

Abstract
D-Galactosamine (GalN) depletes UTP primarily in the liver, resulting in decreased RNA synthesis in hepatocytes. Co-injection of GalN and lipopolysaccharide (LPS) into mice produces fulminant hepatitis with severe hepatic congestion, resulting in rapid death. Although the underlying mechanism is uncertain, GalN enhances the sensitivity to tumour necrosis factor (TNF). Administration of uridine (a precursor of UTP) prior injection of either LPS itself or interleukin-1 (IL-1) reduces the lethality of GalN+LPS. The present study focused on the effects of these agents on TNF production.
Intraperitoneal injection of GalN+LPS into mice greatly elevated serum TNF. Although large doses of LPS alone also greatly elevated serum TNF, LPS itself induced neither hepatic congestion nor rapid death. Administration of a macrophage depletor, liposomes encapsulated with dichloromethylene bisphosphonate, reduced both the TNF production and mortality induced by GalN+LPS.
Uridine, when injected 0.5 h after the injection of GalN+LPS, reduced the production of TNF. Prior injection of LPS, but not of IL-1, also reduced this TNF production.
Serum from LPS-injected mice reduced the TNF production induced by GalN+LPS, but it was less effective at reducing the lethality. Its ability to reduce TNF production was abolished by heat-treatment.
We hypothesize that a factor inhibiting TNF production by macrophages is produced by hepatocytes in response to LPS. Possibly, production of this hepatocyte-derived TNF-down-regulator (TNF-DRh) may be: (i) inhibited by GalN, causing over-production of TNF by macrophages and (ii) stimulated by LPS-pretreatment (and restored by uridine), causing reduced TNF production.
Keywords: Hepatitis; tumour necrosis factor (TNF); interleukin-1 (IL-1); lipopolysaccharide; galactosamine; endotoxin shock, inhibitor of TNF synthesis
Introduction
D-Galactosamine (GalN) is metabolized by the enzymes participating in the galactose pathway, which are most abundant in the liver. Since this pathway consumes uridine nucleotides, administration of GalN leads to a rapid depletion of these nucleotides, primarily in the liver, and to a decrease in RNA synthesis (Decker & Keppler, 1974). Injection of a large dose of this agent into rats induces a hepatic necrosis resembling the effects of human viral hepatitis (Decker & Keppler, 1974), and nucleolar fragmentation is an early event after injection of GalN (Shinozuka et al., 1973a and 1973b). Interestingly, injection of a lower dose of GalN sensitizes experimental animals to the lethal effects of lipopolysaccharides (LPS; endotoxin), so that fulminant hepatitis with severe hepatic congestion occurs after their co-injection, resulting in rapid death within a few hours (Galanos et al., 1979; Tiegs et al., 1989; Endo et al., 1992a). This experimental system is often used as a model of endotoxic shock. Injection of a combination of GalN and heat-killed gram-negative bacteria (instead of LPS) also induces an enhanced lethality, as does injection of a combination of GalN with gram-positive bacteria or with staphylococcal enterotoxin B (SEB), a super antigen (Freudenberg & Galanos, 1991; Miethke et al., 1992). Clarification of the mechanism underlying this sensitization phenomenon should thus help us reach a better understanding of the pathogenesis and pharmacological therapeutics of infectious diseases.
LPS is known to stimulate the production of a variety of cytokines. Of these, tumour necrosis factor (TNF) is considered to be the cytokine responsible for the lethality seen on injection of a combination of GalN with LPS or gram-negative or -positive bacteria, because TNF can be substituted for LPS, and because the lethality is inhibited by anti-TNF antibody (Lehmann et al., 1987; Tiegs et al., 1989; Endo et al., 1992a; Freudenberg & Galanos, 1991). However, Miethke et al., (1992) have reported that GalN produces no statistically significant change in the TNF production induced by SEB. On the basis of these results, GalN is thought to enhance the animal's sensitivity to the effects of TNF, although the underlying mechanism has yet to be elucidated.
Interestingly, administration of uridine (a precursor of uridine nucleotides) (Galanos et al., 1979; Lehmann et al., 1987; Endo et al., 1992a), or prior injection of either LPS itself or IL-1 can reduce the lethal action of GalN+LPS and that of GalN+TNF (Wallach et al., 1988; Libert et al., 1991; Endo et al., 1992a).
On the basis of the background described above, we decided to examine the effect of GalN+LPS on TNF production in mice and the effects of the administration of uridine and of prior injection of LPS or IL-1 on the TNF production and lethality that occur in mice given GalN+LPS.
Methods
Mice and reagents
Female C57BL/6N mice (6–7 weeks old) were obtained from the experimental animals facility of our university. All experiments conformed to national requirements (Japanese law no. 105, notification no. 6) and complied with the Guidelines for Care and Use of Laboratory Animals in Tohoku University. D(+)-Galactosamine (GalN) and uridine were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Recombinant human IL-1α and TNFα were provided by Dainippon Pharmaceutical Co. (Osaka, Japan) (Furutani et al., 1985). Recombinant human IL-1β was from Ohtsuka Pharmaceutical Co. (Tokushima, Japan) (Kikumoto et al., 1987). Mouse IL-12 and human IL-6 were from Genetics Institute (Cambridge, MA, U.S.A.). Human IL-2 and mouse IFNγ were from Shionogi Pharmaceutical Co. (Osaka, Japan). Human IFNα was provided by the Louis Pasteur Center for Medical Research (Kyoto, Japan). A lipopolysaccharide (LPS) from Escherichia coli O55 : B5 prepared by Boivin's method was obtained from Difco Laboratories (Detroit, MI, U.S.A.). Synthetic derivatives of lipid A with low cytotoxicity, namely, ONO-4007 and MPL (a monophosphoryl lipid A), were provided by Ono Pharmaceuticals (Osaka, Japan) and Ribi Immunochem (Hamilton, MT, U.S.A.), respectively. GalN and uridine were dissolved in sterile water, and the pH of each solution was adjusted to 7.0 with NaOH solution: the final concentrations of GalN and uridine were 60 and 50 mg ml−1, respectively. LPS and lipid A derivatives were dissolved in sterile saline. A mixture of LPS and GalN (1 μg and 60 mg, respectively, ml−1) was prepared by adding the LPS solution to the GalN solution. These solutions were injected i.p. (0.1 ml 10 g−1 body weight) except where otherwise noted.
Evaluation of the degree of hepatic congestion
The degree of hepatic congestion was evaluated by macroscopic observation as previously described (Endo et al., 1992a). Briefly, it was scored as follows, (1) no congestion; (2) slight (congestion in up to 20% of the liver); (3) medium (congestion in 20–50% of the liver); (4) severe (congestion in more than 50% of the liver).
Determination of cytokines by enzyme-linked immunosorbent assay (ELISA)
Blood was collected directly into test tubes following the decapitation of unanaesthetized mice. Serum was recovered by centrifugation at 2000×g at 4°C, then stored at −80°C until used. TNFα, IL-1α and IL-1β were assayed using ELISA kits (Endogen, Cambridge, MA, U.S.A.), the assay procedures being performed exactly as described by the manufacturer.
Depletion and detection of macrophages
A suspension of liposomes encapsulating dichloromethylene bisphosphonate (Cl2MBP-liposomes) was prepared by a method similar to that used by Van Rooijen and his coworkers (Van Rooijen & Sanders; 1994), as described previously (Endo et al., 1995; Shibazaki et al., 1998). Briefly, 75 mg of phosphatidylcholine and 11 mg of cholesterol were dissolved in chloroform (20 ml) in a round-bottomed flask (1000 ml). The thin film that formed on the walls of the flask after rotary evaporation at 37°C was dispersed by gentle shaking for 10 min in 10 ml of Cl2 MBP solution (200 mg ml−1) in 10 mM sodium phosphate buffer (PBS, pH 7.4). This suspension was kept for 2 h at room temperature, then sonicated for 3 min (50 Hz) and kept for another 2 h. The resulting liposome layer floating on the aqueous phase was collected using a Pasteur pipette, then suspended in 10 ml PBS and centrifuged at 10,000×g for 30 min. The precipitated liposomes were finally suspended in 4 ml of PBS; this preparation is hereafter referred to as the ‘original suspension' of Cl2MBP-liposomes. This original suspension was diluted as appropriate with PBS, and the resulting suspension was injected intravenously into the tail vein of mice at 0.2 ml mouse−1. Macrophages were detected by immunohistochemical staining of F4/80 antigen as described previously (Endo et al., 1995); this antigen is expressed specifically on the surface of these cells (Austyn & Gordon, 1981; Hirsch et al., 1981).
Data analysis
Experimental values are given as mean±s.d. The statistical significance of differences was analysed using Dunnett's multiple comparison test after ANOVA, P values less than 0.05 being considered significant.
Results
Effect of GalN on lethality in mice injected with LPS
Co-injection of GalN (600 mg kg−1) and LPS (10 μg kg−1) into mice induced rapid death within 5–8 h, severe congestion of the liver being observed in all mice (10/10) that were either moribund or dead. However, GalN by itself, even at a dose of 1500 mg kg−1, caused no death and no obvious ill effects in mice observed for up 3 days. Injection of LPS alone at a dose of 5 mg kg−1 also caused no death and no hepatic congestion, although the mice were considerably weakened. LPS at 10 mg kg−1 produced death in some mice (3/10), although death did not occur for at least 24 h.
Augmented elevation of serum TNFα with GalN+LPS
Injection of LPS (10 μg kg−1) elevated the serum levels of TNFα and IL-1 (α and β) (Figure 1). The elevation of TNFα was more rapid than that of IL-1, the peak levels of TNFα and IL-1 being seen at about 1 and 2–3 h after the injection of LPS, respectively. The peak level of TNFα was greater than that of IL-1β and slightly greater than that of IL-1α. Injection of GalN+LPS produced a much greater increase in serum TNFα than did LPS alone (Figure 2). The peak TNFα level was seen 1.5 h after the injection of GalN+LPS. We had expected that such an increased accumulation of TNFα might rapidly result in death. However, the serum TNFα had returned to its normal level within 3 h, i.e. 2–5 h before death occurred following injection of GalN+LPS (see above). There was no sign of hepatic congestion for at least 3 h after injection of GalN+LPS. At larger doses, LPS itself also greatly elevated the serum level of TNFα. The peak level of TNFα (about 2000 pg ml−1) produced by 1–10 mg kg−1 of LPS (Figure 3) was comparable to that produced by GalN+LPS (Figure 2). However, as described above, these doses of LPS induced neither hepatic congestion nor rapid death.
Figure 1.
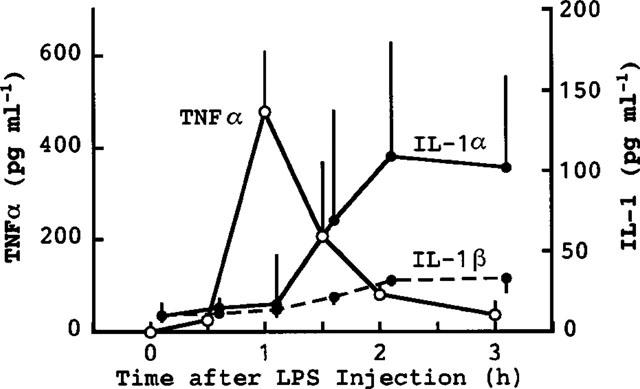
Elevation of serum TNFα, IL-1α and IL-1β following injection of LPS alone. Mice were killed at the indicated times after the injection of LPS (10 μg kg−1, i.p.). Values are mean±s.d. from four mice.
Figure 2.
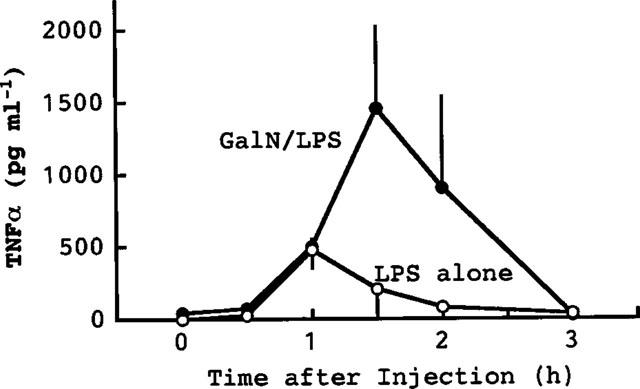
Elevation of serum TNFα following injection of LPS plus GalN. Mice were killed at the indicated times after the co-injection of LPS (10 μg kg−1, i.p.) and GalN (600 mg kg−1, i.p.). The effect on TNFα levels induced by LPS alone (shown in Figure 1) is also shown in this Figure for comparison. Values are mean±s.d. from four mice.
Figure 3.
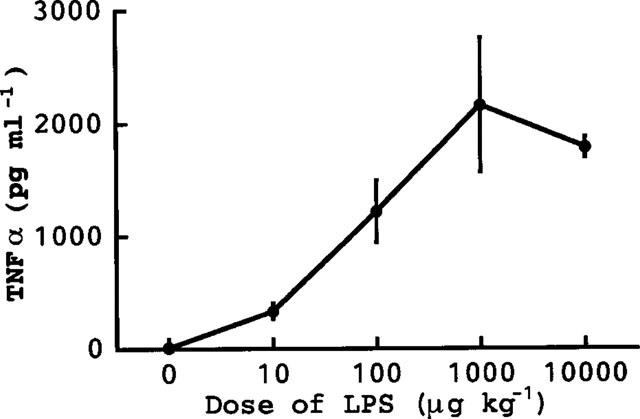
Dose-dependency of the TNFα production induced by LPS alone. The mice were killed 1.5 h after the i.p. injection of various doses of LPS and blood was then collected. Values are mean±s.d. from four mice.
Effect of uridine on the TNFα production induced by GalN+LPS
Administration of uridine can reverse the depletion of uridine nucleotides induced by GalN (Decker & Keppler, 1974). As reported previously, administration of uridine 0.5 h after an injection of GalN+LPS decreased the severity of the hepatic congestion and reduced to zero the lethality caused by GalN+LPS (Endo et al., 1992a). In the present study, it was found that an injection of uridine 0.5 h after GalN+LPS reduced the marked production of TNFα otherwise induced by an injection of GalN+LPS (Figure 4). Figure 4 also shows that GalN itself did not elevate TNFα.
Figure 4.
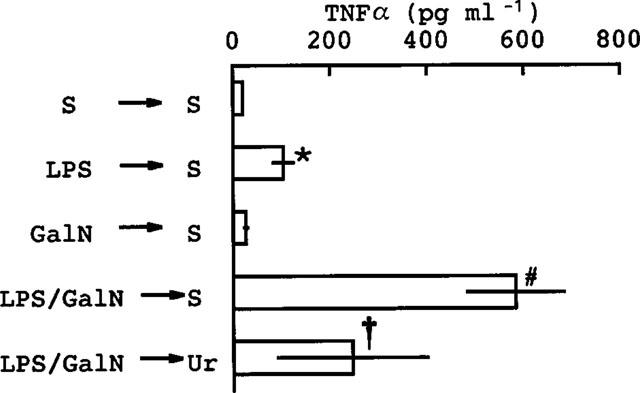
Effect of uridine on the TNFα production induced by co-injection of LPS and GalN. Mice were given a 1st injection (i.p.) of saline (S), LPS (10 μg kg−1), GalN (600 mg kg−1) or LPS plus GalN (10 μg kg−1 and 600 mg kg−1, respectively). Then, 0.5 h later they were given a 2nd injection (i.p.) consisting of either saline or uridine (Ur) (500 mg kg−1). The mice were killed 1.5 h after the 2nd injection and blood was then collected. Values are mean±s.d. from four mice. *P<0.01 vs S→S, #P<0.01 vs LPS→S, †P<0.05 vs LPS/GalN→S.
Effects of prior injection of LPS on the responses to GalN+LPS
We have reported elsewhere that LPS-pretreatment reduces the lethality associated with GalN+LPS injection (Endo et al., 1992a). The magnitude of this protective effect of LPS depends on the time at which LPS is injected (Table 1): it did not occur when LPS was injected 1 or 72 h before the injection of GalN+LPS, but it did occur when LPS was injected 2–48 h before such an injection. Lipid A is thought to be the active centre of LPS molecules. Although larger doses were required than of LPS, two synthetic derivatives of lipid A (ONO4007 and MPL) given separately also protected against GalN+LPS lethality (Table 2). In another experiment on ten mice, LPS-pretreatment completely prevented hepatic congestion. As shown in Figure 5, prior injection of LPS greatly reduced the production of TNFα induced by GalN+LPS. In this experiment, it was also found that LPS-pretreatment reduced the TNFα production induced by saline+LPS (Figure 5). On the other hand, neither GalN nor LPS-pretreatment resulted in detectable effects on the production of IL-1α.
Table 1.
Time-dependent protective effect of pretreatment with LPS before injection of GalN+LPS
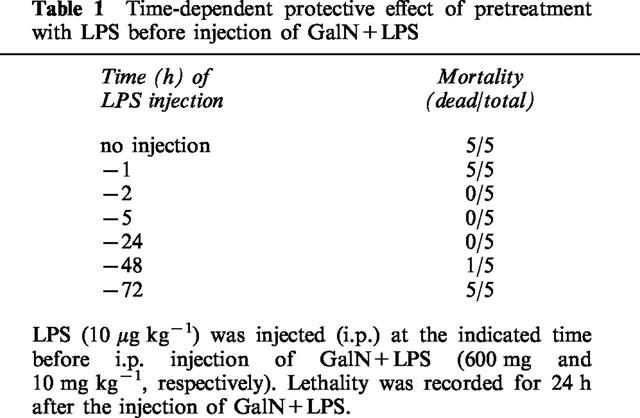
Table 2.
Effects of pretreatment with LPS or cytokines on the lethal results of co-injection of LPS and GalN
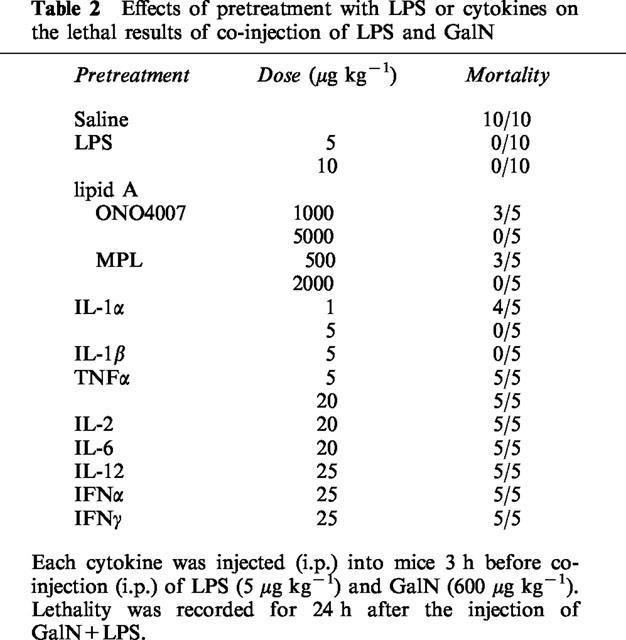
Figure 5.
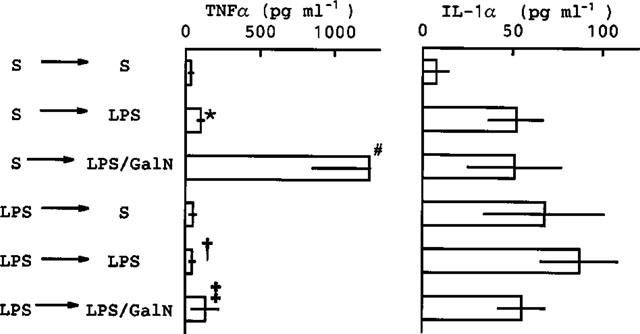
Effects of LPS-pretreatment on the TNFα and IL-1α production induced by GalN+LPS. Mice were given (i.p.) saline (S) or LPS (10 μg kg−1), then 3 h later they were given a 2nd i.p. injection consisting of saline, LPS (10 μg kg−1) or LPS plus GalN (10 μg kg−1 and 600 mg kg−1, respectively). The mice were killed 1.5 h after the 2nd injection and blood was then collected. Values are mean±s.d. from four mice. *P<0.01 vs S→S, #P<0.01 vs S→LPS, †P<0.05 vs S→LPS, ‡P<0.01 vs S→LPS/GalN.
Effects of prior injection of cytokines on the action of GalN+LPS
LPS is known to stimulate the production of various cytokines. For this reason, we examined the effect of pretreatment with cytokines on the lethal actions of GalN+LPS (Table 2). Among the cytokines tested, only IL-1 (both α and β) was effective. IL-1-pretreatment (5 μg kg−1) also bestowed complete protection (mortality 0/5) against the lethal action of GalN+TNFα (600 and 100 μg kg−1, respectively), as previously shown by Libert et al. (1991), and there was no hepatic congestion in these mice. Prior injection of either IL-1α or IL-1β at a dose of 5 μg kg−1, which was sufficient to protect completely against the lethality of GalN+LPS, did not produce a significant suppression of the TNFα production induced by GalN+LPS (Figure 6).
Figure 6.
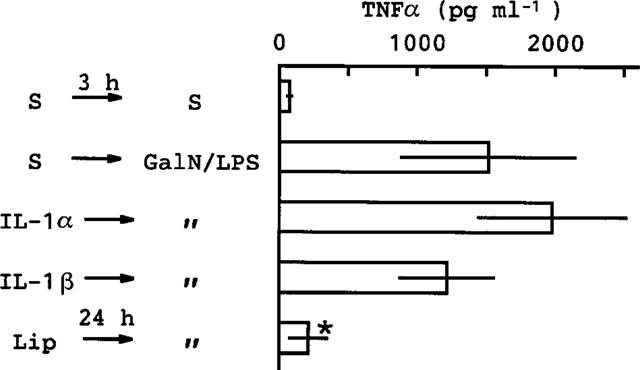
Effects of pretreatment with IL-1 or Cl2MBP-liposomes on the TNFα production induced by GalN+LPS. Mice were given (i.p.) saline (S) or IL-1 (5 μg kg−1) then 3 h later they were given a 2nd i.p. injection consisting of either saline or GalN+LPS (600 mg and 10 μg kg−1, respectively). The mice were killed 1.5 h after the 2nd injection and blood was then collected. In some mice, an injection of Cl2MBP-liposomes (Lip) (original suspension) was given i.v. 24 h before the injection of GalN+LPS. Values are mean±s.d. from four mice. *P<0.01 vs S→GalN/LPS.
Effect of a macrophage depletor on the actions of GalN+LPS
Cl2 MBP-liposome, a depletor of phagocytic macrophages, when injected (i.v.) 24 h before the injection of GalN+LPS, markedly suppressed the production of TNFα induced by GalN+LPS (Figure 6). This treatment also produced a considerable reduction in mortality (from 8/8 to 5/8).
Effects of serum from mice given LPS on the action of GalN+LPS or LPS alone
As to the mechanism underlying the suppressive effect of LPS-pretreatment on the TNFα production induced by GalN+LPS (Figure 5), we hypothesized the two possibilities that (i) a substance(s) that binds to TNFα and interferes with its assay or (ii) a substance(s) that down-regulates TNFα production might be present in the serum of the mice given LPS. To test these possibilities, mice were injected with LPS and their serum was taken 3 or 24 h later.
To test the first possibility, the serum taken from mice 1.5 h after injection of GalN+LPS was mixed with serum of normal mice or LPS-injected mice and kept at room temperature for 30 min, and then TNFα in each mixture was determined (Table 3). However, we could not detect the presence of such TNF-binding substances in the sera tested, suggesting that this possibility is unlikely.
Table 3.
The effect of serum taken from normal mice or from mice given LPS on the level of immunoreactive TNFα in the serum from mice given GalN+LPS

To test the second possibility, normal serum or serum taken from mice given LPS 24 h before (abbreviated as LPS-serum) was injected (i.v.) into mice and, 10 min later, GalN and LPS were co-injected. The LPS-serum markedly suppressed the elevation of TNFα induced by GalN+LPS (Figure 7). However, this treatment was less effective in reducing the mortality induced by GalN+LPS (from 8/8 to 6/8).
Figure 7.
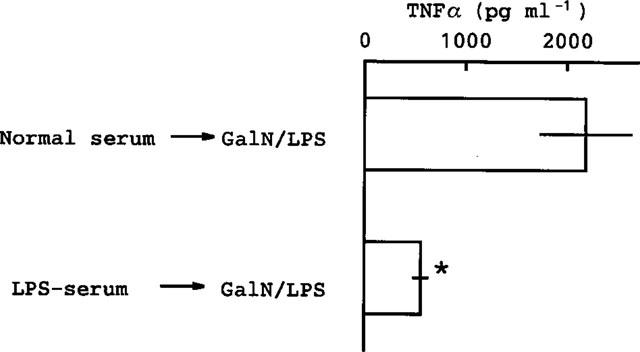
Effect of serum from mice given LPS on the TNFα production induced by GalN+LPS. Serum (0.1 ml per mouse) taken from normal mice or from mice injected with LPS (0.5 mg kg−1, i.p.) 24 h previously (LPS-serum) was injected (i.v.) into other mice, and 10 min later GalN+LPS was injected. The mice were killed 1.5 h after the second injection and blood was then collected. Values are mean±s.d. from four mice. *P<0.01 vs normal serum→GalN/LPS.
Finally, we examined the effect of the LPS-serum on the TNF production induced by LPS alone and the effect of heat-treatment of the LPS-serum on its activity. As shown in Figure 8, the LPS-serum, even when diluted to 1/27, inhibited the TNF production induced by LPS alone, while heat-treatment destroyed its activity.
Figure 8.
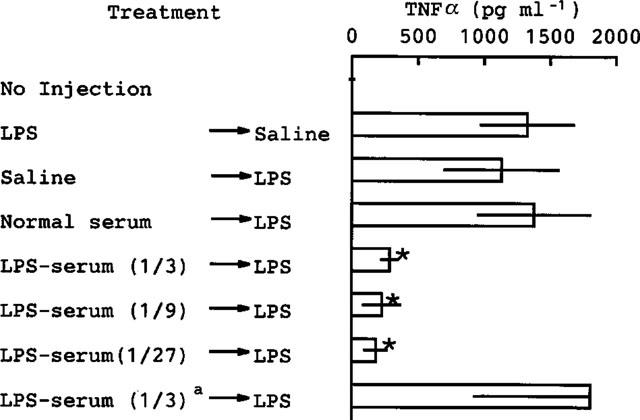
Effect of serum from mice given LPS on the TNFα production induced by LPS alone. Serum taken from normal mice (normal serum) was used without dilution, while serum taken from mice injected with LPS (0.5 mg kg−1, i.p.) 24 h previously (LPS-serum) was diluted with saline as indicated in parenthesis. The normal serum, diluted LPS-serum or saline was injected into other mice (0.1 ml/mouse, i.v.) and, 10 min later, LPS was injected (0.1 mg kg−1, i.v.). The mice were killed 1.5 h after the second injection and blood was then collected. In some experiments (indicated by the superscript a), the LPS-serum diluted to 1/3 was exposed to boiling water for 10 min and then cooled to 37°C in water. Values are mean±s.d. from four mice. *P<0.01 vs normal serum→LPS.
Discussion
The results obtained in the current study and their implications can be summarized as follows. (1) GalN augmented the LPS-induced elevation of serum TNF, even though GalN itself did not elevate serum TNF. (2) At larger doses, LPS alone elevated serum TNF to a level comparable to that induced by GalN+LPS, but produced neither hepatic congestion nor rapid death. (3) The elevated level of serum TNF induced by GalN+LPS had returned to its initial level within 3–4 h, indicating that the mechanism that removes or degrades TNF was operating adequately in mice given GalN. (4) In the first 4 h after the injection of GalN+LPS, neither hepatic congestion nor death had occurred (even though the TNF response ended within this period), suggesting that TNF triggers a secondary reaction(s) responsible for the lethality of GalN+LPS. (5) Administration of uridine (a precursor of uridine nucleotides) after the injection of GalN+LPS partially prevented the increase in TNF production and prevented the hepatic congestion and death. (6) Pretreatment with LPS greatly reduced the increase in TNF production induced by GalN+LPS, and completely prevented the hepatic congestion and death. This effect was dependent on the interval between the injections of LPS and GalN+LPS, an interval of more than 1 h but less than 72 h being required. (7) Of the cytokines tested as a pretreatment, only IL-1 reduced the lethality of GalN+LPS, but it did not significantly suppress the induced production of TNF. (8) Depletion of phagocytic macrophages decreased the TNF production induced by GalN+LPS, indicating that the cells primarily responsible for producing TNF after the injection of GalN+LPS are phagocytic macrophages. Such macrophage depletion considerably reduced the lethality of GalN+LPS. (9) Injection (i.v.; 10 min before GalN+LPS) of serum taken from mice previously given an i.p. injection of LPS (24 h before blood collection) reduced the enhancement of TNF production induced by GalN+LPS, but was weak or not effective at reducing the mortality induced by GalN+LPS. The effects of the various treatments are summarized in Table 4. In the following sections, we discuss the mechanism that might underlie the augmented TNF production induced by GalN+LPS; the augmented lethality of GalN+LPS; and the protective effect bestowed by pretreatment with LPS or IL-1.
Table 4.
Effects of various pretreatments on the TNF production and the lethality produced by GalN+LPS

Mechanisms underlying the augmentation of LPS-induced TNF production by GalN and the reduction of this augmentation by LPS-pretreatment
The GalN-induced impaired synthesis of mRNA in hepatocytes, which is due to selective depletion of UTP (Decker & Keppler, 1974), and the results described above lead us to propose the existence of an unidentified factor(s), tentatively called hepatocyte-derived TNF-down-regulator (TNF-DRh). Our hypothesis requires that this factor is produced by hepatocytes in response to LPS and then acts to prevent the over-production of TNF by macrophages. LPS (but not IL-1) would stimulate hepatocytes directly or indirectly to produce TNF-DRh. By contrast, the GalN-induced impairment of RNA synthesis in hepatocytes would decrease the production of TNF-DRh. Our results showing a suppression of TNF production by uridine and by the serum taken from LPS-treated mice are consistent with this idea.
Czaja et al. (1994) have reported that in the spleen and liver, the increase in TNF-mRNA is much the same whether GalN alone, LPS alone or a combination of the two is given: they unfortunately did not try to measure TNF itself because they believed that TNF would be undetectable in the serum. Using our hypothesis as a basis, their results might be explained as follows. TNF-DRh may be produced in normal mice in small amounts and act so as to prevent TNF production being induced by minor stimuli (e.g. by a small amount of enterobacterial LPS reaching the liver via the portal vein). The decrease in TNF-DRh induced by GalN may itself be sufficient to sensitize macrophages to produce TNF-mRNA but perhaps not to produce or release a detectable amount of TNF protein.
Schade et al. (1996) have reported that an inhibitor of TNF synthesis appeared in the plasma when LPS was injected into LPS-tolerant mice, but not into normal mice. They showed that this inhibitor is produced by macrophages and that it is detectable in the plasma as early as 30 min after LPS injection (Flach & Schade, 1997). Therefore, this inhibitor may be different from our TNF-DRh.
As a mechanism to prevent excessive actions of TNF, it is known that soluble TNF receptors are released in response to LPS (Van Zee et al., 1992). These soluble receptors are the extracellular domains of TNF receptors, and they are shed from cell surface in response to TNF (Higuchi & Aggarwal, 1994; Moshage, 1997). The binding of TNF to the soluble TNF receptors has been shown to reduce its immunoactivity for its antibody, as determined by ELISA (Van Zee et al., 1992). Therefore, it is expected that such soluble receptors may be involved in our observations that LPS-pretreatment or the serum taken from LPS-treated mice suppressed TNF elevation induced by GalN+LPS or by LPS itself. However, as shown in Table 3, we could not detect the presence of such substances. We think that such soluble TNF receptors mostly had already been bound to TNF. In any case, the ability of the LPS-serum shown in Figure 7 and 8 to reduce the elevation of TNF levels induced by GalN+LPS or by LPS itself seems to be not attributable to such soluble TNF-receptors.
Lethality induced by GalN+LPS
When given alone, larger doses of LPS produced a large amount of TNF, an amount comparable to that produced by GalN+LPS. However, LPS alone produced neither hepatic congestion nor rapid death, at least at the doses used in this study. Indeed, injection of TNF itself, even at a dose as large as 500 μg kg−1 does not produce hepatic congestion or death (Endo et al., 1992a). Co-injection of GalN+TNF induces death, and this lethality is abolished by uridine and by treatment with an anti-TNF antibody (Freudenberg & Galanos; 1991). Although IL-1, IL-6 and TNF share a number of biological activities (Dinarello, 1989, 1991), neither GalN+IL-1 nor GalN+IL-6 induced the lethality seen with GalN+TNF: indeed, IL-1 (even 100 μg kg−1)+GalN did not induce hepatic congestion or death (Endo et al., 1992a). These results indicate (a) that LPS or TNF induces lethality only in the presence of GalN or when hepatic RNA synthesis is suppressed (as indicated by the fact that administration of uridine protects against the lethality of GalN+LPS) and (b) that some property of TNF that is lacking in these other cytokines is responsible for the lethality.
We have little available data capable of explaining what actual events are involved in (a) and (b). With regard to (a), we have previously proposed the idea that a polyamine-insufficiency due to an inhibition by GalN of the induction of ornithine decarboxylase (ODC) may explain, at least partly, the way in which lethality is induced by GalN+LPS or GalN+TNF (Endo et al., 1992a). Polyamines bind to RNA and DNA and play important roles in the synthesis of mRNA and proteins (Pegg & McCann, 1982; Tabor & Tabor, 1984). ODC itself is induced through the formation of its mRNA (Tabor & Tabor, 1984). Shinozuka et al. (1973a, 1973b) have shown that GalN induces a rapid nucleolar fragmentation in hepatocytes. Recently, an inhibition of polyamine synthesis was implicated in apoptosis in several cell types (Grassilli et al., 1995; Desiderio et al., 1995; Mori et al., 1997), a finding that is consistent with our hypothesis. With regard to (b), we found some years ago that TNF induces an accumulation of platelets in the liver (Endo, 1991; Endo & Nakamura, 1992, 1993) and GalN has been shown to augment the LPS-induced accumulation of platelets (Shibazaki et al., 1996). In addition, Libert et al. (1996) found that platelet-activating factor is involved in the effect of GalN+TNF. These results suggest the possibility that the action of LPS or TNF on platelets might be involved, at least in part, in the lethal actions of GalN+LPS and GalN+TNF.
Mechanisms underlying the protective effect of LPS
Injection of LPS at 2–48 h before the injection of GalN+LPS prevented death. Among the cytokines tested, only IL-1 (both α and β) shared this protective effect with LPS. IL-1 can also protect against the lethality induced by GalN+TNF (Libert et al., 1991). These results suggest that IL-1 may play an important role in the protective effect of LPS, although other cytokines may also contribute to it. It is also possible that LPS exerts its protective effect without mediation by cytokines. As shown in Table 4, an inhibition of TNF production does not necessarily accompany a prevention of the lethality induced by GalN+LPS. Indeed, while LPS pretreatment reduced both TNF production and lethality, IL-1 exerted its protective effect against lethality without suppressing the production of TNF.
There are a few reports to help us deduce what mechanisms might be involved in the protective effect of LPS or IL-1. It is known that LPS can induce ODC at very low doses (10 μg kg−1 or less) (Endo, 1982 and 1984) and, among the cytokines tested, only IL-1 can induce ODC in the liver at low doses (5 μg kg−1 or less) (Endo, 1989; Endo et al., 1992b). Since LPS and IL-1 were the only pretreatments to reduce the lethality of GalN+LPS, it is possible that the ODC induced by pretreatment with LPS or IL-1 is involved, at least in part, in mediating their protective action (Endo et al., 1992a). Recently, Van Molle et al. (1997) showed that the acute phase proteins α1-acid glycoprotein and α1-antitrypsin reduce the lethality of GalN+TNF, and they proposed the idea that the inhibitory effects of these proteins on the aggregation of platelets might be involved in mediating their protective actions. It would be of interest to know whether our TNF-DRh is identical to these acute phase proteins, because acute phase proteins are produced by hepatocytes in response to LPS (Steel & Whitehead, 1994).
Finally, the above discussion may be summarized as follows: (1) The over-production of TNF induced by GalN+LPS and the decrease in this TNF production induced by LPS-pretreatment may be associated with the absence or presence, respectively, of an unidentified hepatocyte-derived factor that inhibits the production of TNF by macrophages; (2) Both a suppression of hepatic RNA synthesis by GalN and an unidentified action(s) of TNF seem to be involved in the lethal action of GalN+LPS; (3) The protective effect of LPS-pretreatment may be mediated by at least two different mechanisms: suppression of TNF production and IL-1-mediated changes in hepatocytes.
Acknowledgments
This work was supported in part by a grant from the T-cell Institute and a Grant-in-Aid for Scientific Research from the Ministry of Education of Japan (No. 09470409).
Abbreviations
- Cl2MBP
dichloromethylene bisphosphonate
- GalN
galactosamine
- IFN
interferon
- IL-1
interleukin-1
- LPS
lipopolysaccharide
- Lip
liposome
- TNF
tumour necrosis factor
- TNF-DRh
hepatocyte-derived TNF-down-regulator
- Ur
uridine
References
- AUSTYN J.M., GORDON S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- CZAJA M.J., SCHILSKY M.L., XU Y., SCHMIEDEBERG P., COMPTON A., RIDNOUR L., OBERLEY L.W. Induction of MnSOD gene expression in a hepatic model of TNF-α toxicity does not result in increased protein. Am. J. Physiol. 1994;266:G737–G744. doi: 10.1152/ajpgi.1994.266.4.G737. [DOI] [PubMed] [Google Scholar]
- DECKER K., KEPPLER D. Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev. Physiol. Biochem. Pharmacol. 1974;71:77–106. doi: 10.1007/BFb0027661. [DOI] [PubMed] [Google Scholar]
- DESIDERIO M.A., GRASSILLI E., BELLESIA E., SALOMONI P., FRANCESCHI C. Involvement of ornithine decarboxylase and polyamines in glucocorticoid-induced apoptosis of rat thymocytes. Cell Growth Differ. 1995;6:505–513. [PubMed] [Google Scholar]
- DINARELLO C.A. Interleukin-1 and its biologically related cytokines. Adv. Immunol. 1989;44:153–205. doi: 10.1016/s0065-2776(08)60642-2. [DOI] [PubMed] [Google Scholar]
- DINARELLO C.A. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- ENDO Y. Simultaneous induction of histidine and ornithine decarboxylases and changes in their product amines following the injection of Escherichia coli lipopolysaccharide into mice. Biochem. Pharmacol. 1982;31:1643–1647. doi: 10.1016/0006-2952(82)90394-x. [DOI] [PubMed] [Google Scholar]
- ENDO Y. Induction of ornithine decarboxylase in mouse tissues following the injection of mitogenic substances. Biochem. Pharmacol. 1984;33:2123–2127. doi: 10.1016/0006-2952(84)90582-3. [DOI] [PubMed] [Google Scholar]
- ENDO Y. Induction of histidine and ornithine decarboxylase activities in mouse tissues by recombinant interleukin-1 and tumor necrosis factor. Biochem. Pharmacol. 1989;38:1287–1292. doi: 10.1016/0006-2952(89)90335-3. [DOI] [PubMed] [Google Scholar]
- ENDO Y. Parallel relationship between the increase in serotonin in the liver and hypoglycaemia induced in mice by interleukin-1 and tumour necrosis factor. Immunol. Lett. 1991;27:75–80. doi: 10.1016/0165-2478(91)90248-9. [DOI] [PubMed] [Google Scholar]
- ENDO Y., KIKUCHI T., NAKAMURA M. Ornithine and histidine decarboxylase activities in mice sensitized to endotoxin, interleukin-1 or tumour necrosis factor by D-galactosamine. Br. J. Pharmacol. 1992a;107:888–894. doi: 10.1111/j.1476-5381.1992.tb14542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENDO Y., KIKUCHI T., TAKEDA Y., NITTA Y., RIKIISHI H., KUMAGAI K. GM-CSF and G-CSF stimulate the synthesis of histamine and putrescine in the hematopoietic organs in vivo. Immunol. Lett. 1992b;33:9–14. doi: 10.1016/0165-2478(92)90086-4. [DOI] [PubMed] [Google Scholar]
- ENDO Y., NAKAMURA M. The effect of lipopolysaccharide, interleukin-1 and tumour necrosis factor on the hepatic accumulation of 5-hydroxytryptamine and platelets in the mouse. Br. J. Pharmacol. 1992;105:613–619. doi: 10.1111/j.1476-5381.1992.tb09028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENDO Y., NAKAMURA M. Active translocation of platelets into sinusoidal and Disse spaces in the liver in response to lipopolysaccharide, interleukin-1 and tumor necrosis factor. General Pharmacol. 1993;24:1039–1053. doi: 10.1016/0306-3623(93)90348-2. [DOI] [PubMed] [Google Scholar]
- ENDO Y., NAKAMURA M., NITTA Y., KUMAGAI K. Effects of macrophage depletion on the induction of histidine decarboxylase by lipopolysaccharide, interleukin 1 and tumour necrosis factor. Br. J. Pharmacol. 1995;114:187–193. doi: 10.1111/j.1476-5381.1995.tb14924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLACH R., SCHADE F.U. Peritoneal macrophages from endotoxin-tolerant mice produce an inhibitor of tumor necrosis factor α synthesis and protect against endotoxin shock. J. Endo. Res. 1997;4:241–250. [Google Scholar]
- FREUDENBERG M.A., GALANOS G. Tumor necrosis factor alpha mediates lethal activity of killed gram-negative and gram-positive bacteria in D-galactosamine-treated mice. Infec. Immun. 1991;59:2110–2115. doi: 10.1128/iai.59.6.2110-2115.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUTANI Y., NOTAKE M., YAMANISHI M., YAMAGISHI J., NOMURA H., OHUE M., FURUTA R., FUKUI T., YAMADA M., NAKAMURA S. Cloning and characterization of the cDNAs for human and rabbit interleukin-1 precursor. Nucleic Acids Res. 1985;13:5869–5882. doi: 10.1093/nar/13.16.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALANOS C., FREUDENBERG M.A., REUTTER W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc. Natl. Acad. Sci. U.S.A. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRASSILLI E., DESIDERIO M.A., BELLESIA E., SALOMONI P., BENATTI F., FRANCESCHI C. Is polyamine decrease a common feature of apoptosis? Evidence from gamma rays- and heat shock-induced cell death. Biochim. Biophys. Res. Commun. 1995;216:708–714. doi: 10.1006/bbrc.1995.2679. [DOI] [PubMed] [Google Scholar]
- HIGUCHI M., AGGARWAL B.B. TNF induces internalization of the p60 receptor and shedding of the p80 receptor. J. Immunol. 1994;152:3550–3558. [PubMed] [Google Scholar]
- HIRSCH S., AUSTYN J.M., GORDON S. Expression of the macrophage-specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J. Exp. Med. 1981;154:713–725. doi: 10.1084/jem.154.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIKUMOTO Y., HONG Y., NISHIDA T., NAKAI S., MASUI Y., HARAI Y. Purification and characterization of recombinant human interleukin-1 produced in Escherichia coli. Biochem. Biophys. Res. Commun. 1987;147:315–321. doi: 10.1016/s0006-291x(87)80123-7. [DOI] [PubMed] [Google Scholar]
- LEHMANN V., FREUDENBERG M.A., GALANOS C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J. Exp. Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIBERT C., VAN BLADEL S., BROUCKAERT P., SHAW A., FIERS W. Involvement of the liver, but not of IL-6, in IL-1-induced desensitization to the lethal effects of tumor necrosis factor. J. Immunol. 1991;146:2625–2632. [PubMed] [Google Scholar]
- LIBERT C., VAN MOLLE W., BROUCKAERT P., FIERS W. Platelet-activating factor is a mediator in tumor necrosis factor/galactosamine-induced lethality. J. Inflamm. 1996;46:139–143. [PubMed] [Google Scholar]
- MIETHKE T., WAHL C., HEEG K., ECHTENACHER B., KRAMMER P.H., WAGNER H. T cell-mediated lethal shock triggered in mice by the superantigen Staphylococcal enterotoxin B: critical role of tumor necrosis factor. J. Exp. Med. 1992;175:91–98. doi: 10.1084/jem.175.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOSHAGE H. Cytokines and the hepatic acute phase response. J. Pathol. 1997;181:257–266. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- MORI K., HIBASAMIN H., SATOH N., SONODA J., YAMASAKI T., TAJIMA M., HIGUCHI S., WAKABAYASHI H., UCHIDA A., NAKASHIMA K. Induction of apoptic cell death in three human osteosarcoma cell lines by a polyamine synthesis inhibitor, methylglyoxal bis (cyclopentylamidinohydrazine) (MGBCP) Anticancer Res. 1997;17:2385–2389. [PubMed] [Google Scholar]
- PEGG A.E., MCCANN P.P. Polyamine metabolism and function. Am. J. Physiol. 1982;243:C212–C221. doi: 10.1152/ajpcell.1982.243.5.C212. [DOI] [PubMed] [Google Scholar]
- SCHADE F.U., SCHLEGEL J., HOFMANN K., BRADE H., FLACH R. Endotoxin-tolerant mice produce an inhibitor of tumor necrosis factor synthesis. J. Endotoxin. Res. 1996;3:455–462. [Google Scholar]
- SHIBAZAKI M., NAKAMURA M., ENDO Y. Biphasic, organ-specific, and strain-specific accumulation of platelets induced in mice by a lipopolysaccharide from Escherichia coli and its possible involvement in shock. Infect. Immun. 1996;64:5290–5294. doi: 10.1128/iai.64.12.5290-5294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBAZAKI M., NAKAMURA M., NITTA Y., ENDO Y. Displacement of platelets from blood to spleen following intravenous injection of liposomes encapsulating dichloromethylene bisphosphonate. Immunopharmacol. 1998;39:1–7. doi: 10.1016/s0162-3109(97)00092-1. [DOI] [PubMed] [Google Scholar]
- SHINOZUKA H., FARBER J.L., KONISHI Y., ANUKARAHANONTA T.D. Galactosamine and acute liver cell injury. Fed. Proc. 1973;32:1516–1526. [PubMed] [Google Scholar]
- SHINOZUKA H., MARTIN J.T., FARBER J.L. The induction of fibrillar nucleoli in rat liver cells by D-galactosamine and their subsequent re-formation into normal nucleoli. J. Ultrastruc. Res. 1973;44:279–292. doi: 10.1016/s0022-5320(73)80061-9. [DOI] [PubMed] [Google Scholar]
- STEEL D.M., WHITEHEAD A.S. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol. Today. 1994;15:81–88. doi: 10.1016/0167-5699(94)90138-4. [DOI] [PubMed] [Google Scholar]
- TABOR C.W., TABOR H. Polyamines. Ann. Rev. Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- TIEGS G., WOLTER M., WENDEL A. Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem. Pharmacol. 1989;38:627–631. doi: 10.1016/0006-2952(89)90208-6. [DOI] [PubMed] [Google Scholar]
- VAN MOLLE W., LIBERT C., FIERS W., BROUCKAERT P. α1-Acid glycoprotein and α1-antitrypsin inhibit TNF-induced but not anti-Fas-induced apoptosis of hepatocytes in mice. J. Immunol. 1997;159:3555–3564. [PubMed] [Google Scholar]
- VAN ROOIJEN N., SANDERS A. Liposome-mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Meth. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- VAN ZEE K.J., KOHNO T., FISCHER E., ROCK C.R., MOLDAWER L.L., LOWRY S.F. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor α in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4845–4849. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACH D., HOLTMANN D., ENGELMANN H., NOPHAR Y. Sensitization and desensitization to lethal effects of tumor necrosis factor and IL-1. J. Immunol. 1988;140:2994–2999. [PubMed] [Google Scholar]