

Abstract
We investigated whether dopamine plays a role in the neurodegeneration of 5-hydroxytryptamine (5-HT) nerve endings occurring in Dark Agouti rat brain after 3,4-methylenedioxymethamphetamine (MDMA or ‘ecstasy') administration.
Haloperidol (2 mg kg−1 i.p.) injected 5 min prior and 55 min post MDMA (15 mg kg−1 i.p.) abolished the acute MDMA-induced hyperthermia and attenuated the neurotoxic loss of 5-HT 7 days later. When the rectal temperature of MDMA+haloperidol treated rats was kept elevated, this protective effect was marginal.
MDMA (15 mg kg−1) increased the dopamine concentration in the dialysate from a striatal microdialysis probe by 800%. L-DOPA (25 mg kg−1 i.p., plus benserazide, 6.25 mg kg−1 i.p.) injected 2 h after MDMA (15 mg kg−1) enhanced the increase in dopamine in the dialysate, but subsequent neurodegeneration was unaltered. L-DOPA (25 mg kg−1) injected before a sub-toxic dose of MDMA (5 mg kg−1) failed to induce neurodegeneration.
The MDMA-induced increase in free radical formation in the hippocampus (indicated by increased 2,3- and 2,5-dihydroxybenzoic acid in a microdialysis probe perfused with salicylic acid) was unaltered by L-DOPA.
The neuroprotective drug clomethiazole (50 mg kg−1 i.p.) did not influence the MDMA-induced increase in extracellular dopamine.
These data suggest that previous observations on the protective effect of haloperidol and potentiating effect of L-DOPA on MDMA-induced neurodegeneration may have resulted from effects on MDMA-induced hyperthermia.
The increased extracellular dopamine concentration following MDMA may result from effects of MDMA on dopamine re-uptake, monoamine oxidase and 5-HT release rather than an ‘amphetamine-like' action on dopamine release, thus explaining why the drug does not induce degeneration of dopamine nerve endings.
Keywords: 3,4-methylenedioxymethamphetamine; ecstasy; dopamine; free radicals; neurodegeneration; clomethiazole; 5-hydroxytryptamine; L-DOPA, haloperidol
Introduction
The commonly misused recreational drug 3,4-methylenedioxymethamphetamine (MDMA or ‘ecstasy') has two well established phases of action in experimental animals. The acute phase consists of a rapid release of 5-hydroxytryptamine (5-HT) from neuronal stores, thereby producing many of the acute behavioural changes, including behavioural excitation and hyperthermia, that are seen in both experimental animals and humans (see reviews of White et al., 1996; Green et al., 1995; Steele et al., 1994).
The second action of MDMA in the brain of experimental animals is to produce a long lasting neurotoxic loss of 5-HT nerve terminals in several areas of the brain, an effect demonstrated both histologically (O'Hearn et al., 1988; Molliver et al., 1990) and biochemically (Battaglia et al., 1987; Sharkey et al., 1991; Hewitt & Green, 1994). The mechanisms involved in producing this neurodegeneration are not fully understood at present, but recent data have given substantial evidence to support the contention that increased free radical production is involved (see Colado et al., 1997b).
Studies on both the pharmacology of MDMA and putative neuroprotective agents have also indicated a role for dopamine in the neurodegenerative process. For example Stone et al., (1988) proposed a role for dopamine based on their studies which showed that the long term depletion of cerebral 5-HT which follows administration of MDMA was attenuated by the dopamine synthesis inhibitor α-methyl p-tyrosine or the monoamine depletor reserpine. Schmidt et al. (1990b) reported that injection into the striatum of the selective dopamine neurotoxin 6-hydroxydopamine (6-OHDA) blocked the neurotoxic effects of MDMA on 5-HT neurones not only in the striatum but also other forebrain regions such as the hippocampus and cortex. In addition Stone et al. (1988) reported that the dopamine uptake inhibitor GBR 12909 was neuroprotective. Finally, both Schmidt et al. (1990b) and Hewitt & Green (1994) have shown that acute administration of the dopamine antagonist haloperidol together with MDMA provides effective protection against neurotoxic damage. It is acknowledged that none of these studies demonstrate authoritatively the involvement of dopamine in the MDMA-induced neurodegeneration because none of the compounds used in these investigations (haloperidol, 6-OHDA, α-methyl p-tyrosine) selectively alter dopamine function. Nevertheless, when taken with other evidence (see below) that MDMA administration alters dopamine biochemistry in the brain, the interpretation that dopamine was involved in the mechanisms by which MDMA causes neurodegeneration of 5-HT nerve terminals appeared plausable.
Whilst MDMA has generally been reported to not produce neurotoxic degeneration of dopamine neurones (Stone et al., 1986; Schmidt, 1987; Schmidt & Kehne, 1990; Colado et al., 1997a, 1997b), both in vitro and in vivo studies have found that MDMA enhances dopamine efflux from cerebral tissue. For example MDMA increases release of [3H]-dopamine from striatal slices (Johnson et al., 1986; Schmidt et al., 1987) and the drug has also been reported to inhibit [3H]-dopamine uptake into striatal synaptosomes (Steele et al., 1987). Use of in vivo voltammetry (Yamamoto & Spanos, 1988) and in vivo microdialysis (Hiramatsu & Cho, 1990; Nash, 1990; Nash & Brodkin, 1991) has demonstrated that MDMA increases dopamine release and these results are supported by the observation that MDMA produces an acute increase in striatal dopamine content and a decrease in the dihydroxyphenylacetic acid (DOPAC) content (Schmidt et al., 1986; Johnson et al., 1991; Colado & Green 1994).
These data led us to suggest that the reason that drugs which enhanced GABA function such as pentobarbitone (Colado & Green, 1994) and clomethiazole (Colado et al., 1993) were neuroprotective was because their administration resulted in an inhibition of dopamine release (Colado & Green, 1994; Green et al., 1995). Recently however it has been apparent that some re-evaluation is necessary of reports on the neuroprotective action of various compounds. This is because it is now clear that some drugs attenuate damage solely because they either induce hypothermia (Farfel & Seiden, 1995a, 1995b; Malberg et al., 1996) or merely prevent the MDMA-induced hyperthermia (Colado et al., 1998b). In particular the report that α-methyl p-tyrosine is neuroprotective because of its hypothermic effect (Malberg et al., 1996) and our recent observation that pentobarbitone is not protective when the animals are kept warm (Colado et al., 1998a) has encouraged us both to re-examine our earlier data on haloperidol and also to try and clarify further the possible role of dopamine in the neurodegenerative changes which follow MDMA administration.
Methods
Animals, drugs and reagents
Adult male Dark Agouti rats (Interfauna, Barcelona, Spain) weighing 150–170 g were used. They were housed in groups of five, in conditions of constant temperature (21°C±2°C) and a 12 h light/dark cycle (lights on: 07 h 00 min) and given free access to food and water.
The following drugs were used: (±) 3,4-methylenedioxymethamphetamine HCl (Ministry of Health, Spain), clomethiazole edisylate (Astra Arcus, Södertälje, Sweden), haloperidol (Sigma-Aldrich Química S.A., Spain), L-3,4-dihydroxyphenylalanine methyl esther (Methyl L-DOPA, Sigma-Aldrich Química S.A., Spain) and benserazide (Sigma-Aldrich Química S.A., Spain).
In all the experiments using L-DOPA, the dose of L-DOPA was preceded 1 min earlier by an injection of benserazide, a peripheral DOPA decarboxylase inhibitor, at a dose of 6.25 mg kg−1 i.p. in order to avoid the peripheral metabolism of L-DOPA.
All compounds except haloperidol were dissolved in 0.9% w/v NaCl (saline). Haloperidol was suspended in peanut oil. Control animals were injected with the appropriate vehicle. All drugs were injected i.p. in a volume of 1 ml kg−1. Doses are always quoted in terms of the base.
Measurement of rectal temperature
Temperature was measured by use of a digital readout thermocouple (Type K thermometer, Portec, U.K.) with a resolution of±0.1°C and accuracy of±0.2°C attached to a CAC-005 Rodent Sensor which was inserted 2.5 cm into the rectum, the rat being lightly restrained by holding in the hand. A steady readout was obtained within 10 s of probe insertion.
Elevation of body temperature with a homeothermic blanket
In one experiment rats were injected with MDMA+ haloperidol and had their rectal temperature kept elevated to near that seen in rats given MDMA alone. This was achieved by placing the rats in a cage with a Harvard Homeothermic Blanket (Model 50-7087) covering the base.
Measurement of monoamines and their metabolites in cerebral tissue
Rats were killed by cervical dislocation and decapitation, the brains rapidly removed and cortex, hippocampus and striatum dissected out on ice. Tissue was homogenized and 5-hydroxytryptamine (5-HT), 5-hydroxyindoleacetic acid (5-HIAA), dopamine, homovanillic acid (HVA) and 3,4-dihydroxyphenylacetic acid (DOPAC) measured by high performance liquid chromatography (h.p.l.c.). Briefly, the mobile phase consisted of: KH2PO4 0.05 M, octanesulphonic acid 0.16 mM, EDTA 0.1 mM and methanol (16%), and was adjusted to pH 3 with phosphoric acid, filtered and degassed. The flow rate was 1 ml min−1 and the working electrode potential was set at 0.85 V.
The h.p.l.c. system consisted of a pump (Waters 510) linked to an automatic sample injector (Loop 200 μl, Waters 712 WISP), a stainless steel reversed-phase column (Spherisorb ODS2, 5 μm, 150×4.6 mm) with a precolumn and an amperometric detector (Waters M460). The current produced was monitored by using an integrator (Waters M745).
[3H]-Paroxetine binding in tissue homogenates
[3H]-Paroxetine binding was measured by the method described in detail by Hewitt & Green (1994). The animals were killed, the brain rapidly removed and dissected on ice within 2 min. Tissue from individual animals was homogenized in ice-cold Tris-HCl (50 mM; pH 7.4) containing (in mM): NaCl 120 and KCl 5, using an Ultra-Turrax. The homogenate was centrifuged at 30,000×g for 10 min at 4°C. The supernatant was discarded and the wash procedure repeated twice more. The pellet was finally resuspended in the Tris buffer at a concentration of 10 mg tissue ml−1. In order to obtain an estimate of the maximal density of [3H]-paroxetine-labelled 5-HT uptake sites the assay solution (1 ml) contained a saturating concentration of [3H]-paroxetine (1 nM) and 800 μl tissue preparation with the addition of 5-HT (100 μM) for determination of non specific binding. Incubation was for 60 min at room temperature. Assays were terminated by rapid filtration and counting of the radioactivity by scintillation spectrometry. Protein concentrations were measured by the method of Lowry et al. (1951).
Implantation of microdialysis probe in the striatum
Rats were anaesthetized with sodium pentobarbitone (Euta-Lender, 40 mg kg−1 i.p.) and secured in a Kopf stereotaxic frame with the tooth bar at −3.3 mm below the interaural zero. The dialysis probe (3.5 mm×200 μm: Cuprophan) was implanted in the right striatum +7.9 mm from the interaural line, −2.5 mm lateral and −8 mm below the surface of the brain (König & Klippel, 1963). Probes were secured to the skull as described by Baldwin et al. (1994).
Measurement of dopamine and their metabolites in the striatal dialysate
Twenty-four hours after implantation, probes were perfused with artificial cerebrospinal fluid (in mM): KCl 2.5, NaCl 125, MgCl2.6H2O 1.18, CaCl2.2H2O 1.26, at a rate of 1 μl min−1 and samples collected from the freely moving animals at 30 min intervals in tubes containing 5 μl of a solution composed of HClO4 (0.01 M), cysteine (0.2%) and sodium metabisulphite (0.2%). The first 60 min sample was discarded and the next three 30 min baseline samples collected.
Dopamine, DOPAC and HVA were measured in the dialysate by h.p.l.c. and electrochemical detection. The mobile phase consisted of KH2PO4 (0.05 M), octanesulphonic acid (1 mM), EDTA (0.1 mM) and methanol (14%), and was adjusted to pH 3 with phosphoric acid, filtered and degassed. The flow rate was 1 ml min−1. The h.p.l.c. system consisted of a pump (Waters 510) linked to manual sample injector (Loop 20 μl Rheodyne), a stainless steel reversed-phase column (Spherisorb ODS2, 5 μm, 150×4.6 mm) with a precolumn and a coulometric detector (Coulochem 5100A) with a 5011 analytical cell. The working electrode potential was set at 400 mV with 500 nA gain. The current produced was monitored by using a computer data handling system (AXXIOM 747).
The limit of the sensitivity of these assays was approximately 8 pg (dopamine), 30 pg (DOPAC) and 125 pg (HVA). The per cent recovery of a solution containing dopamine (1 pg μl−1), DOPAC (100 pg μl−1) and HVA (150 pg μl−1) was 26.5±5.5% (n=4) for dopamine, 25.8±1.7% (n=4) for DOPAC and 28.0±1.4% (n=4) for HVA. The percentage of recovery is maintained constant for the following ranges of concentrations in the external medium: dopamine 1–27 pg μl−1, DOPAC 100–2700 pg μl−1, and HVA 150–4050 pg μl−1. These ranges were comparable to the amounts of dopamine and metabolites recovered from the brain in vivo.
Implantation of microdialysis probe in the hippocampus
Rats were anaesthetized with sodium pentobarbitone (‘Euta-Lender', 40 mg kg−1 i.p.) and secured in a Kopf stereotaxic frame with the tooth bar at −3.3 mm below the interaural zero. The dialysis probe (3.5 mm×200 μm: Cuprophan) was implanted in the right hippocampus +2.2 mm from the interaural line, −4.3 mm lateral and −8 mm below the surface of the brain (König & Klippel, 1963). Probes were secured to the skull as described by Baldwin et al. (1994).
Measurement of free radical formation in vivo using microdialysis
Free radical formation in the brain in vivo was determined by the method recently described in detail by Colado et al. (1997b). The method relies on the fact that hydroxyl free radicals react with salicylic acid to generate 2,3- and 2,5-dihydroxybenzoic acids (2,3-DHBA and 2,5-DHBA) and this reaction is utilized by measuring the formation of 2,3- and 2,5-DHBA in the dialysate of a microdialysis probe implanted in the hippocampus (see above) and which is being perfused with salicylic acid (see Chieuh et al., 1992; Giovanni et al., 1995). Twenty-four hours after implantation, probes were perfused with artificial cerebrospinal fluid (in mM): KCl 2.5, NaCl 125, MgCl2.6H2O 1.18, CaCl2.2H2O 1.26, containing salicylic acid (0.5 mM) at a rate of 1 μl min−1 and samples collected from the freely moving animals at 30 min intervals. The first 60 min sample was discarded and the next three 30 min baseline samples collected.
The two compounds 2,3-DHBA and 2,5-DHBA were measured by h.p.l.c. and electrochemical detection. The mobile phase consisted of KH2PO4 (0.025 M), acetonitrile (20%) and methanol (10%) and was adjusted to pH 3.7 with phosphoric acid, filtered and degassed. The flow rate was 1 ml min−1.
The h.p.l.c. system consisted of a pump (Waters 510) linked to manual sample injector (Loop 20 μl Rheodyne), a stainless steel reversed-phase column (250×4.6 mm, 5 μm C8 Ultracarb, Phenomenex) with a precolumn (30×4.60 mm, 5 μm C8 Ultracarb, Phenomenex) and a coulometric detector (Coulochem 5100A) with a 5011 analytical cell. The working electrode potential was set at 400 mV with 1 μA gain. The current produced was monitored by using a computer data handling system (AXXIOM 747).
Statistics
Statistical analyses of the microdialysis experiments were performed using the statistical computer package BMDP/386 Dynamic (BMDP Statistical Solutions, Cork, Eire). Data were analysed by analysis of variance (ANOVA) with repeated measures (program 2 V) or, where missing values occurred, an unbalanced repeated measure model (program 5 V) was used. Both used treatment as the between subjects factor and time as the repeated measure. ANOVA was performed on both pre-treatment and post-treatment data. Temperature data were also analysed by ANOVA with repeated measures. Data from monoamine and [3H]-paroxetine binding experiments were analysed by the Newman-Keuls test.
Results
The effect of haloperidol on rectal temperature of MDMA-treated rats kept at normal ambient temperature
Administration of MDMA (15 mg kg−1 i.p.) produced a clear and sustained hyperthermia (Figure 1a). In contrast, when haloperidol (2 mg kg−1 i.p.) was injected 5 min before and 55 min after the MDMA injection the hyperthermic response was totally abolished (Figure 1a). Administration of haloperidol (2 mg kg−1 i.p.) alone had no effect on rectal temperature (Figure 1b).
Figure 1.
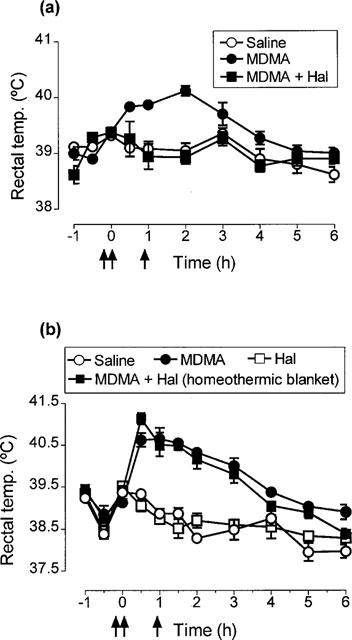
Effect of haloperidol (Hal) on MDMA-induced hyperthermia. (a) Rectal temperature of rats injected with saline or MDMA (15 mg kg−1 i.p.) at time zero and rats injected with haloperidol (2 mg kg−1 i.p.) at −5 min and +55 min and treated with MDMA at time zero. There was no difference in the basal temperature of the groups. MDMA produced a significant rise in body temperature (F(1,10)=19.89, P<0.01) compared with the saline injected group. Administration of haloperidol to MDMA-treated rats prevented the MDMA-induced hyperthermia (F(1,11)=27.16, P<0.001). (b) Effect of haloperidol on MDMA-induced hyperthermia when rats injected with haloperidol+MDMA were kept at high ambient temperature. Temperature of rats given haloperidol and saline is also shown. There was no difference in basal temperature of the groups. Using the homeothermic blanket, the rectal temperature of the haloperidol+MDMA-treated rats was significantly higher than that of the saline group (F(1,10)=141.69, P<0.001). All results shown as means±s.e.mean of n=4–6 rats.
The effect of haloperidol on the cerebral monoamine content and [3H]-paroxetine binding in MDMA-treated rats kept at normal ambient temperature
MDMA (15 mg kg−1 i.p.) administration produced a substantial loss in 5-HT and 5-HIAA concentration in the three brain areas examined and also in [3H]-paroxetine binding values in the cortex (Figure 2) 7 days later. Haloperidol administration provided significant protection against this neurotoxic degeneration (Figure 2).
Figure 2.
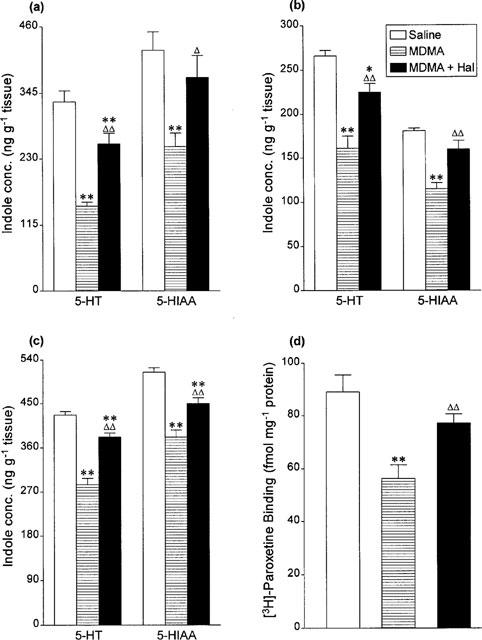
Indole concentrations in (a) hippocampus, (b) cortex and (c) striatum and (d) density of 3H-paroxetine labelled 5-HT uptake sites in cortex 7 days following saline or haloperidol (Hal, 2 mg kg−1 i.p.) 5 min before and 55 min after MDMA (15 mg kg−1 i.p.). Results shown as means±s.e.mean, n=4–7. Different from saline-treated: *P<0.05, **P<0.01. Different from MDMA-treated: ΔP<0.05, ΔΔP<0.01.
The effect of haloperidol on rectal temperature of MDMA-treated rats kept at high ambient temperature
The experiment above was repeated except that the rats which were injected with both MDMA and haloperidol were kept at high ambient temperature (see Methods) in order to prevent the decrease in rectal temperature that occurred when this group was kept at normal ambient temperature. This aim was achieved (Figure 1b).
The effect of haloperidol on the cerebral monoamine content and [3H]-paroxetine binding in MDMA-treated rats kept at high ambient temperature
Administration of MDMA (15 mg kg−1 i.p.) again produced a substantial loss in 5-HT and 5-HIAA concentration and [3H]-paroxetine binding in the brain 7 days later (Figure 3). However little or no protection was seen in the group given haloperidol plus MDMA when the temperature of the rats had been kept elevated to match the temperature seen in the rats given only MDMA (Figure 3).
Figure 3.
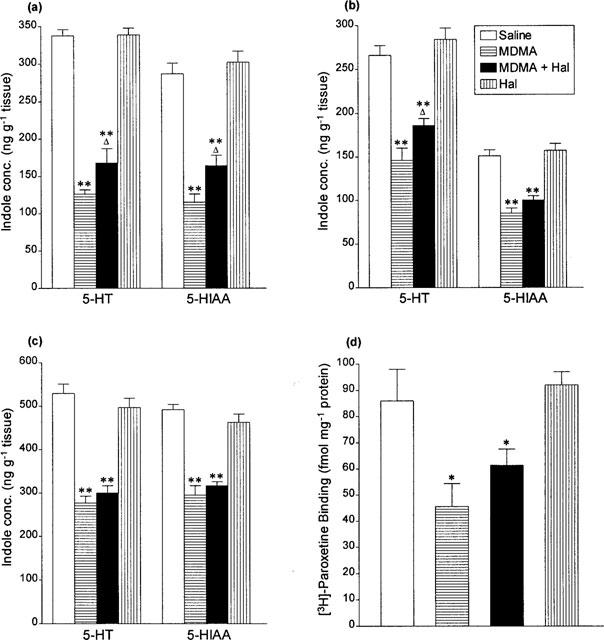
Indole concentrations in (a) hippocampus, (b) cortex and (c) striatum and (d) density of 3H-paroxetine labelled 5-HT uptake sites in cortex 7 days following saline or haloperidol (Hal, 2 mg kg−1 i.p.) 5 min before and 55 min after saline or MDMA (15 mg kg−1 i.p.). The MDMA+haloperidol group was kept at high ambient temperature during the 6 h after the first haloperidol injection. Results shown as means±s.e.mean, n=4–6. Different from saline-treated: *P<0.05, **P<0.01. Different from MDMA-treated: ΔP<0.05.
Effect of L-DOPA on rectal temperature of MDMA treated rats
Administration of L-DOPA (25 mg kg−1+benserazide 6.25 mg kg−1) had no effect on rectal temperature (Figure 4). However, when this dose was administrated 2 h after MDMA (15 mg kg−1) it extended the duration of the major MDMA-induced hyperthermia (Figure 4). Higher doses of L-DOPA or administering this dose of the compound at the same time as MDMA resulted in death of the animals, apparently as the result of severe hyperthermia so such protocols could not be investigated further.
Figure 4.
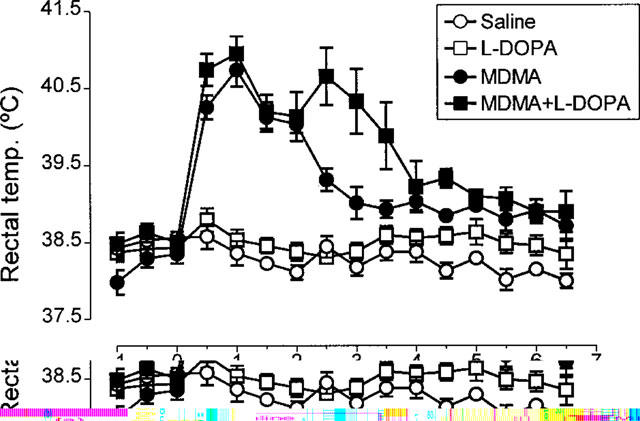
Rectal temperature of rats receiving L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) 2 h after MDMA (15 mg kg−1 i.p.) or saline. Also shown are the rectal temperature of animals injected with saline instead of L-DOPA and administered 2 h before saline or MDMA. There was no difference in the basal temperature of the groups. MDMA produced a significant rise in body temperature (F(1,9)=129.36, P<0.001) compared with the saline injected group. Administration of L-DOPA to MDMA-treated rats produced a prolongation of the hyperthermic response induced by MDMA (F(1,11)=4.12, P<0.05).
Effect of MDMA and L-DOPA on dopamine metabolism
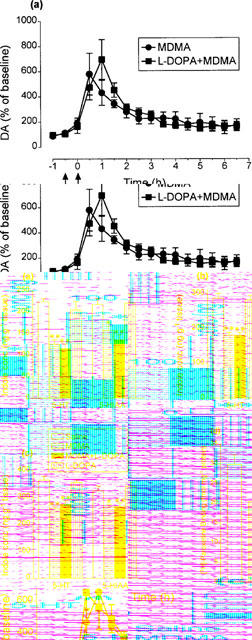
Injection of MDMA (15 mg kg−1) produced a rapid and substantial increase in extracellular dopamine in the striatum, as measured by microdialysis (Figure 5a). This peak was followed by a sustained decrease in both HVA and DOPAC, the decrease in the latter starting earlier than that of HVA (1 h after MDMA, the DOPAC and HVA levels compared to basal values were 23±2% and 60±3%, respectively, P<0.01) (Figure 5b and c). Injection of L-DOPA (25 mg kg−1+benserazide 6.25 mg kg−1) alone produced no change in the concentration of dopamine in the dialysate, but did produce a sustained and large increase in both HVA and DOPAC (Figure 5b and c). When L-DOPA was injected 2 h after MDMA it did produce a clear increase in dopamine, thereby enhancing the size of the total dopamine response (Figure 5a). A prolonged increase in both HVA and DOPAC was also seen, thereby reversing the MDMA-induced decrease in the concentration of these two metabolites (Figure 5b and c).
Figure 5.
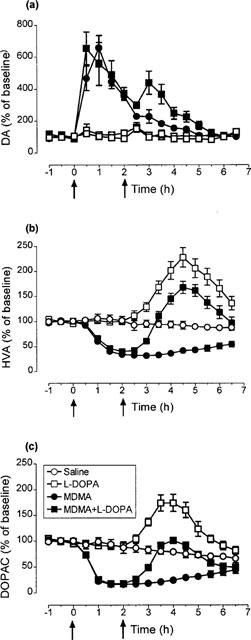
Changes in the levels of (a) dopamine (DA), (b) homovanillic acid (HVA) and (c) 3,4-dihydroxyphenylacetic acid (DOPAC) in the striatal dialysate of rats treated with L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) or saline 2 h after MDMA (15 mg kg−1 i.p.). MDMA produced an increase in the content of DA (F(1,9)=74.34, P<0.001) and a decrease in the levels of HVA (F(1,9)=81.07, P<0.001) and DOPAC (F(1,9)=52.61, P<0.001). Administration of L-DOPA enhanced the effect of MDMA on extracellular dopamine (F(1,10)=9.57, P<0.001) and reversed the MDMA-induced decrease in HVA (F(1,10)=66.72, P<0.001) and DOPAC (F(1,10)=43.10, P<0.001). L-DOPA, when given alone, increased the extracellular levels of HVA (F(1,10)=27.30, P<0.001) and DOPAC (F(1,10)=24.54, P<0.001). Values are expressed as a percentage of the mean of three measurements before drug administration. Each value is the means±s.e.mean of 5–7 experiments. The basal concentrations in saline-treated rats were: DA (0.83±15 pg μl−1), HVA (246±16 pg μl−1) and DOPAC (252±23 pg μl−1).
Effect of MDMA and L-DOPA on free radical formation in the brain
In confirmation of previous findings (see Discussion) MDMA injection increased the concentration of 2,3- and 2,5-DHBA in the dialysate from a microdialysis probe implanted in the hippocampus and perfused with salicylic acid (Figure 6). L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) alone did not produced significant changes in the concentration of 2,3 DHBA or 2,5 DHBA (Figure 6). The increase in formation of 2,3- and 2,5-DHBA resulting from MDMA injection was unaltered by an injection of L-DOPA (25 mg kg−1) 2 h later (Figure 6).
Figure 6.

Changes in the levels of (a) 2,3 dihydroxybenzoic acid (DHBA) and (b) 2,5 DHBA in the hippocampal dialysate of rats treated with L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) or saline 2 h after MDMA (15 mg kg−1 i.p.). MDMA increased the extracellular levels of 2,3 DHBA (F(1,11)=10.34, P<0.001) and 2,5 DHBA (F(1,11)=4.66, P<0.05). This effect was not modified by L-DOPA injection 2 h after MDMA. L-DOPA, when given to saline-treated rats did not significantly modify the extracellular levels of 2,3 DHBA or 2,5 DHBA. Values are expressed as a percentage of the mean of three measurements before drug administration. Each value is the means±s.e. mean of 6–10 experiments. The basal concentrations in saline-treated rats (n=6) were: 2,3 DHBA (10.1±1.1 pg μl−1) and 2,5 DHBA (11.6±1.22 pg μl−1).
The effect of L-DOPA on MDMA-induced degeneration of 5-HT neurones
A single injection of MDMA (15 mg kg−1) resulted in a major loss of 5-HT and 5-HIAA in the hippocampus, cortex and striatum 7 days later and a similar loss of [3H]-paroxetine binding in the cortex (Figure 7). The degenerative loss of 5-HT was similar in rats injected with MDMA and those given MDMA plus L-DOPA, while L-DOPA alone was without effect on the concentration of either the indole concentration or [3H]-paroxetine binding in the brain (Figure 7). MDMA injection either alone or with L-DOPA also had no effect on the concentration of dopamine or DOPAC in the striatum 7 days later (Table 1).
Figure 7.
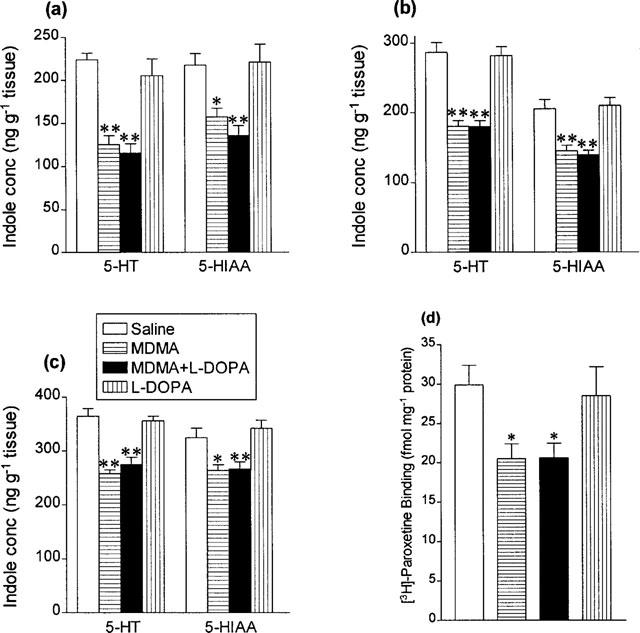
Indole concentrations in (a) hippocampus, (b) cortex and (c) striatum and (d) density of 3H-paroxetine labelled 5-HT uptake sites in cortex 7 days following saline or L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) 2 h after saline or MDMA (15 mg kg−1 i.p.). Results shown as means±s.e. mean, n=5–8. Different from saline-treated: *P<0.05, **P<0.01.
Table 1.
The concentration of dopamine and DOPAC in striatum 7 days after injection of MDMA (15 mg kg−1 i.p.) followed 2 h later by L-DOPA (25 mg kg−1 i.p.)

Effect of low dose MDMA and L-DOPA on rectal temperature, dopamine metabolism and subsequent neurotoxicity of cerebral 5-HT neurones
Rats were injected with L-DOPA (25 mg kg−1+benserazide 6.25 mg kg−1) followed 30 min later with a sub-neurotoxic dose of MDMA (5 mg kg−1) to examine whether pretreatment with the amino acid would result in MDMA now producing toxicity. Pretreatment with L-DOPA did not enhance the hyperthermic effect of MDMA (Figure 8). Although L-DOPA did not enhance the MDMA-induced increase in dopamine in the striatal dialysate, it induced marked increases in both HVA and DOPAC in place of the decrease in the concentration of these metabolites produced by MDMA alone (Figure 9). The low dose of MDMA did not result in neurodegeneration occurring 7 days later, nor did the pretreatment with L-DOPA result in this dose of MDMA producing neurodegeneration (Figure 10).
Figure 8.
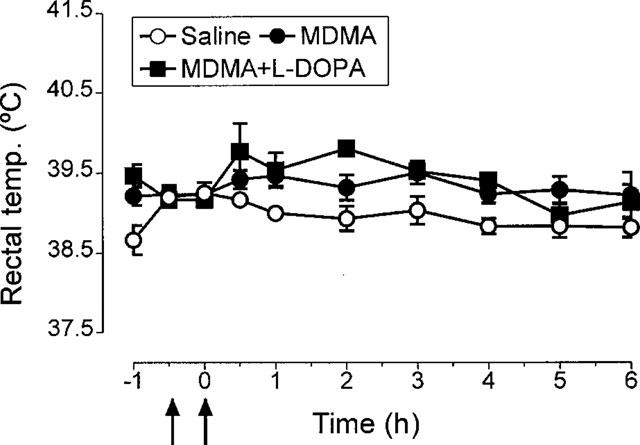
Rectal temperature of rats receiving L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) 30 min before MDMA (5 mg kg−1 i.p.). Also shown are the rectal temperature of animals injected with saline instead of L-DOPA and administered saline or MDMA 30 min later. There was no difference in the basal temperature of the groups. MDMA produced a rise in body temperature (F(1,10)=7.15, P<0.05). Pretreatment with L-DOPA did not alter the hyperthermic response induced by MDMA.
Figure 9.
Changes in the levels of (a) dopamine (DA), (b) homovanillic acid (HVA) and (c) 3,4-dihydroxyphenylacetic acid (DOPAC) in the striatal dialysate of rats treated with L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) 30 min before MDMA (5 mg kg−1 i.p.). L-DOPA did not modify the effect of MDMA on extracellular dopamine (F(1,8)=0.0908, n.s.) but reversed the MDMA-induced decrease in HVA (F(1,9)=47.82, P<0.001) and DOPAC (F(1,10)=20.55, P<0.001). Values are expressed as a percentage of the mean of two measurements before drug administration. Each value is the means±s.e. mean of 4–6 experiments.
Figure 10.
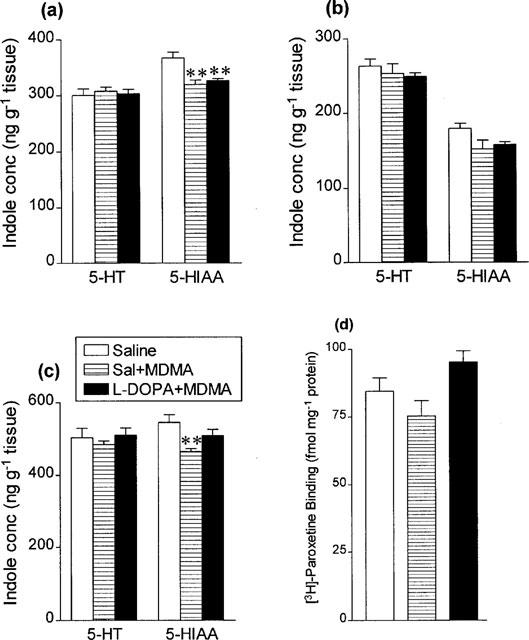
Indole concentrations in (a) hippocampus, (b) cortex and (c) striatum and (d) density of 3H paroxetine labelled 5-HT uptake sites in cortex 7 days following saline or L-DOPA (25 mg kg−1 i.p.+benserazide 6.25 mg kg−1 i.p.) 30 min before MDMA (5 mg kg−1 i.p.). Results shown as means±s.e. mean, n=5–8. Different from saline-treated: **P<0.01.
Effect of clomethiazole on the MDMA-induced change in dopamine metabolism in vivo
The increase in dopamine and decrease in HVA and DOPAC induced by MDMA (15 mg kg−1) was totally unaltered by administration of clomethiazole (50 mg kg−1) 5 min prior and 55 min post MDMA injection (Figure 11).
Figure 11.

Changes in the levels of dopamine (DA) and the metabolites (shown in the insert) in the striatal dialysate of rats treated with clomethiazole (CMZ, 50 mg kg−1 i.p.) or saline 5 min before and 55 min after MDMA (15 mg kg−1 i.p.). Clomethiazole did not modify the effect of MDMA on DA, DOPAC and HVA extracellular levels. Values are expressed as a percentage of the mean of three measurements before drug administration. Each value is the means±s.e. mean of 6–7 experiments.
Discussion
Both Schmidt et al. (1990b) and Hewitt & Green (1994) have reported that administration of haloperidol concurrently with MDMA attenuates the neurodegeneration of 5-HT terminals in the brain produced by this neurotoxic amphetamine and this observation was confirmed by the current study. Schmidt et al. (1990b) did not measure body temperature but Hewitt & Green (1994) observed that the combination of haloperidol and MDMA, whilst not producing hypothermia, did markedly attenuate the hyperthermia normally produced by the drug, an observation also confirmed by the present study. We therefore repeated the experiment on the neuroprotective effect of haloperidol but used a homeothermic blanket, thereby keeping the rectal temperature of the animals elevated to a similar level to that seen in the group given only MDMA (Figure 1). The protective effect of haloperidol in this hyperthermic group was marginal. These data support the findings in our other recent investigations (Colado et al., 1998a, 1998b) which have indicated strongly that drugs can provide substantial neuroprotection not only by producing hypothermia as has previously been demonstrated by others (Farfel & Seiden 1995a, 1995b; Malberg et al., 1996; Colado et al., 1998a) but also by merely preventing the MDMA-induced hyperthermia (Colado et al., 1998b). Haloperidol therefore can be concluded to be protective because of its effect on temperature and not because of a selective neuroprotective mechanism.
This finding in turn raised the question as to the validity of earlier proposals that dopamine plays a key role in the expression of MDMA-induced neurotoxicity. Various observations have been used to support this tenet, including the fact that MDMA alters dopamine release (Johnson et al., 1986; Schmidt et al., 1987; Hiramatsu & Cho, 1990; Nash, 1990; Nash & Brodkin, 1991) and the observation that L-DOPA administration potentiates MDMA-induced damage (Schmidt et al., 1991). We therefore re-investigated both these claims and also investigated whether L-DOPA administration alters the increase in free radical formation which has been shown to follow MDMA injection (Colado et al., 1997b; 1998c).
With regard to the alteration of dopamine metabolism which follows MDMA injection, both Johnson et al. (1986) and Schmidt et al. (1987) noted that MDMA released [3H]-dopamine from striatal slices and Steele et al. (1987) reported that the drug inhibited [3H]-dopamine uptake into striatal synaptosomes. Subsequent studies using either in vivo voltammetry (Yamamoto & Spanos, 1988) or microdialysis (Nash, 1990; Hiramatsu & Cho, 1990; Gough et al., 1991) found that MDMA administration resulted in a rapid rise in extracellular dopamine and decrease in DOPAC and HVA. Both the size and the time course of these changes as reported by Nash (1990) and Gough et al. (1991) were replicated in the current study. The size of the dopamine increase and DOPAC decrease are considerably greater than those observed in postmortem tissue 4 h after a single injection of MDMA (Colado & Green, 1994).
The current investigation found that following MDMA the concentration of HVA in the dialysate was decreased in a similar fashion to DOPAC, but that the change in the extracellular level of HVA lagged behind the DOPAC change. The difference in the time courses of changes in the extracellular DOPAC and HVA levels following MDMA may reflect the fact that HVA is a secondary metabolite of dopamine, being predominantly derived from intraneuronally formed DOPAC but also from the released dopamine via 3-methoxytyramine (Westerink & Spaan, 1982). Therefore, the delayed decrease in HVA compared to DOPAC might be due to an initial rise in 3-methoxytyramine and its subsequent conversion to HVA by catechol O-methyl transferase. These data suggest that MDMA exerts two separate actions on dopaminergic activity in vivo, one causing an increase in dopamine release and another producing a decrease in the intraneuronal dopamine metabolism (reflected by the extracellular levels of DOPAC and HVA). These changes on dopamine metabolites are probably the results of MDMA blocking dopamine re-uptake (causing less dopamine to be metabolized to DOPAC) and also by inhibiting monoamine oxidase (MAO). Dopamine is preferentially metabolized by MAO-A in the rat striatum in vivo (Kato et al., 1986) and MDMA acts as a competitive inhibitor of MAO-A activity (Leonardi & Azmitia, 1994). An additional explanation of the decrease in DOPAC and HVA would be a reduction in dopamine synthesis following MDMA. However this hypothesis is unlikely since MDMA does not alter tyrosine hydroxylase activity in the striatum (Stone et al., 1986).
The data raise a further point; that is, the nature of the mechanism of MDMA-induced dopamine release. It has often been assumed that the dopamine release is by a carrier-mediated exchange mechanism as occurs with other amphetamine-like drugs (Raiteri et al., 1979; Fischer & Cho, 1979; also see McMillen, 1983). However it is worth noting that several investigators have provided convincing evidence that 5-HT2 agonists enhance MDMA-induced dopamine release while 5-HT2 antagonists inhibit release (Nash, 1990; Schmidt et al., 1990a, 1994; Yamamoto et al., 1995; Gudelsky et al., 1994). MDMA-induced 5-HT release therefore appears to play a major role in the release of dopamine. When one adds the known effects of MDMA on dopamine uptake inhibition (Steele et al., 1987) and MAO inhibition (Leonardi & Azmitia, 1994) then it becomes plausible to suggest that carrier-mediated exchange need not be a major determinant in the increase of extracellular dopamine which follows MDMA administration, a suggestion supported by recent in vitro studies (Crespi et al., 1997). Further support for this proposal comes from our study on the effect of clomethiazole. This drug, which enhances GABAA receptor activity (see Green, 1998), inhibited the increase in extracellular dopamine which follows administration of methamphetamine, a known carrier-mediated mechanism (McMillen, 1983). Therefore, the failure of this drug to alter the MDMA-induced increase in extracellular dopamine may reflect a difference in the release mechanism between these two amphetamine derivatives. The implications of this to the selective neurotoxic action of MDMA are discussed later.
Interestingly the size of the increase in extracellular dopamine in the striatum following MDMA seen in vivo was similar in magnitude to that seen after a high dose of methamphetamine (Baldwin et al., 1993) which raises questions as to why the locomotor and behavioural activity seen after MDMA administration appears to be 5-HT rather than dopamine receptor mediated, having a different profile to that seen after amphetamine (Callaway et al., 1990; Callaway & Geyer, 1992). It could be that the serotonin-mediated behaviours predominate over any dopamine-mediated effects. While MDMA releases dopamine in both the accumbens (Callaway & Geyer, 1992) and striatal regions (Nash, 1990; this paper) the accumbens is the locus for locomotor activity following amphetamine administration (Kelly et al., 1975). However, recent data suggest that the predominant action of MDMA in this region may be that of inhibiting dopamine neuronal firing (Gifford et al., 1996). Since the drug can induce Ca2+ dependent dopamine release (Crespi et al., 1997) dopamine mediated function would be suppressed, thereby allowing the expression of 5-HT mediated behaviours including the serotonin syndrome (Spanos & Yamamoto, 1989; Colado et al., 1993; Green et al., 1995).
When L-DOPA was given 2 h after MDMA we observed that extracellular dopamine was increased above that seen in the MDMA alone treated group for approximately 3 h and that HVA and DOPAC concentrations also increased markedly over a similar period. These changes presumably reflect increased dopamine synthesis and release. Despite this clear in vivo indication of increased dopamine release and possibly also function, no enhancement of MDMA neurotoxicity was observed 7 days later. These data are in contradiction of the report of Schmidt et al. (1991) that MDMA-induced neurotoxicity is enhanced by L-DOPA. While Schmidt and colleagues used higher doses of L-DOPA these proved impossible to administer in the current study as they proved fatal. This is probably because of the use of the Dark Agouti strain of rat in our study since we have previously found that neurotoxicity occurs in these animals after much lower doses of MDMA and p-chloroamphetamine (PCA) than are required with other strains (see Colado et al., 1995; 1997b; Murray et al., 1996). Nevertheless using the highest practical dose of L-DOPA, no potentiation of MDMA-induced toxicity was detected. We were also unable to induce neurotoxicity in rats given a sub-toxic dose of MDMA by also administering L-DOPA, as was also reported by Schmidt et al. (1991).
In order to investigate further this apparent discrepancy between our data and that of Schmidt et al. (1991) we also examined whether L-DOPA altered MDMA-induced free radical formation. The MDMA-induced increase in the dialysate concentration of 2,3-and 2,5-DHBA that has previously been reported to occur after MDMA administration (Colado et al., 1997b; 1998c) was again seen in the current study. While the concentration of 2,5-DHBA appeared to be increased by L-DOPA, this change was not statistically significant. In any event, as Halliwell et al. (1991) have pointed out, a 2,3-DHBA increase is the only reliable indicator of increased free radical formation because of enzyme-induced conversion of salicylate to 2,5-DHBA. Nevertheless we found no evidence that the MDMA-induced increase in either 2,3- or 2,5-DHBA was even modestly enhanced by L-DOPA administration. Thus one plausible explanation for the results obtained by Schmidt et al. (1991), namely that tissue damage resulting from free radicals produced by MDMA metabolism (see Colado et al., 1997b) would be further potentiated by administration of L-DOPA, a compound that through its metabolism to dopamine would also increase free radical production through auto-oxidation (see Chiueh et al., 1992; 1993), could not be supported.
What does remain a possible explanation for the discrepant findings is the degree of hyperthermia produced by addition of L-DOPA to MDMA-treated rats. As we and others have shown, sustained hyperthermia markedly enhances the neurotoxic damage produced by MDMA (Colado et al., 1998b; Broening et al., 1995) presumably because hyperthermia is conducive to enhancing free radical production (Kil et al., 1996). If L-DOPA produced a sustained hyperthermia in the strain of rats used by Schmidt et al. (1990) rather than the more transient effect we observed, then neurotoxic damage would probably be increased. However, such a mechanism does not indicate that dopamine has a core role in MDMA-induced neurodegeneration as has been proposed by others (Stone et al., 1988; Schmidt et al., 1991). In this regard it is also worth pointing out that in our current study the increase in extracellular dopamine was almost the same whether a neurotoxic dose of MDMA of 15 mg kg−1 or a non-toxic dose of 5 mg kg−1 was given.
Finally we examined the effect of injecting clomethiazole on MDMA-induced changes in dopamine metabolism. Previously in an in vivo study we found that clomethiazole inhibited both the increase in dopamine and reduction in HVA and DOPAC which followed an injection of methamphetamine (Baldwin et al., 1993). In contrast, clomethiazole appeared to have no effect on MDMA-induced changes in dopamine metabolism. This finding is perhaps suprising in the light of a report suggesting that MDMA-induced dopamine release is regulated by the activity of a GABAergic input into the substantia nigra (Yamamoto et al., 1995). Clomethiazole is one of the few compounds to have been shown unequivocally to protect against MDMA-induced damage by a mechanism which does not involve a reduction in body temperature (Colado et al., 1998b). It is also a compound that has been shown to have a powerful neuroprotective action in a wide variety of animal models of acute ischaemic stroke (Green, 1998). These data suggest that dopamine is not involved in the neuroprotective effect of clomethiazole against MDMA-induced neurotoxicity.
Finally it is worth pointing out that a major conundrum is raised by the current study. In rats, but not mice (see Laverty & Logan 1990), MDMA produces a neurodegenerative loss of 5-HT nerve terminals, whilst sparing dopamine terminals (Schmidt & Kehne, 1990; Colado et al., 1997a, 1997b), a fact again supported by the findings in this current study (see Table 1). The question arises as to the mechanisms involved in this selectivity. It is noteworthy that methamphetamine which also increases extracellular 5-HT and dopamine (see for example Baldwin et al., 1993) and also increases free radical formation (Giovanni et al., 1995) produces neurotoxic degeneration of both 5-HT and dopamine neurones (Gibb et al., 1990; Green et al., 1992; Baldwin et al., 1993). If we are correct and the extracellular dopamine increase does not reflect carrier-mediated exchange then relatively little MDMA may be entering the dopamine nerve ending and being metabolised to the neurotoxic metabolites which are then auto-oxidised thereby producing free radicals (see Colado et al., 1997b). This may, in part, explain its lack of neurotoxic action on dopamine nerve endings since it would be reasonable to assume that MDMA would be metabolized in the same way in dopamine and 5-HT neurones. Against this hypothesis however there is good evidence that dopamine uptake inhibitors markedly inhibit MDMA-induced dopamine release (Nash & Brodkin, 1991). This observation would strongly support the idea that MDMA acts to release dopamine by a facilitated exchange mechanism which utilises the dopamine uptake carrier mechanism (see Raiteri et al., 1979; Fischer & Cho, 1979; McMillen, 1983).
Another possible explanation for the selective damage which follows MDMA is that the neurotoxic metabolites of methamphetamine and MDMA differ in their pattern of damage. A major postulated neurotoxic metabolite of MDMA, namely dihydroxymethylamphetamine (Hiramatsu et al., 1990; Colado et al., 1995, 1997b), has been reported not to be toxic to dopamine neurones (Johnson et al., 1992).
In conclusion, the current results demonstrate that MDMA has a major acute effect on dopamine metabolism in the brain but we suggest that there is little evidence to indicate that the changes reported are crucially involved in the long term neurotoxic degeneration of 5-HT neurones produced by this commonly used recreational drug.
Acknowledgments
M.I. Colado thanks CICYT (SAF 98-0074) and Astra Arcus for financial support. The authors are grateful to Servicio de Restriccion de Estupefacientes, Ministry of Health, Spain for supplying MDMA.
Abbreviations
- ANOVA
analysis of variance
- CMZ
chlomethiazole
- DA
dopamine
- DHBA
dihydroxybenzoic acid
- DOPAC
dihydroxyphenylacetic acid
- Hal
haloperidol
- 5-HIAA
5-hydroxyindoleacetic acid
- h.p.l.c.
high performance liquid chromatography
- 5-HT
5-hydroxytryptamine
- HVA
homovanillic acid
- MAO
monoamine oxidase
- MDMA
(±) 3,4-methylenedioxymethamphetamine
- 6-OHDA
6-hydroxydopamine
- PCA
p-chloroamphetamine
References
- BALDWIN H.A., COLADO M.I., MURRAY T.K., DE SOUZA R.J., GREEN A.R. Striatal dopamine release in vivo following neurotoxic doses of methamphetamine and effect of the neuroprotective drugs, chlormethiazole and dizocilpine. Br. J. Pharmacol. 1993;108:590–596. doi: 10.1111/j.1476-5381.1993.tb12847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALDWIN H.A., WILLIAMS J.L., SNARES M., FERREIRA T., CROSS A.J., GREEN A.R. Attenuation by chlormethiazole administration of the rise in extracellular amino acids following focal ischaemia in the cerebral cortex of the rat. Br. J. Pharmacol. 1994;112:188–194. doi: 10.1111/j.1476-5381.1994.tb13050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATTAGLIA G., YEH S.Y., O'HEARN E., MOLLIVER M.E., KUHAR K.J., DE SOUZA E.B. 3,4-methylenedioxyamphetamine destroys serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]-paroxetine-labelled serotonin uptake sites. J. Pharmacol. Exp. Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- BROENING H.W., BOWYER J.F., SLIKKER W., JR Age dependent sensitivity of rats to the long term effects of the serotonergic neurotoxicant (±)-3,4-methylenedioxymethamphetamine (MDMA) correlates with the magnitude of the MDMA-induced thermal response. J. Pharmacol. Exp. Ther. 1995;275:325–333. [PubMed] [Google Scholar]
- CALLAWAY C.W., GEYER M.A. Stimulant effects of 3,4-methylenedioxymethamphetamine in the nucleus accumbens of rats. Eur. J. Pharmacol. 1992;214:45–51. doi: 10.1016/0014-2999(92)90094-k. [DOI] [PubMed] [Google Scholar]
- CALLAWAY C.W., WING L.L., GEYER M.A. Serotonin release contributes to the locomotor stimulant effects of 3,4-methylenedioxymethamphetamine in rats. J. Pharmacol. Exp. Ther. 1990;254:456–464. [PubMed] [Google Scholar]
- CHIUEH C.C., KRISHNA G., TULSI P., OBATA T., LANG K., HUANG S.J., MURPHY D.L. Intracranial microdialysis of salicylic acid to detect hydroxyl radical generation through dopamine autooxidation in the caudate nucleus: effect of MPP+ Free Rad. Biol. Med. 1992;13:581–583. doi: 10.1016/0891-5849(92)90151-6. [DOI] [PubMed] [Google Scholar]
- CHIUEH C.C., MIYAKE H., PENG M.-T. Role of dopamine autoxidation, hydroxyl radical generation and calcium overload in underlying mechanisms involved in MPTP-induced Parkinsonism. Adv. Neurol. 1993;60:251–258. [PubMed] [Google Scholar]
- COLADO M.I., ESTEBAN B., O'SHEA E., GRANADOS R., GREEN A.R.Studies on the protective effect of pentobarbitone on MDMA-induced neurodegeneration Psychopharmacology 1998a(In press) [DOI] [PubMed]
- COLADO M.I., GRANADOS R., O'SHEA E., ESTEBAN B., GREEN A.R. Role of hyperthermia in the protective action of clomethiazole against MDMA (‘ecstasy')-induced degeneration, comparison with the novel NMDA channel blocker AR- R15896. Br. J. Pharmacol. 1998b;124:479–484. doi: 10.1038/sj.bjp.0701859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., ESTEBAN B., GRANADOS R., GREEN A.R.In vivo evidence against clomethiazole being neuroprotective against MDMA (‘ecstasy')-induced degeneration of rat brain 5-HT nerve terminals by a free radical scavenging mechanism Neuropharmacology 1998c(In press) [DOI] [PubMed]
- COLADO M.I., GREEN A.R. A study of the mechanism of MDMA (‘ecstasy')-induced neurotoxicity of 5-HT neurones using chlormethiazole, dizocilpine and other protective compounds. Br. J. Pharmacol. 1994;111:131–136. doi: 10.1111/j.1476-5381.1994.tb14034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., MURRAY T.K., GREEN A.R. 5-HT loss in rat brain following 3,4-methylenedioxymethamphetamine (MDMA), p-chloroamphetamine and fenfluramine administration and effects of chlormethiazole and dizocilpine. Br. J. Pharmacol. 1993;108:583–589. doi: 10.1111/j.1476-5381.1993.tb12846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., MISRA A., MURRAY T.K., GREEN A.R. A study of the neurotoxic effect of MDMA (‘ecstasy') on 5-HT neurones in the brains of mothers and neonates following administration of the drug during pregnancy. Br. J. Pharmacol. 1997a;121:827–833. doi: 10.1038/sj.bjp.0701201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., MURRAY T.K., WILLIAMS J.L., GREEN A.R. In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA (“Ecstasy”) and p-chloroamphetamine but not the degeneration following fenfluramine. Br. J. Pharmacol. 1997b;121:889–900. doi: 10.1038/sj.bjp.0701213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., WILLIAMS J.L., GREEN A.R. The hyperthermic and neurotoxic effects of ‘Ecstasy' (MDMA) and 3,4-methylenedioxyamphetamine (MDA) in the Dark Agouti (DA) rat, a model of the CYP2D6 poor metaboliser phenotype. Br. J. Pharmacol. 1995;115:1281–1289. doi: 10.1111/j.1476-5381.1995.tb15037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRESPI D., MENNINI T., GOBBI M. Carrier dependent and Ca2+ dependent 5-HT and dopamine release induced by (+)-amphetamine, 3,4-methylenedioxyamphetamine, p-chloroamphetamine and fenfluramine. Br. J. Pharmacol. 1997;121:1735–1743. doi: 10.1038/sj.bjp.0701325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARFEL C.M., SEIDEN L.S. Role of hypothermia in the mechanism of protection against serotonergic toxicity. I. Experiments with 3,4-methylenedioxymethamphetamine, dizocilpine, CGS 19755 and NBQX. J. Pharmacol. Exp. Ther. 1995a;272:860–867. [PubMed] [Google Scholar]
- FARFEL C.M., SEIDEN L.S. Role of hypothermia in the mechanism of protection against serotonergic toxicity. II. Experiments with methamphetamine, p-chloroamphetamine, fenfluramine, dizocilpine and dextromethorphan. J. Pharmacol. Exp. Ther. 1995b;272:868–875. [PubMed] [Google Scholar]
- FISCHER J.F., CHO A.K. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J. Pharmacol. Exp. Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- GIBB J.W., JOHNSON M., STONE D., HANSON G.R. MDMA: historical perspectives. Ann. N.Y. Acad. Sci. 1990;600:601–611. doi: 10.1111/j.1749-6632.1990.tb16913.x. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., MINABE Y., TOOR A., WANG R.Y., ASHBY C.R., JR Examination of the action of 3,4-methylenedioxymethamphetamine on rat A10 dopamine neurons. Synapse. 1996;23:52–57. doi: 10.1002/(SICI)1098-2396(199605)23:1<52::AID-SYN6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- GIOVANNI A., LIANG L.P., HASTINGS T.G., ZIGMOND M.J. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J. Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- GOUGH B., ALI S.F., SLIKKER W., JR, HOLSON R.R. Acute effects of 3,4-methylenedioxymethamphetamine (MDMA) on monomines in rat caudate. Pharmacol. Biochem. Behav. 1991;39:619–623. doi: 10.1016/0091-3057(91)90137-q. [DOI] [PubMed] [Google Scholar]
- GREEN A.R. Clomethiazole (Zendra®) in acute ischemic stroke: basic pharmacology and biochemistry and clinical efficacy. Pharmacol. Ther. 1998;80:123–147. doi: 10.1016/s0163-7258(98)00024-2. [DOI] [PubMed] [Google Scholar]
- GREEN A.R., CROSS A.J., GOODWIN G.M. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or ‘Ecstasy') Psychopharmacology. 1995;119:247–260. doi: 10.1007/BF02246288. [DOI] [PubMed] [Google Scholar]
- GREEN A.R., DE SOUZA R.J., WILLIAMS J.L., MURRAY T.K., CROSS A.J. The neurotoxic effects of methamphetamine on 5-hydroxytryptamine and dopamine in brain: evidence for the protective effect of chlormethiazole. Neuropharmacology. 1992;31:315–321. doi: 10.1016/0028-3908(92)90062-t. [DOI] [PubMed] [Google Scholar]
- GUDELSKY G.A., YAMAMOTO B.K., NASH J.F. Potentiation of 3,4-methylenedioxymethamphetamine-induced dopamine release and serotonin neurotoxicity by 5-HT2 receptor agonists. Eur. J. Pharmacol. 1994;264:325–330. doi: 10.1016/0014-2999(94)90669-6. [DOI] [PubMed] [Google Scholar]
- HALLIWELL B., KAUR H., INGELMAN-SUNDBERG M. Hydroxylation of salicylate as an assay for hydroxyl radicals: a cautionary note. Free Rad. Biol. Med. 1991;10:439–441. doi: 10.1016/0891-5849(91)90052-5. [DOI] [PubMed] [Google Scholar]
- HEWITT K.E., GREEN A.R. Chlormethiazole, dizocilpine and haloperidol prevent the degeneration of serotonergic nerve terminals induced by administration of MDMA (‘Ecstasy') Neuropharmacology. 1994;33:1589–1595. doi: 10.1016/0028-3908(94)90134-1. [DOI] [PubMed] [Google Scholar]
- HIRAMATSU M., CHO A.K. Enantiomeric differences in the effects of 3,4-methylenedioxymethamphetamine on extracellular monoamines and metabolites in the striatum of freely-moving rats: an in vivo microdialysis study. Neuropharmacology. 1990;29:269–275. doi: 10.1016/0028-3908(90)90012-g. [DOI] [PubMed] [Google Scholar]
- HIRAMATSU M., KUMAGAI Y., UNGER S.E., CHO A.K. Metabolism of methylenedioxymethamphetamine: formation of dihydroxymethamphetamine and a quinone identified as its glutathione adduct. J. Pharmacol. Exp. Ther. 1990;254:521–527. [PubMed] [Google Scholar]
- JOHNSON M., ELAYAN I., HANSON G.R., FOLTZ R.L., GIBB J.W., LIM H.K. Effects of 3,4-dihydroxymethamphetamine and 2,4,5-trihydroxymethamphetamine, two metabolites of 3,4-methylenedioxymethamphetamine, on central serotonergic and dopaminergic systems. J. Pharmacol. Exp. Ther. 1992;261:447–453. [PubMed] [Google Scholar]
- JOHNSON M.P., HOFFMAN A.J., NICHOLS D.E. Effects of the enantiomers of MDA, MDMA and related analogues on [3H]-serotonin and [3H]-dopamine release from superfused rat brain slices. Eur. J. Pharmacol. 1986;132:269–276. doi: 10.1016/0014-2999(86)90615-1. [DOI] [PubMed] [Google Scholar]
- JOHNSON M.P., HUANG X., NICHOLS D.E. Serotonin neurotoxicity in rats after combined treatment with a dopaminergic agent followed by a non-neurotoxic 3,4-methylenedioxymethamphetamine (MDMA) analogue. Pharmacol. Biochem. Behav. 1991;40:915–922. doi: 10.1016/0091-3057(91)90106-c. [DOI] [PubMed] [Google Scholar]
- KATO T., DONG B., ISHII K., KINEMUCHI H. Brain dialysis: In vivo metabolism of dopamine and serotonin by monoamine oxidase A but not B in the striatum of unrestrained rats. J. Neurochem. 1986;46:1277–1282. doi: 10.1111/j.1471-4159.1986.tb00650.x. [DOI] [PubMed] [Google Scholar]
- KELLY P.H., SEVIOUR P.H., IVERSEN S.D. Amphetamine and apomorphine responses in the rat following 6-OHDA lesions of the nucleus accumbens septi and corpus striatum. Brain Res. 1975;94:507–522. doi: 10.1016/0006-8993(75)90233-4. [DOI] [PubMed] [Google Scholar]
- KIL H.Y., ZAHNG J., PIANTADOSI L.A. Brain temperature alters hydroxyl radical production during cerebral ischemia/reperfusion in rats. J. Cereb. Blood Flow Metab. 1996;16:100–106. doi: 10.1097/00004647-199601000-00012. [DOI] [PubMed] [Google Scholar]
- KÖNIG J.F.R., KLIPPEL R.A.The rat brain. A Stereotaxic atlas of the forebrain and lower parts of the brain stem 1963. New York, Robert E Krieger Publishing Co. Inc
- LAVERTY R., LOGAN B.J. Protection by MK801 and other drugs of methylenedioxymethamphetamine (MDMA) neurotoxicity in rats and mice. Eur. J. Pharmacol. 1990;183:451–452. [Google Scholar]
- LEONARDI E.T.K., AZMITIA E.C. MDMA (Ecstasy) inhibition of MAO type A and type B: Comparisons with fenfluramine and fluoxetine (Prozac) Neuropsychopharmacology. 1994;10:231–238. doi: 10.1038/npp.1994.26. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MALBERG J.E., SABOL K.E., SEIDEN L.S. Co-administration of MDMA with drugs that protect against MDMA neurotoxicity produces different effects on body temperature in the rat. J. Pharmacol. Exp. Ther. 1996;278:258–267. [PubMed] [Google Scholar]
- MCMILLEN B.A. CNS stimulants: two distinct mechanisms of action for amphetamine-like drugs. Trends Pharmacol. Sci. 1983;4:429–432. [Google Scholar]
- MOLLIVER M.E., BERGER U.V., MANOUNAS L.A., MOLLIVER D.L., O'HEARN E., WILSON M.A. Neurotoxicity of MDMA and related compounds: anatomic studies. Ann. N.Y. Acad. Sci. 1990;600:640–661. doi: 10.1111/j.1749-6632.1990.tb16916.x. [DOI] [PubMed] [Google Scholar]
- MURRAY T.K., WILLIAMS J.L., MISRA A., COLADO M.I., GREEN A.R. The spin trap reagent PBN attennuates degeneration of 5-HT neurons in rat brain induced by p-chloroamphetamine but not fenfluramine. Neuropharmacology. 1996;35:1615–1620. doi: 10.1016/s0028-3908(96)00118-9. [DOI] [PubMed] [Google Scholar]
- NASH J.F. Ketanserin pretreatment attenuates MDMA-induced dopamine release in the striatum as measured by in vivo microdialysis. Life Sci. 1990;47:2401–2408. doi: 10.1016/0024-3205(90)90484-9. [DOI] [PubMed] [Google Scholar]
- NASH J.F., BRODKIN J. Microdialysis studies on 3,4-methylenedioxymethamphetamine-induced dopamine release: effect of dopamine uptake inhibitors. J. Pharmacol. Exp. Ther. 1991;259:820–825. [PubMed] [Google Scholar]
- O'HEARN E., BATTAGLIA G., DE SOUZA E.B., KUHAR M.J., MOLLIVER M.E. Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause ablation of serotonergic nerve terminals in forebrain: immunocytochemical evidence. J. Neurosci. 1988;8:2788–2803. doi: 10.1523/JNEUROSCI.08-08-02788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAITERI M., CERRITO F., CERVONI A.M., LEVI G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J. Pharmacol. Exp. Ther. 1979;208:195–202. [PubMed] [Google Scholar]
- SCHMIDT C.J. Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J. Pharmacol. Exp. Ther. 1987;240:1–7. [PubMed] [Google Scholar]
- SCHMIDT C.J., ABBATE G.M., BLACK C.K., TAYLOR V.L. Selective 5-hydroxytryptamine2 receptor antagonists protect against the neurotoxicity of methylenedioxymethamphetamine in rats. J. Pharmacol. Exp. Ther. 1990a;255:478–483. [PubMed] [Google Scholar]
- SCHMIDT C.J., BLACK C.K., TAYLOR V.L. Antagonism of the neurotoxicity due to a single administration of methylenedioxymethamphetamine. Eur. J. Pharmacol. 1990b;181:59–70. doi: 10.1016/0014-2999(90)90245-2. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., BLACK C.K., TAYLOR V.L. L-DOPA potentiation of the serotonergic deficits due to a single administration of 3,4-methylenedioxymethamphetamine to rats. Eur. J. Pharmacol. 1991;203:41–49. doi: 10.1016/0014-2999(91)90788-r. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., KEHNE J.H. Neurotoxicity of MDMA: neurochemical effects. Ann. N.Y. Acad. Sci. 1990;600:665–681. doi: 10.1111/j.1749-6632.1990.tb16917.x. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., RITTER J.K., SONSALLA P.K., HANSON G.R., GIBB J.W. Role of dopamine in the neurotoxic effects of methamphetamine. J. Pharmacol. Exp. Ther. 1987;233:539–544. [PubMed] [Google Scholar]
- SCHMIDT C.J., SULLIVAN C.K., FADAYEL G.N. Blockade of striatal 5-hydroxytryptamine2 receptors reduced the increase in extracellular concentrations of dopamine produced by the amphetamine analogue 3,4-methylenedioxymethamphetamine. J. Neurochem. 1994;62:1382–1389. doi: 10.1046/j.1471-4159.1994.62041382.x. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., WU L., LOVENBERG W. Methylenedioxy methamphetamine: a potentially neurotoxic amphetamine analogue. Eur. J. Pharmacol. 1986;124:175–178. doi: 10.1016/0014-2999(86)90140-8. [DOI] [PubMed] [Google Scholar]
- SHARKEY J., MCBEAN D.E., KELLY P.A.T. Alterations in hippocampal function following repeated exposure to the amphetamine derivative methylenedioxymethamphetamine (“Ecstasy”) Psychopharmacology. 1991;105:113–118. doi: 10.1007/BF02316872. [DOI] [PubMed] [Google Scholar]
- SPANOS L.J., YAMAMOTO B.K. Acute and subchronic effects of methylenedioxymethamphetamine [(±)MDMA] on locomotion and serotonin syndrome behavior in the rat. Pharmacol. Biochem. Behav. 1989;32:835–840. doi: 10.1016/0091-3057(89)90044-0. [DOI] [PubMed] [Google Scholar]
- STEELE T.D., MCCANN U.D., RICAURTE G.A. 3,4-methylenedioxymethamphetamine (MDMA, ‘Ecstasy'); pharmacology and toxicology in animals and humans. Addiction. 1994;89:539–551. doi: 10.1111/j.1360-0443.1994.tb03330.x. [DOI] [PubMed] [Google Scholar]
- STEELE T.D., NICHOLAS D.E., YIM G.K.W. Stereochemical effects of 3,4-methylenedioxymethamphetamine (MDMA) and related amphetamine derivatives on inhibition of uptake of [3H]-monoamines into synaptosomes from different regions of rat brain. Biochem. Pharmacol. 1987;36:2297–2303. doi: 10.1016/0006-2952(87)90594-6. [DOI] [PubMed] [Google Scholar]
- STONE D.M., JOHNSON M., HANSON G.R., GIBB J.W. Role of endogenous dopamine in the central serotonergic deficits induced by 3,4-methylenedioxymethamphetamine. J. Pharmacol. Exp. Ther. 1988;247:79–87. [PubMed] [Google Scholar]
- STONE D.M., STAHL D.C., HANSON G.R., GIBB J.W. The effects of 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-methylenedioxyamphetamine (MDA) on monoaminergic systems in the rat brain. Eur. J. Pharmacol. 1986;128:41–48. doi: 10.1016/0014-2999(86)90555-8. [DOI] [PubMed] [Google Scholar]
- WESTERINK B.H.C., SPAAN S.J. On the significance of endogenous 3-methoxytyramine for the effects of centrally acting drugs on dopamine release in the rat brain. J. Neurochem. 1982;38:680–686. doi: 10.1111/j.1471-4159.1982.tb08685.x. [DOI] [PubMed] [Google Scholar]
- WHITE S.R., OBRADOVIC T., IMEL K.M., WHEATON M.J. The effects of methylenedioxymethamphetamine (MDMA, ‘ecstasy') on monoaminergic neurotransmission in the central nervous system. Progr. Neurobiol. 1996;49:455–479. doi: 10.1016/0301-0082(96)00027-5. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO B.K., NASH J.F., GUDELSKY G.A. Modulation of methylenedioxymethamphetamine-induced striatal dopamine release by the interaction between serotonin and γ-aminobutyric acid in the substantia nigra. J. Pharmacol. Exp. Ther. 1995;273:1063–1070. [PubMed] [Google Scholar]
- YAMAMOTO B.K., SPANOS L.J. The acute effects of methylenedioxymethamphetamine on dopamine release in the awake-behaving rat. Eur. J. Pharmacol. 1988;148:195–203. doi: 10.1016/0014-2999(88)90564-x. [DOI] [PubMed] [Google Scholar]