

Abstract
CHO-K1 cells that were stably transfected with the gene for the human AT1 receptor (CHO-AT1 cells) were used for pharmacological studies of non-peptide AT1 receptor antagonists.
In the presence of 10 mM LiCl, angiotensin II caused a concentration-dependent and long-lasting increase of inositol phosphates accumulation with an EC50 of 3.4 nM. No angiotensin II responses are seen in wild-type CHO-K1 cells.
[3H]-Angiotensin II bound to cell surface AT1 receptors (dissociates under mild acidic conditions) and is subject to rapid internalization.
Non-peptide selective AT1 antagonists inhibited the angiotensin II (0.1 μM) induced IP accumulation and the binding of [3H]-angiotensin II (1 nM) with the potency order: candesartan > EXP3174 > irbesartan > losartan. Their potencies are lower in the presence of bovine serum albumin.
Preincubation with the insurmountable antagonist candesartan decreased the maximal angiotensin II induced inositol phosphate accumulation up to 94% and, concomitantly, decreased the maximal binding capacity of the cell surface receptors. These inhibitory effects were half-maximal for 0.6 nM candesartan and were attenuated by simultaneous preincubation with 1 μM losartan indicating a syntopic action of both antagonists.
Losartan caused a parallel rightward shift of the angiotensin II concentration-response curves and did not affect the maximal binding capacity. EXP3174 (the active metabolite of losartan) and irbesartan showed a mixed-type behavior in both functional and binding studies.
Reversal of the inhibitory effect was slower for candesartan as compared with EXP3174 and irbesartan and it was almost instantaneous for losartan, suggesting that the insurmountable nature of selective AT1 receptor antagonists in functional studies was related to their long-lasting inhibition.
Keywords: Angiotensin II, AT1 receptors, CHO cells, calcium transients, extracellular acidification, inositol trisphosphate, antagonist, candesartan, surmountable, insurmountable
Introduction
Angiotensin II, the effector peptide of the renin-angiotensin system, is an important regulator of the cardiovascular system which stimulates contraction of vascular smooth muscle cells and promotes their growth (Timmermans et al., 1992). These biological actions of angiotensin II are mediated by receptors of the AT1 subtype (Hodges et al., 1992).
The effect of antagonists on AT1 receptor function is traditionally studied by measuring their ability to antagonize angiotensin II-induced contraction of rabbit aorta rings/strips, a system with very small receptor reserve (Zhang et al., 1993). These studies include a preincubation step, in which the tissue is equilibrated with the antagonist, and an incubation step which comprises the consecutive addition of increasing concentrations of angiotensin II to generate a concentration-response curve. These experiments have shed light on the existence of different categories of selective AT1 receptor antagonists. Most antagonists produce parallel rightward shifts of the angiotensin II concentration-response curve, without affecting the maximal response. These are classified as surmountable antagonists (Gaddum et al., 1955) and losartan is a typical example (Mochizuki et al., 1995). Insurmountable antagonists are able to depress the maximal response. The extent of this decline varies considerably from one antagonist to another and it seems to be unrelated to the potency of the antagonist to produce rightward shifts of the concentration-response curve (Liu et al. 1992; Cazaubon et al., 1993; Noda et al., 1993; Mochizuki et al., 1995). The molecular basis for insurmountable antagonism is still a matter of debate. Although the longevity of the antagonist-receptor complex is retained as the cause in many studies, insurmountable antagonism has also been explained by the presence of allosteric binding sites on the receptor (Timmermans et al., 1991); slowly interconverting receptor conformations (de Chaffoy de Courcelles et al., 1986; Robertson et al., 1994); slow removal of the antagonist from tissue compartments, cells or matrix surrounding the receptor (Robertson et al., 1992; Panek et al., 1995) and even by the ability of the antagonist to modulate the amount of internalized receptors (Liu et al., 1992).
Compared to contraction studies with rabbit aortic strips, cell lines expressing the transfected human AT1 receptor may offer additional advantages for the investigation of the fundamental properties of antagonist-AT1 receptor interactions. First, radioligand binding and functional experiments can be performed on a single, intact cell system and under the same experimental conditions. Second, the presence of the human AT1 receptor also minimizes potential species-related differences when comparing the in vitro effects of antagonists with their clinical action. Third, the untransfected parent cells may serve as useful controls for the detection of receptor-unrelated phenomena and, finally, experiments are possible with various receptor mutants which lack typical properties such as activation and internalization.
The aim of the present study is to use CHO-K1 cells transfected with the gene coding for the human AT1 receptor (CHO-AT1 cells) to distinguish between surmountable and full or partially insurmountable antagonists by functional assays and radioligand binding.
Methods
Drugs used
Candesartan (CV-11974; 2-ethoxy-1-[(2′-(1H-tetrazol-5-yl)biphenyl -4 -yl)methyl] -1H -benzimidazoline -7 -carboxylic acid) (Shibouta et al., 1993; Noda et al., 1993), EXP 3174 (2-n-butyl-4-chloro-1-[(2′-(1H-tetrazol-5-yl) biphenyl-4-yl) methyl]imidazole-5-carboxylic acid) (Wong et al., 1990), losartan (DuP 753; 2-n-butyl-4-chloro-5-hydroxymethyl-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole) (Wong et al., 1990) and irbesartan (SR 47436; 2-n-butyl-4-spirocyclopentane-1- [(2′-(1H -tetrazol -5 -yl) biphenyl -4 -yl) methyl] 2 -imidazolin -5-one) (Cazaubon et al., 1993) were obtained from Astra Hässle (Mölndal, Sweden). Unlabelled angiotensin II, bovine serum albumin (BSA, Fraction V), (±)-sulphinpyrazone and ionomycin were obtained from Sigma (St. Louis, MO, U.S.A.). [3H]-Angiotensin II (50 Ci mmol−1 was obtained from New England Nuclear (Boston, U.S.A.) and myo-[3H]-inositol (20 Ci mmol−1) was from Pharmacia/Amersham/Biotech (Buckinghamshire, U.K). Fura-2-acetoxymethylester and pluronic acid were from Molecular Probes Inc. (Eugene, U.S.A). All other chemicals were of the highest grade commercially available.
Cell culture
Wild-type Chinese Hamster Ovary cells (CHO-K1) were kindly donated by Dr H. Verschueren (Pasteur Institute, Brussels, Belgium). CHO-K1 cells were stably transfected with the cDNA for the human angiotensin II AT1 receptor (denoted as CHO-AT1 cells) as follows. Primers corresponding to the 5′-(ATGATTCTCAACTCTTCTACT) and 3′-(TCACTCAACCTCAAAACATGG) ends of the AT1 receptor sequence (Furuta et al., 1992) were used to perform PCR on cDNA from human adrenal gland (Clontech, U.S.A.). The PCR was done in a final volume of 50 μl and contained 1 ng cDNA, 1× AmpliTaq DNA polymerase buffer, 200 μM of each deoxynucleotide, 1 μM of each primer and 1 unit of AmpliTaq DNA polymerase (Perkin Elmer, U.S.A.). The PCR thermal profile used was 93°C for 45 s, 60°C for 20 s and 72°C for 60 s for a total of 30 cycles. Resulting PCR product, containing the entire coding region of human AT1 receptor was cloned in PCR3 vector (InVitrogen) and sequenced using the sequenase kit (Amersham, U.K.). The entire experiment starting with PCR was done on three occasions, and the clone containing the exact sequence as earlier published (Furuta et al., 1992) was chosen for further study. The cloned human AT1 receptor DNA was transfected in CHO-K1 cells using Lipofectin (LifeTechnologies, U.S.A.) according to the manufacturer's instructions. After 72 h of transfection, cells were harvested and replated in medium containing 1 mg ml−1 geneticin. Individual clones were collected, grown and analysed for binding to [125I]-angiotensin II. The clone with maximum binding was grown further in medium containing 0.5 mg ml−1 geneticin, and used in present studies.
Cells were cultivated in 75 cm2 flasks in Dulbecco's Modified Essential Medium (DMEM) which was supplemented with 2 mM L-glutamine, 2% of a stock solution containing 5000 I.U. ml−1 penicilline and 5000 μg ml−1 streptomycin (Life Technologies, Merelbeke, Belgium) and 10% foetal bovine serum (Life Technologies, Merelbeke, Belgium). The cells were grown in 5% CO2 at 37°C until confluent.
[3H]-Angiotensin II binding
Cells were plated in 24 well plates and cultured until confluent. Before the experiment, the cells were washed three times with 0.5 ml per well of DMEM at room temperature and left for 15 min 37°C in 400 μl DMEM. Preincubations were initiated by addition of 50 μl DMEM either alone (total binding) or containing 1 μM unlabelled angiotensin II (non-specific binding) or unlabelled competitor and proceeded at 37°C for 30 min or the indicated periods of time (kinetic experiment, Figure 9). Incubations were started by adding 50 μl of DMEM containing [3H]-angiotensin II (final concentration: 0.5–10 nM for saturation binding as in Figure 8 and 1 nM for other studies) was added and the plates incubated at 37°C for 30 min or for the indicated time periods (kinetic experiment, Figure 6). At the end of the incubation, the cells were placed on ice and washed three times with ice-cold PBS containing (in gl−1) CaCl2.2H2O, 0.132, KCl 0.2, KH2PO4 0.2, MgCl2.6H2O 0.1, NaCl 8 and Na2HPO4.2H2O 1.44). The cell surface binding of [3H]-angiotensin II was extracted by mild acid treatment: i.e. a 5 min incubation with 0.5 ml ice cold 50 mM glycine buffer (pH 3) containing 125 mM NaCl. This step was repeated and the radioactivity in the pooled fractions was counted after addition of 7 ml of scintillation liquid (Optisafe of Wallac, Turku, Finland) for 3 min in a liquid scintillation counter. To measure internalized radioligand, 0.5 ml 1 M NaOH was added to the acid-treated cells and the solubilized radioactivity counted as above.
Figure 9.
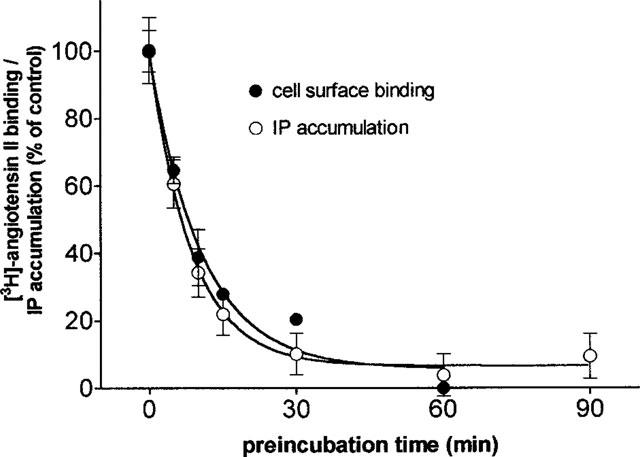
Comparison of the effect of candesartan on cell surface binding of [3H]-angiotensin II and IP accumulation. Cells are pretreated for 30 min with 1.5 nM candesartan for the indicated periods of time. Then 1 nM [3H]-angiotensin II is added and the incubation is continued for 30 min before measuring its cell surface binding. Alternatively, 0.1 μM angiotensin II is added and the incubation is continued for 5 min before measuring IP accumulation. Data are the average±s.e.mean of three independent experiments (triplicate) and are expressed as per cent of control binding or IP accumulation (i.e. with no candesartan pretreatment).
Figure 8.
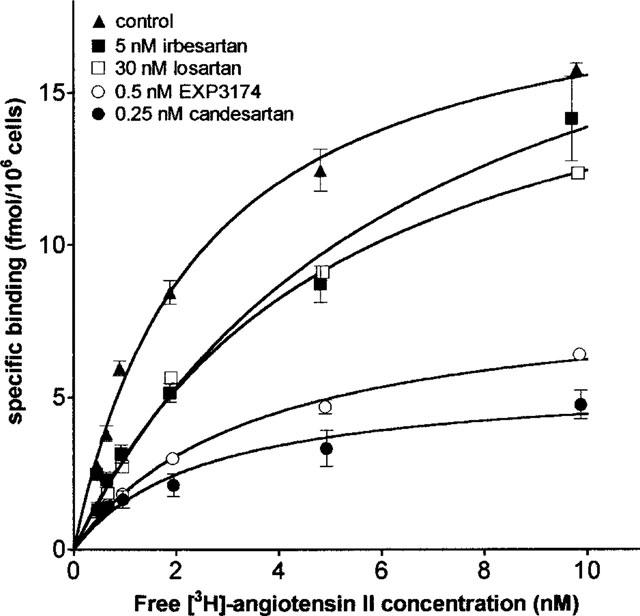
Cell surface saturation binding of [3H]-angiotensin II after pretreatment of the cells with buffer (control), candesartan, EXP3174, irbesartan or losartan. Cell surface binding is measured by mild acid treatment. Data are the average±s.e.mean of triplicate determinations of one representative experiment.
Figure 6.
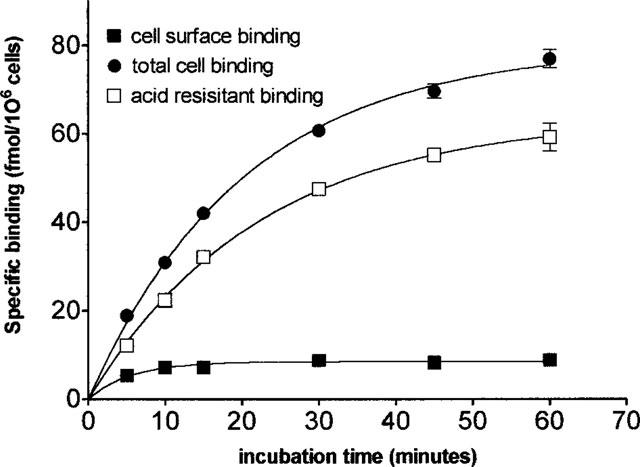
Distinction between cell surface binding of [3H]-angiotensin II and internalization of the radioligand in CHO-AT1 cells. Binding is measured after incubation of the cells with 1 nM radioligand for the indicated periods of time and removal of the free radioligand by washing with PBS at 4°C (see Methods section). Cell surface binding is determined by mild acid treatment after which internalized radioligand is determined by NaOH treatment. Total cell binding is measured in cells treated with NaOH only.
IP accumulation
The cells were plated in 24 well plates and cultured until confluent. The medium was replaced by DMEM containing 1 μCi ml−1 myo-[3H]-inositol and the cells were grown further for 20 h in 5% CO2 at 37°C. Just before the incubation, the cells were washed two times with DMEM (0.5 ml per well), subsequently 400 μl of DMEM containing 10 mM LiCl added to each well and the cells left for 15 min at 37°C. Preincubations were initiated by addition of 50 μl DMEM containing 10 mM LiCl without (controls) or with antagonists and proceed at 37°C for 30 min or the indicated periods of time (kinetic experiment, Figure 9). When candesartan was tested in the presence of 1 μM losartan (Figure 4b) both antagonists were added simultaneously. Subsequent incubations were initiated by adding 50 μl of DMEM containing 10 mM LiCl alone (basal accumulation) or containing the indicated concentration angiotensin II and the plates incubated at 37°C for 5 min (or for 30 min in Figure 4a) or for the indicated time periods (kinetic experiment, Figure 1c).
Figure 4.
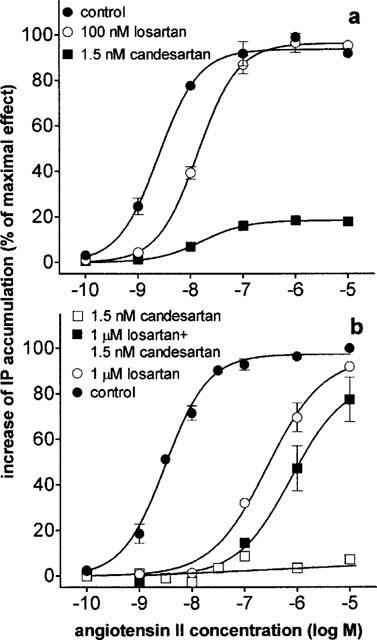
Concentration-response curves of angiotensin II mediated IP accumulation in CHO-AT1 cells pretreated with antagonists. Data are the average±s.e.mean of three independent experiments and expressed as outlined in Figure 3. (a) Incubation with angiotensin II for 30 min (control) or after pretreatment losartan or candesartan. (b) Incubation with angiotensin II for 5 min (control) or after pretreatment with losartan, candesartan or both.
Figure 1.
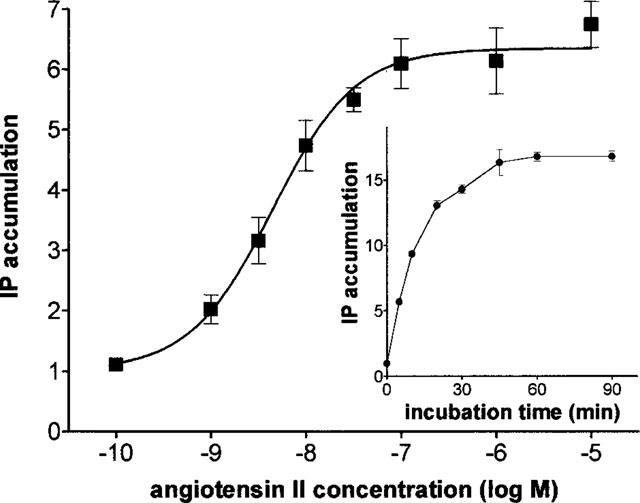
Concentration-response curve of angiotensin II mediated increase of IP accumulation in CHO-AT1 cells. Each data point is the average±s.e.mean of 11 independent experiments (each performed in duplicate) and are given as fold stimulation i.e. d.p.m after addition of the indicated angiotensin II concentration/d.p.m without angiotensin II. Basal and angiotensin II (10 μM) induced accumulation correspond to 318±24 and 2002±168 d.p.m respectively. Inset : Fold stimulation of basal IP accumulation in CHO-AT1 cells by addition of 0.1 μM angiotensin II and incubation for different periods of time.
For the recovery experiments (Figure 6), cells were preincubated with 500 μl DMEM either alone (controls) or containing antagonist at 37°C for 30 min. Then the cells were washed three times with 500 μl DMEM, left for the indicated periods of time at 37°C, washed three times again and left for 15 minutes at 37°C in 400 μl DMEM containing 10 mM LiCl. Incubations were started by adding 50 μl of DMEM either alone (basal accumulation) or containing 0.1 μM angiotensin II and the plates incubated at 37°C for 5 min.
The accumulation of IP was determined using a method modified from Seeuwen et al. (1988). Briefly, at the end of the incubation, the 24 well plates were placed on ice and IP formation stopped by aspiration of the medium and by addition of 750 μl of 10 mM formic acid at 4°C. After 30 min this solution was applied to a column containing 1 ml Bio-Rad anion exchange resin (AG 1×8, formate form, 200–400 mesh) and free inositol was eluted by 3 ml of 0.1 M formic acid. The inositol mono-, bis- and trisphosphates (denoted as IP accumulation) were eluted with 3 ml 0.1 M formic acid containing 0.8 M ammonium formate and the radioactivity measured in a liquid scintillation counter after addition of 10 ml of scintillation liquid (Ultima-flo-AF, Packard, Groningen, The Netherlands).
Measurement of [Ca2+]i in CHO-AT1 cells
Cells were plated on round glass coverslips (13 mm diameter), placed in 12 well plates and cultured until confluent. Then, the coverslips were washed four times with 2 ml per well of loading buffer containing (mM): HEPES 10, MgSO4 1, KCl 5, glucose 10, NaCl 145, CaCl21. (±)Sulphinpyrazone (0.25 mM) was added to the buffer to block the leakage of fura-2 through non-selective anion channels. After the last wash step, the coverslips were placed in loading buffer containing 3 μM fura-2-acetoxymethylester and 0.06 % (w v−1) pluronic acid and incubated for 2 h in the dark at room temperature. The coverslips were then washed three times with loading buffer and kept in the dark at room temperature for at least 20 min. Just before measurement the coverslips were washed again two times and placed diagonally in 5 ml polymethacrylate cuvettes (Kartell, Milano, Italy) containing 1.5 ml loading buffer in a thermostatized holder at 37°C in a fluorimeter (Photon Technology International Inc., Ontario, Canada), with the coverslip 45° with respect to the excitation light beam. The light source was a 75 W Power ArcTM Xenon lamp (Photon Technology International Inc., Ontario, Canada). After 5 min of equilibration fura-2, fluorescence was excited alternatively at 340 and 380 nm via a monochromator and the intensity of the light emitted through a 510 nm bandpass filter was measured using a SX-R928PT photomultiplier tube (Photon Technology International Inc., Ontario, Canada). The ratio of 340 and 380 nm excitation was calculated each 2 s with FelixTM software version 1.1 (Photon Technology International Inc., Ontario, Canada). The intracellular free calcium concentration was calculated according to Grynkiewicz et al. (1985) and the compounds were applied directly into the cuvette by injection of 50 μl from a stock solution in loading buffer. Compensation for background fluorescence was performed by subtracting the autofluorescence measured in cells that were not loaded with fura-2-acetoxymethylester. After the measurements, the fluorescence values at saturating [Ca2+]i were determined for each coverslip by application of 10 μM ionomycin. Subsequently, minimal calcium levels were determined by application of 8 mM EGTA and 0.2 M NaOH.
Extracellular acidification rate
Extracellular acidification rates were measured using a Cytosensor microphysiometer (Molecular Devices Corp., Menlo Park, CA, U.S.A.). Cells were seeded 16–18 h before the experiment into 12 mm diameter disposable polycarbonate cell capsule cups (Molecular Devices Corp.) at 3×105 cells in each cup and incubated at 37°C in 5% CO2 in growth medium (DMEM supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 units ml−1 penicillin and 100 mg ml−1 streptomycin). The capsule cups were loaded into the sensor chambers of the microphysiometer and perfused with running medium (growth medium without serum and without NaHCO3) either alone (basal acidification levels) or containing the indicated concentrations of angiotensin II. Flow rate was set to 100 ml min−1 pump cycles were 90 s. All experimental conditions were controlled by the Cytosoft software running on a Macintosh computer. For each cycle, cells were perfused with medium for the first 60 s and the pump was switched off for the remaining 30 s. The pH of the medium was recorded during the 65–90 s interval. The acidification rate of the buffer was calculated by the Cytosoft programme as derived from pH-measurement of the last 30 s of the cycle and rates were reported as percent of control; i.e. as per cent of the average acidification rate during the four cycles immediately preceding the addition of angiotensin II.
Data analysis
All experimental values represented the means±s.e.mean of at least three independent experiments (each performed in duplicate or triplicate). IC50 values from competition binding experiments, the KD and the Bmax values from saturation binding curves, EC50 values and corresponding maximal effects from concentration-response curves and kinetic constants from time curves were calculated by non-regression analysis by GraphPad Prism (San Diego, CA, U.S.A.) based on a one-site bimolecular reaction obeying the mass action law (the corresponding equation is [L-R]=[Rtot].[L].([L]+KD)−1) and for functional assays as described by Limbird (1996). The pKB value of losartan (−logKD) was calculated by Schild regression analysis according to Arunlakshana & Schild (1959). Statistical significance was assessed by a non-paired Mann-Whitney test with two-tailed P-values.
Results
Angiotensin II mediated functional responses in CHO-AT1 cells
Angiotensin II increased the intracellular free calcium concentration ([Ca2+]i) and the extracellular acidification rate in CHO-AT1 cells but not in the untransfected CHO-K1 cells (data not shown). These effects of angiotensin II were concentration dependent with EC50 values of approximately 3 nM. The rise in [Ca2+]i (up to 1000 nM) was rapid and was already declined to basal values (40–150 nM) after 1 min. The increase in extracellular acidification rate (up to 44% above baseline values) reached a maximum within less than 90 s and decreased rapidly thereafter.
In the presence of 10 mM LiCl, 0.1 μM angiotensin II caused a time-dependent accumulation of IP in myo-[3H]-inositol loaded CHO-AT1 cells (Figure 1, insert) but not in the untransfected CHO-K1 cells (data not shown). The accumulation increased linearly with the incubation time for the first 10 min and then gradually levelled off to attain a plateau value at 30 min. At this time point, cells still contained about 70% of the original amount of incorporated myo-[3H]-inositol. Unless stated otherwise, incubations for 5 min were adopted for all subsequent experiments. As shown in Figure 1, angiotensin II elicited a concentration-dependent increase of the IP accumulation with an EC50 of 3.4±0.7 nM (n=11). Maximal stimulation was produced at 0.1 μM angiotensin II and corresponded to a 6.6±0.12 fold stimulation over the basal IP level. This assay allowed the most accurate, reproducible and time-efficient determinations and was adopted for the measurement of angiotensin II-mediated responses in subsequent experiments.
Effect of selective AT1 receptor antagonists on IP accumulation
As shown in Figure 2, preincubation of the cells for 30 min with selective AT1 receptor antagonists caused a concentration-dependent inhibition of angiotensin II (0.1 μM) induced IP accumulation. The order of potency of the tested antagonists was: candesartan > EXP3174 > irbesartan > losartan (Table 1). The concentration-response curve of angiotensin II was not affected when adding 0.1% BSA in the medium (data not shown) but the inhibition curve of some of the antagonists was shifted to the right (Table 1). In the subsequent experiments BSA was omitted.
Figure 2.
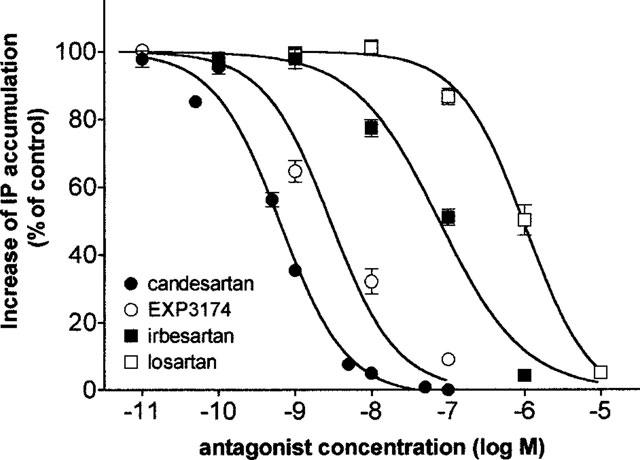
Inhibition curves of angiotensin II (0.1 μM, 5 min incubation) IP accumulation in CHO-AT1 cells after preincubating the cells for 30 min with increasing concentrations of candesartan, EXP3174, irbesartan or losartan. Data points are the average±s.e.mean of three independent experiments (duplicate) and are expressed as per cent of the control angiotensin II effect by preincubation with medium alone.
Table 1.
Potency of antagonist inhibition of [3H]-angiotensin II binding and IP accumulation

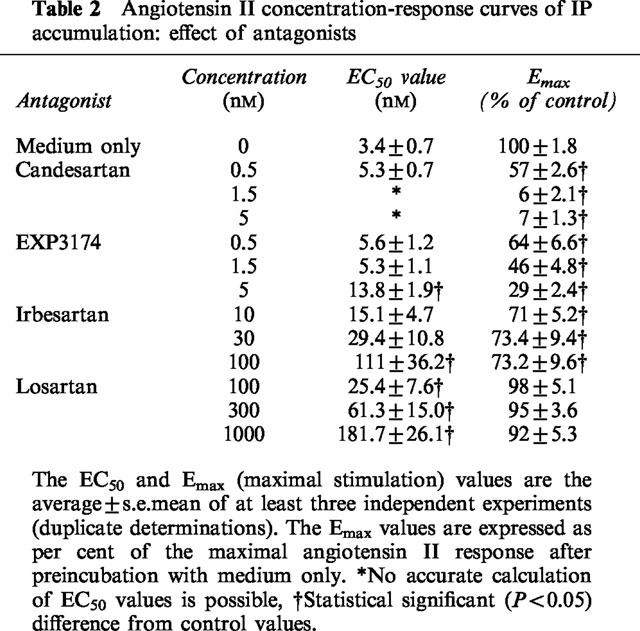
The effect of these antagonists on angiotensin II concentration-response curves was depicted in Figure 3 and the corresponding quantitative parameters were given in Table 2. The maximal response to angiotensin II declined by half when the cells were preincubated with 0.5 nM candesartan (Figure 3a). The decline was near to complete at 1.5 and 5 nM candesartan. Preincubation of the cells with 0.5–5 nM EXP3174 produced a partial reduction in the maximal response, along with a rightward shift of the angiotensin II concentration-response curve (Figure 3b). At concentrations between 10 and 100 nM, irbesartan produced a concentration-dependent, rightward shift of the concentration-response curve but a similar (±27%) reduction of the maximal response (Figure 3c). In contrast, preincubation of the cells with 100–1000 nM losartan only caused a progressive rightward shift of the concentration response curve without altering the maximal response (Figure 3d). Schild regression analysis of these data allowed to calculate the pKB for losartan was 7.90±0.06 with a slope of 0.92±0.06. As shown in Figure 4a for losartan and candesartan, the pattern of inhibition was retained when the incubation time with angiotensin II was increased to 30 min.
Figure 3.
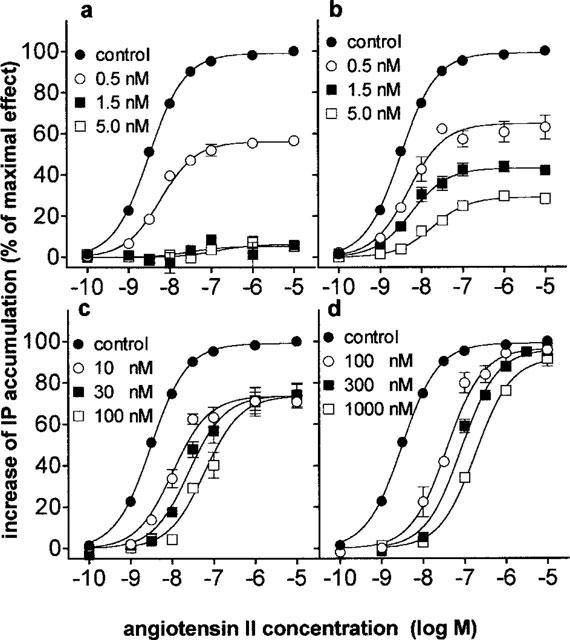
Concentration-response curves of angiotensin II mediated IP accumulation after preincubating CHO-AT1 cells for 30 min with increasing concentrations of candesartan (a), EXP3174 (b), irbesartan (c) or losartan (d). Data are the average±s.e.mean of at least three independent experiments (each performed in duplicate) and are expressed as per cent of the maximal angiotensin II response after preincubation with medium alone (control). The corresponding EC50 values are given in Table 2.
Table 2.
Angiotensin II concentration-response curves of IP accumulation: effect of antagonists
An angiotensin II concentration-response curve was obtained after preincubation of the cells with both candesartan (1.5 nM) and losartan (1000 nM) and compared with the curve with candesartan (1.5 nM) or losartan (1000 nM) pretreatment alone (Figure 4b). The maximal angiotensin II effect (Emax values) were 96±6% for losartan and 85±9% (extrapolated values) for losartan and candesartan and 7±1.5 (stimulation at 10 μM angiotensin II) for candesartan alone. The corresponding −logEC50 values were 8.50±0.04 nM for the control curve, 6.57±0.12 nM with losartan alone and 6.11±0.16 nM for the combination of losartan and candesartan.
Washout experiments were performed to compare the duration of the inhibitory effect of the different antagonists. For this purpose, cells were incubated for 30 min with antagonist, washed and left in fresh medium for the desired periods of time before measuring the (0.1 μM) angiotensin II-mediated response. As shown in Figure 5a 0.5 and 5 nM candesartan produced a 64±6 and 93±2% decrease of the response before washout. This decrease was minimally affected by the washing step (of 5 min) and was only slowly alleviated during the subsequent incubation step in fresh medium. After 4 h of incubation the response to angiotensin II was still attenuated by 36±3 and 60±3%, respectively. In contrast, the recovery of the angiotensin II response was much faster for EXP3174-, irbesartan- and losartan-pretreated cells; the inhibitory effect of EXP3174- and irbesartan was completely reversed after 4 and 1.5 h incubation in fresh medium, respectively (Figure 5b,c), and the inhibition by losartan completely reversed within less than 30 min (Figure 5d).
Figure 5.
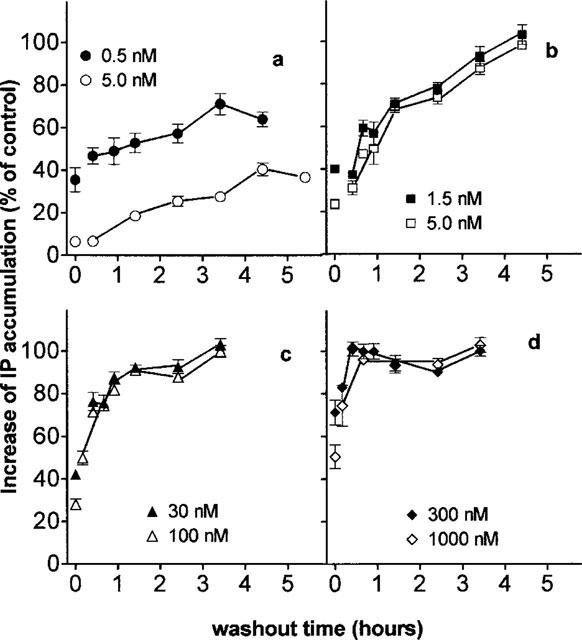
Recovery of angiotensin II (0.1 μM, 5 min incubation) induced IP accumulation from CHO-AT1 cells preincubated with candesartan (a), EXP3174 (b), irbesartan (c) or losartan (d). Cells are preincubated with antagonists for 30 min, washed and incubated for the indicated periods of time (washout time) in fresh medium before addition of angiotensin II. Data are the average±s.e.mean of at least three independent determinations (performed in triplicate) and are expressed as per cent of the control angiotensin II effect after treating the cells with medium alone under corresponding conditions.
Effect of selective AT1 receptor antagonists on the binding of [3H]-angiotensin II
Whereas control CHO-K1 cells did not display any specific [3H]-angiotensin II binding (data not shown) its binding to intact CHO-AT1 cells was shown in Figure 6. [3H]-angiotensin II bound initially to the cell surface receptors, followed by rapid internalization of the angiotensin II-receptor complexes. Surface-bound and internalized radioligand could be separated from each other: the former was readily released by mild acid treatment (see Methods) of the cells and the latter, acid-resistant binding, could be solubilized using 0.1 M NaOH (Crozat et al., 1986; Conchon et al., 1994; Hein et al., 1997). When CHO-AT1 cells were incubated at 37°C with 1 nM [3H]-angiotensin II, there was a steady increase of the binding with the incubation time (between 5 and 90 min). Although acid-resistant radioligand accounted for most of the binding at all the incubation times (90% after 5 min to 95% after 90 min), it was almost exclusively responsible for the time-dependent increase in total binding. Only acid-displaceable (cell surface) binding was considered in the subsequent experiments and the incubation time was set at 30 min.
Preincubation of the cells for 30 min with selective AT1 receptor antagonists caused a concentration-dependent inhibition of the cell-surface binding of 1 nM [3H]-angiotensin II (Figure 7). The order of potency of the antagonists was the same as for the functional experiments: i.e. candesartan > EXP3174 > irbesartan > losartan. The antagonists displaced the binding to the same extent as unlabelled angiotensin II and, as shown in Table 1, addition of 0.1% BSA in the incubation medium decreased the IC50 values of candesartan (2.7 fold), EXP3174 (4.7 fold) and losartan (3.0 fold) but not of irbesartan and angiotensin II.
Figure 7.
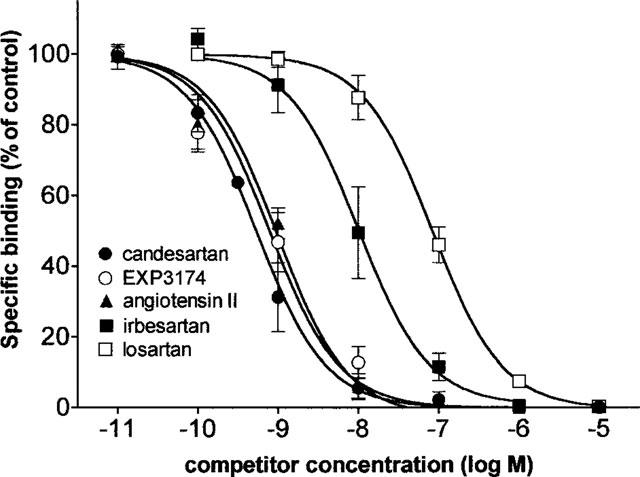
Inhibition of cell surface binding of 1 nM [3H]-angiotensin II by preincubation of the cells with candesartan, angiotensin II, EXP3174, irbesartan or losartan. Cell surface binding is measured by mild acid treatment and each data point represents the average ±s.e.mean of three independent experiments (performed in duplicate) and is expressed as per cent of control binding (i.e. preincubation with medium only).
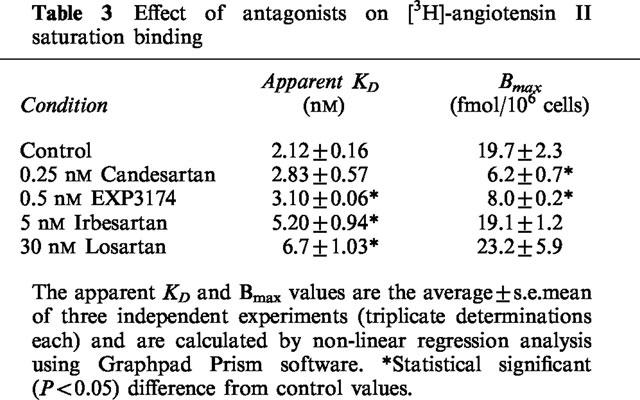
Figure 8 and Table 3 show the effect of the antagonists on the maximal binding capacity (Bmax) of [3H]-angiotensin II. In these experiments, cells were preincubated for 30 min with the antagonist, after which the binding of increasing concentrations of [3H]-angiotensin II to cell surface receptors was measured. Non-linear regression analysis of the saturation curves (Figure 8 and Table 3) revealed that 0.25 nM candesartan decreased the Bmax without significantly affecting the apparent KD. EXP3174 (0.5 nM) also decreased the Bmax and slightly increased the apparent KD. In contrast, 30 nM losartan and 5 nM irbesartan increased the apparent KD and left the Bmax value unaffected.
Table 3.
Effect of antagonists on [3H]-angiotensin II saturation binding
Time-dependency of the inhibitory effect of candesartan
In Figure 9 the time-dependency of the inhibition by candesartan of (a) the angiotensin II-mediated IP accumulation and (b) the binding of [3H]-angiotensin II to the cell surface receptors is shown. In these experiments, CHO-AT1 cells were preincubated with 1.5 nM candesartan for increasing periods of time up to 60 min, after which the response to 0.1 μM angiotensin II and the binding of 1 nM [3H]-angiotensin II were measured. Both were inhibited by candesartan in a matching, time-dependent fashion. The inhibition was almost complete after 60 min preincubation and the calculated apparent first-order rate constants for this process were 0.118±0.007 min−1 for the angiotensin II-mediated IP accumulation and 0.094±0.019 min−1 for the binding of [3H]-angiotensin II to the cell surface receptors.
Discussion
Cell lines which have been transfected with the gene encoding for wild-type and mutant AT1 receptors have proven to be particularly useful for investigating phenomena such as receptor-desensitization and internalization. Their utility in pharmacological studies was initially evoked by Perlman et al. (1995) and is further supported by the observations in the present report.
AT1 receptors are a member of the seven transmembrane guanyl nucleotide-binding protein (G-protein)-coupled receptor superfamily and biochemical studies have established that, in primary cultures of vascular smooth muscle cells, they stimulate the phosphoinositide signalling system leading to an elevation of the intracellular free calcium concentration ([Ca2+]i). This, in turn, triggers cell growth and constriction (Vincentini & Villereal, 1986). The initial AT1 receptor-evoked responses can conveniently be measured in primary cell cultures and in cell lines (Dickinson et al., 1994; Koh et al., 1994; Panek et al., 1995; Perlman et al., 1995; Balmforth et al., 1997) and appear to be common to the biological actions of angiotensin II. In addition, it was also observed by Dickinson et al. (1994) that angiotensin II increases the rate at which rabbit aortic smooth muscle cells acidify their environment with products of their energy metabolism. This latter response is commonly observed for G-protein coupled receptors and it is claimed to occur irrespective of the initial cellular events (McConnell et al., 1992).
In the present study, CHO-K1 cells were permanently transfected with the gene coding for the human AT1 receptor (CHO-AT1 cells). In this transfected cell line, angiotensin II concentration-response curves yielded comparable potencies for the IP production, rise in [Ca2+]i and increase in extracellular acidification rate. The two latter responses were transient and completely faded within less than 1–2 min. On the other hand, IP accumulation increased almost linearly during the first 10 min, after which it gradually levelled off to come to a standstill after about 30 min. Since only 30% of the cell's original amount of [3H]-phosphatidyl-inositol had been consumed, it is likely that the discontinued response is related to the well-known desensitization of the AT1 receptor. As the data from the phosphatidyl inositol turnover assay showed the least variability, this assay was selected for the ensuing investigation of selective AT1 receptor antagonists. As in rabbit aortic strip contraction studies, CHO-AT1 cells were preincubated with the investigated antagonist (30 min), and then challenged with angiotensin II.
Under these conditions, the selective AT1 receptor antagonists varied in the type of inhibition from almost completely insurmountable (candesartan, producing an over 90% decline of the maximal response) to completely surmountable (losartan, only producing a leftward shift of the agonist concentration-response curve). The same pattern for candesartan and losartan was observed when the IP accumulation was measured after a 5 or 30 min period, an observation in agreement with earlier rabbit aortic strip contraction studies (Cazaubon et al., 1993; Noda et al., 1993; Mochizuki et al., 1995). Kinetic experiments (Figure 9 and Noda et al., 1993) indicate that the inhibitory effect of candesartan increases with the preincubation time and that optimal inhibition is obtained after prolonged incubation. Since the preincubation times vary considerably from one report to another, particular attention should be paid to this factor when comparing with data in the literature. Yet, under comparable experimental conditions (i.e. also after a preincubation of 30 min), candesartan reduced the maximal response to angiotensin II by half at about 0.5 nM in the present study on CHO-AT1 cells, and at about 0.3 nM in rabbit aortic strip contraction experiments (Noda et al., 1993). On the other hand, the inhibitory effect antagonists such as losartan was the same regardless of the preincubation time (data not shown). This suggests that the equilibrium binding between this antagonist and the receptors is already attained after a very short incubation time. In agreement, the slope of the Schild regression analysis for losartan is close to unity (0.92±0.05) and the pKB-value (7.90±0.06) is comparable to those (8.2–8.5) reported in earlier contraction studies (Robertson et al., 1992; Renzetti et al., 1995; Mochizuki et al., 1995).
EXP3174, the active metabolite of losartan and irbesartan exhibited intermediary properties with only a partial suppression of the maximal response curve (respectively about 70 and 27%) along with a leftward shift of the concentration-response curve. Comparable values have been reported earlier for rabbit aortic strip contraction studies (Cazaubon et al., 1993; Mochizuki et al., 1995).
In agreement with the functional experiments, surmountable and insurmountable AT1 antagonists could also be discriminated by binding studies with [3H]-angiotensin II. Pretreatment of the CHO-AT1 cells with candesartan produced a net decrease in the number of [3H]-angiotensin II-labelled receptors at the cell surface. Kinetic experiments, involving different preincubation times, pointed out that this decrease in receptor number coincides with the decline in the angiotensin II-mediated IP accumulation. In contrast, losartan was only capable of increasing the KD of [3H]-angiotensin II without affecting the total number of AT1 receptors at the cell surface. Taken together, these data suggest that, at least for CHO-AT1 cells, there is a close relationship between the number of cell surface receptors that are available for [3H]-angiotensin II binding and the extent of the angiotensin II-mediated IP accumulation.
The degree of insurmountable antagonism was also reflected by the difference between the antagonist IC50 values for inhibiting the angiotensin II-mediated IP accumulation and [3H]-angiotensin II binding (Table 1). This is related to the larger agonist concentration in the functional experiment as in radioligand binding (100 versus 1 nM) and to the fact that inhibition curves for surmountable/competitive antagonists are shifted to the right when the angiotensin II concentration increases, whereas the curves for insurmountable antagonists are unaffected by this (data not shown). BSA, which is well known to bind selective AT1 receptor antagonists (Beauchamp et al., 1994), also produced rightward shifts of the inhibition curves (Table 1) but no distinction could be made between surmountable and insurmountable antagonists.
Although the molecular basis for insurmountable antagonism is still a matter of debate, it has been advanced that the longevity of the antagonist-receptor complex is responsible for the insurmountable effect of candesartan and several other selective AT1 receptor antagonists (Olins et al., 1994; Cirillo et al., 1995; Ojima et al., 1997). This explanation was based on the very slow recovery of the angiotensin II-mediated contraction in candesartan-treated rabbit aortic strips. In agreement, the angiotensin II-mediated IP accumulation in CHO-AT1 cells recovered only slowly after the removal of candesartan. Half-maximal recovery occurred after about 6 h. In contrast, the recovery was appreciably faster for EXP3174 (half-maximal after about 90 min) and still faster in the case of irbesartan and losartan.
The present findings are compatible with a model in which insurmountable antagonists dissociate so slowly from the receptors that they cannot be replaced by subsequently added angiotensin II. However, it cannot be excluded at the present level of investigation that such antagonists produce a long-lasting effect without remaining bound to the receptor. Such situations, in which the interconversion between inactive and active receptor conformations is much slower than the ligand binding, has been proposed in the two-state model by Gero (1983) and Robertson et al. (1994) and in the related coupling model of de Chaffoy de Courcelles et al. (1986). In addition, it was suggested by Panek et al. (1995) that antagonists may be insurmountable in tissues because of their slow removal from tissue compartments, cells or matrix surrounding the receptor, which allows the antagonist to rebind to the receptor. Direct radioligand binding studies with insurmountable antagonists such as candesartan should provide more insight in these remaining issues.
Allosteric modulation of AT1 receptors has been advanced as an alternative explanation for insurmountable antagonism (Wienen et al., 1992). Yet, similar to the experiments on rabbit aortic strips by Liu et al. (1992) and Ojima et al. (1997), we found that in CHO-AT1 cells the insurmountable antagonism by candesartan of angiotensin II-mediated IP accumulation is attenuated in the presence of a high concentration of the surmountable antagonist losartan. This result rather suggests that candesartan antagonism is due to a syntopic action at the AT1 receptor.
In conclusion, [3H]-angiotensin II binding and IP accumulation in intact CHO-AT1 cells can be used to discriminate between insurmountable (e.g. candesartan) and surmountable (e.g. losartan) selective AT1 receptor antagonists. In agreement with previous functional studies in isolated organs, our data suggest that the insurmountable action of candesartan is related to the slow recovery of angiotensin II responses after antagonist pretreatment.
Acknowledgments
G.V. is Onderzoeksdirecteur of the Nationaal Fonds voor Wetenschappelijk Onderzoek, Belgium. We are most obliged to Astra-Hässle Sweden, Astra Belgium and the Queen Elisabeth Foundation Belgium for their kind support. This text presents research results of the Belgian program on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming. The scientific responsibility is assumed by its authors.
Abbreviations
- [Ca++]i
intracellular free calcium concentration
- candesartan
2-ethoxy-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]-1H-benzimidazoline-7-carboxylic acid
- CHO-AT1 cells
Chinese hamster ovary cells expressing human AT1 receptors
- CHO-K1 cells
Wild-type Chinese hamster ovary cells
- DMEM
Dulbecco's Modified Essential Medium
- EXP 3174
2-n-butyl-4-chloro-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole-5-carboxylic acid
- IP
inositol mono-, bis- and trisphosphates
- irbesartan
2-n-butyl-4-spirocyclopentane-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]2-imidazolin-5-one)
- losartan
2-n-butyl-4-chloro-5-hydroxymethyl-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative use of drugs antagonists. Br. J. Pharmacol. 1959;14:48–52. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALMFORTH A.J., LEE A.L., WARBURTON P., DONNELLY D., BALL S.G. The conformational change responsible for ATl receptor activation is dependent upon two juxtaposed asparagine residues on transmembrane HeIices III and VII. J. Biol. Chem. 1997;272:4245–4251. doi: 10.1074/jbc.272.7.4245. [DOI] [PubMed] [Google Scholar]
- BEAUCHAMP H.T., CHANG R.S.L., SIEGL P.K.S., GIBSON R.E. In vivo receptor occupancy of the angiotensin II receptor by nonpeptide antagonists: relationship to in vitro affinities and in vivo pharmacologic potency. J. Pharm. Exp. Ther. 1994;272:612–618. [PubMed] [Google Scholar]
- CAZAUBON C., GOUGAT J., BOUSQUET F., GUIRAUDOU P, , GAYRAUD R., LACOUR C., ROCCON A., GALINDO G., BARTHELEMY G., GAUTRET B., BERNHART C., PERREAUT P., BRELIERE J.-C., LE FUR G., NISATO D. Pharmacological characterization of SR 47436, a new non-peptide AT1 subtype angiotensin II receptor antagonist. J. Pharmacol. Exp. Ther. 1993;265:826–834. [PubMed] [Google Scholar]
- CIRILLO R., RENZETTI A.R., CUCCHI P., GUELFI M., SALIMBENI A., CALIARI S., CASTELLUCCI A., EVANGELISATA S., SUBISSI A, GIACHETTI A. Pharmacology of LR-B/081, a new highly potent, selective and orally active, nonpeptide angiotensin II AT1 receptor antagonist. Br. J. Pharmacol. 1995;114:1117–1124. doi: 10.1111/j.1476-5381.1995.tb13323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONCHON S., MONNOT C., TEUTSCH B., CORVOL P., CLAUSER E. Internalization of the rat AT1a and AT1b receptors: pharmacological and functional requirements. FEBS Lett. 1994;349:365–370. doi: 10.1016/0014-5793(94)00703-9. [DOI] [PubMed] [Google Scholar]
- CROZAT A., PENHOAT A, SAEZ J.M. Processing of angiotensin II (A-II) and (sar1,Ala8)A-II by cultured bovine adrenocortical cells. Endocrinol. 1986;118:2312–2318. doi: 10.1210/endo-118-6-2312. [DOI] [PubMed] [Google Scholar]
- DE CHAFFOY DE COURCELLES D., LEYSEN J.E., ROEVENS P., VAN BELLE H. The Serotonin-S2 Receptor: A receptor, transducer coupling model to explain insurmountable antagonist effects. Drug Dev. Res. 1986;8:173–178. [Google Scholar]
- DICKINSON K.E., COHEN R.B., SKWISH S., DELANEY C.L., SERAFINO R.P., POSS M.A., GU Z., RYONO D.E., MORELAND S., POWELL J.R. BMS-180560, an insurmountable inhibitor of angiotensin II-stimulated responses: comparison with losartan and EXP3174. Br. J. Pharmacol. 1994;113:179–189. doi: 10.1111/j.1476-5381.1994.tb16191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUTA H., GUO D.-F., INAGAMI T. Molecular cloning and sequencing of the gene encoding human angiotensin II type 1 receptor. Biochem. Biophys. Res. Comm. 1992;183:8–13. doi: 10.1016/0006-291x(92)91600-u. [DOI] [PubMed] [Google Scholar]
- GADDUM J.H., HAMEED K.A., HATHAWAY D.E., STEPHENS F.F. Quantitative studies on antagonists for 5-hydroxytryptamine. Q. J. Exp. Physiol. 1955;40:49–74. doi: 10.1113/expphysiol.1955.sp001097. [DOI] [PubMed] [Google Scholar]
- GERO A. Desensitization, two-state receptors and pharmacological parameters. J. Theoret Biol. 1983;103:137–161. doi: 10.1016/0022-5193(83)90204-7. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HEIN L., MEINEL L., PRATT R.E., DZAU V.J., KOBILKA B.K. Intracellular traficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol. Endocrinol. 1997;11:1266–1277. doi: 10.1210/mend.11.9.9975. [DOI] [PubMed] [Google Scholar]
- HODGES J.C., HAMBY J.M., BLANKLEY C.J. Angiotensin II receptor binding inhibitors. Drugs Future. 1992;17:575–593. [Google Scholar]
- KOH E., MORIMOTO S., TOMITA J., RAKUGI H., JIANG B., INOUE T., NABATA T., FUKUO K., OGIHARA T. Effects of an angiotensin II receptor antagonist, CV-11974, on angiotensin II-induced increases in cytosolic free calcium concentration, hyperplasia, and hypertrophy of cultured vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1994;23:175–179. [PubMed] [Google Scholar]
- LIMBIRD L.L.Methods for characterization of receptors based on receptor-mediated responses in tissue or intact cell preparations Cell Surface Receptors: A Short Course on Theory and Methods 1996Boston/Dordrecht/London: Kluwer Academic Publishers; 27–60.ed. Limbird, L.L. pp [Google Scholar]
- LIU Y.J., SHANKLEY N.P., WELSH N.J., BLACK J.W. Evidence that the apparent complexity of receptor antagonism by angiotensin II analogues is due to a reversible and synoptic action. Br. J. Pharmacol. 1992;106:233–241. doi: 10.1111/j.1476-5381.1992.tb14322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCONNELL H.M., OWICKI J.R., PARCE J.W., MILLER D.L., BAXTER G.T., WADA H.G., PITCHFORD S. The cytosensor microphysiometer: biological applications of silicon technology. Science. 1992;257:1906–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- MOCHIZUKI S., SATO T., FURATA K., HASE K., OHKURA Y., FUKAI C., KOSAKAI K., WAKABAYASHI S., TOMIYAMA A. Pharmacological properties of KT3-671, a novel nonpeptide angiotensin II receptor antagonist. J. Cardiovasc. Pharmacol. 1995;25:22–29. doi: 10.1097/00005344-199501000-00005. [DOI] [PubMed] [Google Scholar]
- NODA M., SHIBOUTA Y., INADA Y., OJIMA M., WADA T., SANADA T., KUBO K., KOHARA Y., NAKA T., NISHIKAWA K. Inhibition of rabbit aortic angiotensin II (AII) receptor by CV-11974, a new neuropeptide AII antagonist. Biochem. Pharmacol. 1993;46:311–318. doi: 10.1016/0006-2952(93)90420-2. [DOI] [PubMed] [Google Scholar]
- OJIMA M., INADA Y., SHIBOUTA Y., WADA T., SANADA T., KUBO K., NISHIKAWA K. Candesartan (CV-11974) dissociates slowly from the angiotensin ATl receptor. Eur. J. Pharmacol. 1997;319:137–146. doi: 10.1016/s0014-2999(96)00837-0. [DOI] [PubMed] [Google Scholar]
- OLINS G.M., CHEN S.T., MCMAHON E., PALOMO M.A., REITZ D.B. Elucidation of the insurmountable nature of an angiotensin receptor antagonist, SC-54629. Mol. Pharmacol. 1994;47:115–120. [PubMed] [Google Scholar]
- PANEK R.L., LU G.H., OVERHISSER R.W., MAJOR T.C., HODGES J.C., TAYLOR D.G. Functional studies but not receptor binding can distinguish surmountable from insurmountable ATl antagonism. J. Pharm. Exp. Ther. 1995;273:753–761. [PubMed] [Google Scholar]
- PERLMAN S., SCHAMBYE H.T., RIVERO R.A., GREENLEE W.J., HJORTH S.A., SCHWARTZ T.W. Non-peptide angiotensin agonist. Functional and molecular interaction with the AT1 receptor. J. Biol. Chem. 1995;270:1493–1496. doi: 10.1074/jbc.270.4.1493. [DOI] [PubMed] [Google Scholar]
- RENZETTI A., CUCCHI P., GUELFI M., CIRILLO R., SALIMBENI A., SUBISSI A., GIACHETTI A. Pharmacology of LR-B/057, a novel orally active AT1 receptor antagonist. J. Cardiovasc. Pharmacol. 1995;25:354–360. doi: 10.1097/00005344-199503000-00002. [DOI] [PubMed] [Google Scholar]
- ROBERTSON M.J., BARNES J.C., DREW G.M., CLARK K.L., MARSHALL F.H., MICHEL A., MIDDLEMISS D., ROSS B.C., SCOPES D., DOWLE M.D. Pharmacological profile of GR 117289 in vitro: a novel, potent and specific non-peptide angiotensin AT1 receptor antagonist. Br. J. Pharmacol. 1992;107:1173–1180. doi: 10.1111/j.1476-5381.1992.tb13425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTSON M.J., DOUGALL I.G., HARPER D., MCKECHNIE K.C.W., LEFF P. Agonist-antagonist interactions at angiotensin receptors: application of a two-state receptor model. Trends Pharmacol. Sci. 1994;15:364–369. doi: 10.1016/0165-6147(94)90156-2. [DOI] [PubMed] [Google Scholar]
- SEEUWEN K, , LAGARDE A, POUYSSEGUR J. Deregulation of hamster fibroblast proliferation by mutated ras oncogenes is not mediated by constitutive activation of phosphoinositide-specific phospholipase C. EMBO J. 1988;7:161–168. doi: 10.1002/j.1460-2075.1988.tb02796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBOUTA Y., INADA Y., OJIMA M., WADA T., NODA M., SANADA T., KUBO K., KOHARA Y., NAKA T., NISHIKAWA K. Pharmacological profile of a highly potent & long-acting Angiotensin II receptor antagonist, 2-Ethoxy-l-[[2′-(1H-tetrazol-5-yl)biphenyl - 4- yl] methyl] -1 H-benzimidazole -7- carboxylic Acid (CV-11974), and its prodrug, (±)-1-(Cyclohexyloxycarbonyloxy)-ethyl 2-Ethoxy-1-[[2′-(1H-tetrazol-5-yl)biohenyl-4-yl] methyl] -lH- benzimidazole -7- carboxylate (TCV-116) J. Pharm. Exp. Ther. 1993;266:114–120. [PubMed] [Google Scholar]
- TIMMERMANS P.B., BENFIELD P., CHIU A.T., HERBLIN W.F., WONG P.C. Angiotensin II receptors and functional correlates. Am. J. Hypertens. 1992;5:221S–235S. doi: 10.1093/ajh/5.12.221s. [DOI] [PubMed] [Google Scholar]
- TIMMERMANS P.B.M.W.M., WONG P.C., CHIU A.T., HERBLIN W.F. Nonpeptide angiotensin II receptor antagonists. Trends. Pharmacol. Sci. 1991;12:55–62. doi: 10.1016/0165-6147(91)90498-h. [DOI] [PubMed] [Google Scholar]
- VINCENTINI L.M., VILLEREAL M.L. Inositol phosphates turnover, cytosolic Ca2+ and pH, Putative signals for the control of cell growth. Life Sci. 1986;38:2269–2276. doi: 10.1016/0024-3205(86)90632-6. [DOI] [PubMed] [Google Scholar]
- WIENEN W., MAUZ A.B.M., VAN MEEL J.C.A., ENTZEROTH M. Different type of receptor interaction of peptide and nonpeptide angiotensin II antagonists revealed by receptor binding and functional studies. Mol. Pharmacol. 1992;41:1081–1088. [PubMed] [Google Scholar]
- WONG P.C., PRICE A.W., CHIU A.T., DUNCIA J.V., CARINI D.J., WEXLER R., JOHNSON A., TIMMERMANS P.B.M.W.M. Nonpeptide angiotensin receptor antagonists. XI. Pharmacology of EXP3174: An active metabolite of DuP 753, an orally active antihypertensice agent. J. Pharmacol. Exp. Ther. 1990;255:211–217. [PubMed] [Google Scholar]
- ZHANG J.C., VAN MEEL A., PFAFFENDORF M., VAN ZWIETEN P. Different types of angiotensin II receptor antagonism Induced by BIBS 222 in the rat portal vein and rabbit aorta; the influence of receptor reserve. J. Pharmacol. Exp. Ther. 1993;269:509–514. [PubMed] [Google Scholar]