAN S., LIU S., DALLOB A., EHRICH E., RODGER I.W., CHAN C.C. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm. Res. 1996;45:68–74. doi: 10.1007/BF02265118. [DOI] [PubMed] [Google Scholar]
- CHRISTENSEN S.B., GUIDER A., FORSTER C.J., GLEASON J.G., BENDER P.E., KARPINSKI J.M., DEWOLF W.E., JR, BARNETTE M.S., UNDERWOOD D.C., GRISWOLD D.E., CIESLINSKI L.B., BURMAN M., BOCHNOWICZ S., OSBORN R.R., MANNING C.D., GROUS M., HILLEGAS L.M., BARTUS J.O., RYAN M.D., EGGLESTON D.S., HALTIWANGER R.C., TORPHY T.J. 1,4-Cyclohexanecarboxylates: potent and selective inhibitors of phosophodiesterase 4 for the treatment of asthma. J. Med. Chem. 1998;41:821–835. doi: 10.1021/jm970090r. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARUMIYA S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Re. 1994;46:205–229. [PubMed] [Google Scholar]
- CORTIJO J., VILLAGRASA V., NAVARETTE C., ZANZ C., BERTO L., MICHEL A., BONNET P.A., MORCILLO E.J. Effects of SCA40 on human isolated bronchus and human polymorphonuclear leukocytes: comparison with rolipram, SFK94120 and levcromakalim. Br. J. Pharmacol. 1996;119:99–106. doi: 10.1111/j.1476-5381.1996.tb15682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIDSON E.M., RAE S.A., SMITH M.J. Leukotriene B4, a mediator of inflammation present in synovial fluid in rheumatoid arthritis. Ann. Rheum. Dis. 1983;42:677–679. doi: 10.1136/ard.42.6.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINARELLO C.A., CANNON J.G., WOLFF S.M., BERNHEIM H.A., BEUTLER B., CERAMI A., FIGARI I.S., PALLADINO M.A., JR, O'CONNOR J.V. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J. Exp. Med. 1986;163:1433–1450. doi: 10.1084/jem.163.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOERFLER M.E., DANNER R.L., SHELHAMER J.H., PARRILLO J.E. Bacterial lipopolysaccharides prime human neutrophils for enhanced production of leukotriene B4. J. Clin. Invest. 1989;83:970–977. doi: 10.1172/JCI113983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EIGLER A., GRETEN T.F., SINHA B., HASLBERGER C., SULLIVAN G.W., ENDRES S. Endogenous adenosine curtails lipopolysaccharide-stimulated tumour necrosis factor synthesis. Scand. J. Immunol. 1997;45:132–139. doi: 10.1046/j.1365-3083.1997.d01-377.x. [DOI] [PubMed] [Google Scholar]
- EIGLER A., SINHA B., HARTMANN G., ENDRES S. Taming TNF: strategies to restrain this proinflammatory cytokine. Immunology Today. 1997;8:487–492. doi: 10.1016/s0167-5699(97)01118-3. [DOI] [PubMed] [Google Scholar]
- ESSAYAN D.M., KAGEY-SOBOTKA A., LICHTENSTEIN L.M., HUANG S.K. Differential regulation of human antigen-specific Th1 and Th2 lymphocyte responses by isozyme selective cyclic nucleotide phosphodiesterase inhibitors. J. Pharmacol. Exp. Ther. 1997;282:505–512. [PubMed] [Google Scholar]
- FOEY A.D., PARRY S.L., WILLIAMS L.M., FELDMANN M., FOXWELL B.M., BRENNAN F.M. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-alpha: role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- FONTEH A.N., WINKLER J.D., TORPHY T.J., HERAVI J., UUNDEM B.J., CHILTON F.H. Influence of isoproterenol and phosphodiesterase inhibitors on platelet-activating factor biosynthesis in the human neutrophil. J. Immunol. 1993;151:339–350. [PubMed] [Google Scholar]
- FORD-HUTCHINSON A.W. Leukotriene B4 in inflammation. Crit. Rev. Immunol. 1990;10:1–12. [PubMed] [Google Scholar]
- GORDON J.R., GALLI S.J. Release of both preformed and newly synthesized tumor necrosis factor alpha(TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J. Exp. Med. 1991;174:103–107. doi: 10.1084/jem.174.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRABBE J., CZARNETZKI B.M., ROSENBACH T., MARDIN M. Identification of chemotactic lipoxygenase products of arachidonate metabolism in psoriatic skin. J. Invest. Dermatol. 1984;82:477–479. doi: 10.1111/1523-1747.ep12260985. [DOI] [PubMed] [Google Scholar]
- HAMEL P., RIENDEAU D., BRIDEAU C., CHAN C.C., DESMARAIS S., DELORME D., DUBE D., DUCHARME Y., ETHIER D., GRIMM E., FALGUEYRET J.P., GUAY J., JONES T.R., KWONG E., MCAULIFFE M., MCFARLANE C.S., PIECHUTA H., ROUMI M., TAGARI P., YOUNG R.N., GIRARD Y. Substituted (pyridylmethoxy) naphthalenes as potent and orally active 5-lipoxygenase inhibitors; synthesis, biological profile, and pharmacokinetics of L-739,010. J. Med. Chem. 1997;40:2866–2875. doi: 10.1021/jm970046b. [DOI] [PubMed] [Google Scholar]
- HAN J., THOMPSON P., BEUTLER B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J. Exp. Med. 1990;172:391–394. doi: 10.1084/jem.172.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTMAN D.A., OCHALSKI S.J, CARLSON R.P. The effects of anti-inflammatory and antiallergic drugs on the release of IL-1 beta and TNF-alpha in the human whole blood assay. Agents Actions. 1993;39:C70–C72. doi: 10.1007/BF01972724. [DOI] [PubMed] [Google Scholar]
- HARTMAN D.A., OCHALSKI S.J., CARLSON R.P. The effects of antiinflammatory and antiallergic drugs on cytokine release after stimulation of human whole blood by lipopolysaccharide and zymosan A. Inflamm. Res. 1995;44:269–274. doi: 10.1007/BF02032567. [DOI] [PubMed] [Google Scholar]
- HUGHES B., HOWAT D., LISLE H., HOLBROOK M., JAMES T., GOZZARD N., BLEASE K., HUGHES P., KINGABY R., WARRELLOW G., ALEXANDER R., HEAD J., BOYD E., EATON M., PERRY M., WALES M., SMITH B., OWENS R., CATTERALL C., LUMB S., RUSSELL A., ALLEN R., MERRIMAN M., BLOXHAM D., HIGGS G. The inhibition of antigen-induced eosinophilia and bronchoconstriction by CDP840, a novel stereo-selective inhibitor of phosphodiesterase type 4. Br. J. Pharmacol. 1996;118:1183–1191. doi: 10.1111/j.1476-5381.1996.tb15522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIA S.F., KLEINERMAN E.S. Antitumor activity of TNF-alpha, IL-1, and IFN-gamma against three human osteosarcoma cell lines. Lymph. Cyto. Res. 1991;10:281–284. [PubMed] [Google Scholar]
- KAMINUMA O., MORI A., SUKO M., NISHIZAKI Y., KIKKAWA H., IKEZAWA K., OKUDAIRA H. A novel phosphodiesterase inhibitor, T-440: possible management of eosinophilic inflammation by down-regulation of interleukin-5 production. Int. Arch. Allergy Immunol. 1996;111:16–18. doi: 10.1159/000237407. [DOI] [PubMed] [Google Scholar]
- KRAKAUER T., OPPENHEIM J.J. Il-1 and tumor necrosis factor-alpha each up-regulate both the expression of IFN-gamma receptors and enhance IFN-gamma-induced HLA-DR expression on human monocytes and a human monocytic cell line (THP-1) J. Immunol. 1993;150:1205–1211. [PubMed] [Google Scholar]
- KRUMP E., LEMAY G., BORGEAT P. Adenosine A2 receptor-induced inhibition of leukotriene B4 synthesis in whole blood ex vivo. Br. J. Pharmacol. 1996;117:1639–1644. doi: 10.1111/j.1476-5381.1996.tb15334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARSEN J.S., JACKSON S.K. Antileukotriene therapy for asthma. Am. J. Health. Syst. Pharm. 1996;53:2821–2830. doi: 10.1093/ajhp/53.23.2821. [DOI] [PubMed] [Google Scholar]
- MACKAY F., LOETSCHER H., STUEBER D., GEHR G., LESSLAUER W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells in under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993;177:1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANNING C.D., MCLAUGHLIN M.M., LIVI G.P., CIESLINSKI L.B., TORPHY T.J., BARNETTE M.S. Prolonged beta adrenoceptor stimulation up-regulates cAMP phosphodiesterase activity in human monocytes by increasing mRNA and protein for phosphodiesterase 4A and 4B. J. Pharmacol. Exp. Ther. 1996;276:810–818. [PubMed] [Google Scholar]
- MEJA K.K., BARNES P.J., GIEMBYCZ M.A. Characterization of the prostanoid receptor(s) on human blood monocytes at which prostaglandin E2 inhibits lipopolysaccharide-induced tumour necrosis factor-α generation. Br. J. Pharmacol. 1997;122:149–157. doi: 10.1038/sj.bjp.0701360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOHRI M., SPRIGGS D.R., KUGE D. Effects of lipopolysaccharide on phospholipase A2 activity and tumor necrosis factor expression in HL-60 cells. J. Immunol. 1990;144:2678–2682. [PubMed] [Google Scholar]
- NAKANE A., NUMATA A., MINAGAWA T. Endogenous tumor necrosis factor, interleukin-6, and gamma interferon levels during Listeria monocytogenes infection in mice. Infect. Immun. 1992;60:523–528. doi: 10.1128/iai.60.2.523-528.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIELSON C.P., VESTAL R.E., STURM R.J., HEASLIP R. Effects of selective phosphodiesterase inhibitors on the polymorphonuclear leukocyte respiratory burst. J. Allergy Clin. Immunol. 1990;86:801–808. doi: 10.1016/s0091-6749(05)80186-1. [DOI] [PubMed] [Google Scholar]
- OLLIVIER V., PARRY G.C.N., COBB R.R., DE PROST D., MACKMAN N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J. Biol. Chem. 1996;271:20828–20835. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- OSHIMA M., DINCHUK J.E., KARGMAN S.L., OSHIMA H., HANCOCK B., KWONG E., TRZASKOS J.M., EVANS J.F., TAKETO M.M. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- PATRIGNANI P., PANARA M.R., GRECO A., FUSCO O., NATOLI C., IACOBELLI S., CIPOLLONE F., GANCI A., CREMINON C., MACLOUF J., PATRONO C. Biochemical and pharmacological characterization of the cyclooxygenase activity of human whole blood prostaglandin endoperoxide synthases. J. Pharmacol. Exp. Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- SCHADE F.U., SCHUDT C. The specific type III and IV phosphodiesterase inhibitor zardaverine suppresses formation of tumor necrosis factor by macrophages. Eur. J. Pharmacol. 1993;230:9–14. doi: 10.1016/0014-2999(93)90403-5. [DOI] [PubMed] [Google Scholar]
- SCHEURICH P., THOMA B., UCER U., PFIZENMAIER K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: induction of TNF receptors on human T cells and TNF-alpha-mediated enhancement of T cell responses. J. Immunol. 1987;138:1786–1790. [PubMed] [Google Scholar]
- SCHUDT C., TENOR H., HATZELMANN A. PDE isoenzymes as targets for anti-asthma drugs. Eur. Respir. J. 1995;8:1179–1183. doi: 10.1183/09031936.95.08071179. [DOI] [PubMed] [Google Scholar]
- SCHUDT C., WINDER S., FORDERKUNZ S., HATZELMANN A., ULLRICH V. Influence of selective phosphodiesterase inhibitors on human neutrophil functions and levels of cAMP and Cai. Naunyn Schmiedebergs Arch. Pharmacol. 1991;344:682–690. doi: 10.1007/BF00174752. [DOI] [PubMed] [Google Scholar]
- SCHUTZE S., WIEGMANN K., MACHLEIDT T., KRONKE M. TNF-induced activation of NF-kappa B. Immunobiology. 1995;193:193–203. doi: 10.1016/s0171-2985(11)80543-7. [DOI] [PubMed] [Google Scholar]
- SELDON P.M., BARNES P.J., MEJA K., GIEMBYCZ M.A. Suppression of lipopolysaccharide-induced tumor necrosis factor-alpha generation from human peripheral blood monocytes by inhibitors of phosphodiesterase 4: interaction with stimulants of adenylyl cyclase. Mol. Pharmacol. 1995;48:747–757. [PubMed] [Google Scholar]
- SHARON P., STENSON W.F. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86:453–460. [PubMed] [Google Scholar]
- SOUNESS J.E., GRIFFIN M., MASLEN C., EBSWORTH K., SCOTT L.C., POLLOCK K., PALREYMAN M.N., KARLSSON J.A. Evidence that cyclic AMP phosphodiesterase inhibitors suppress TNF alpha generation from human monocytes by interacting with a ‘low-affinity' phosphodiesterase 4 conformer. Br. J. Pharmacol. 1996;118:649–658. doi: 10.1111/j.1476-5381.1996.tb15450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOUNESS J.E., MASLEN C., WEBBER S., FOSTER M., RAEBURN D., PALFREYMAN M.N., ASHTON M.J., KARLSSON J.A. Suppression of eosinophil function by RP 73401, a potent and selective inhibitor of cyclic AMP-specific phosphodiesterase: comparison with rolipram. Br. J. Pharmacol. 1995;115:39–46. doi: 10.1111/j.1476-5381.1995.tb16317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SURETTE M.E., PALMANTIER R., GOSSELIN J, BORGEAT P. Lipopolysaccharides prime whole human blood and isolated neutrophils for the increased synthesis of 5-lipoxygenase products by enhancing arachidonic acid availability: Involvement of the CD14 antigen. J. Exp. Med. 1993;178:1347–1355. doi: 10.1084/jem.178.4.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TALPAIN E., ARMSTRONG R.A., COLEMAN R.A., VARDEY C.J. Characterization of the PGE receptor subtype mediating inhibition of superoxide production in human neutrophils. Br. J. Pharmacol. 1995;114:1459–1465. doi: 10.1111/j.1476-5381.1995.tb13370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORPHY T.J., CHRISTENSEN S.B., BARNETTE M.S., BURMAN M., CIESLINSKI L.B., DEWOLF W.E. Molecular basis for an improved therapeutic index of SB 207499, a second generation phosphodiesterase 4 inhibitor (Abstract) Eur. Respir. J. 1997;10:313s. [Google Scholar]
- TORPHY T.J., UNDEM B.J. Phosphodiesterase inhibitors: new opportunities for the treatment of asthma. Thorax. 1991;46:512–523. doi: 10.1136/thx.46.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORPHY T.J., ZHOU H.L., CIESLINSKI L.B. Stimulation of beta adrenoceptors in a human monocyte cell line (U937) up-regulates cyclic AMP-specific phosphodiesterase activity. J. Pharm. Exp. Ther. 1992;263:1195–1205. [PubMed] [Google Scholar]
- TRACEY K.J., BEUTLER B., LOWRY S.F., MERRYWEATHER J., WOLPE S., MILSARK I.W., HARIRI R.J., FAHEY T.J., III, ZENTELLA A., ALBERT J.D., SHIRES T., CERAMI A. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- TREFZER U., BROCKHAUS M., LOTSCHER H., PARLOW F., BUDNICK A., GREWE M., CHRISTOP H., KAPP A., SCHOPF E., LUGER T.A., KRUTMANN J. The 55-kD tumor necrosis factor receptor on human keratinocytes is regulated by tumor necrosis factor-alpha and by ultraviolet B radiation. J. Clin. Invest. 1993;92:462–470. doi: 10.1172/JCI116589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNDERWOOD D.C., KOTZER C.J., BOCHNOWICZ S., OSBORN R.R., LUTTMANN M.A., HAY D.W., TORPHY T.J. Comparison of phosphodiesterase III, IV and dual III/IV inhibitors on bronchospasm and pulmonary eosinophil influx in guinea pigs. J. Pharmacol. Exp. Ther. 1994;270:250–259. [PubMed] [Google Scholar]
- VALYI-NAGY I., JENSEN P.J., ALBELDA S.M., RODECK U. Cytokine-induced expression of transforming growth factor-alpha and the epidermal growth factor receptor in neonatal skin explants. J. Invest. Dermatol. 1992;99:350–356. doi: 10.1111/1523-1747.ep12616672. [DOI] [PubMed] [Google Scholar]
- VILLAGRASA V., NAVARETE C., SANZ C., BERTO L., PERPINA M., CORTIJO J., MORCILLO E.J. Inhibition of phosphodiesterase IV and intracellular calcium levels in human polymorphonuclear leukocytes. Methods Find. Exp. Clin. Pharmacol. 1996;18:239–245. [PubMed] [Google Scholar]
- WARREN J.S., WARD P.A., JOHNSON K.J. Tumor necrosis factor: a plurifunctional mediator of acute inflammation. Mod. Pathol. 1988;1:242–247. [PubMed] [Google Scholar]
- WILHELM R.S., FATHEREE P.R. 1994. WO patent 22852