

Abstract
We have investigated the role of kinin B1 receptor induction in the endotoxemic rat heart and elucidated the mechanisms underlying B1 receptor-mediated coronary vasodilation. We also investigated the role of these receptors in endotoxin-induced hypotension.
Endotoxin treatment induced cardiac B1 receptor mRNA expression and promoted a coronary vasodilation response to des-Arg9bradykinin (DABK; ED50=149.4 pmol, n=9) ex vivo peaking at 6 h. The B1 receptor antagonist des-Arg9-[Leu8]-BK (DALBK, 30 nM) significantly (P<0.05) inhibited the DABK-induced response (pA2=8.4, n=5) whilst HOE140 (B2 receptor antagonist, 10 nM) was inactive (n=4).
Removal of the endothelium or infusion with indomethacin (5 μM), but not L-NAME (300 μM) or ODQ (1 μM), inhibited (>85%, P<0.05, n=5) the DABK-induced response. DABK caused a dose-dependent release of the prostacyclin metabolite, 6-keto-PGF1a (Emax=0.3 ng ml−1, n=6).
In vitro perfusion of hearts with endotoxin (1 μg ml−1, n=6) or interleukin-1β (5 ng ml−1, n=6) induced B1 receptor mRNA expression and promoted a time-dependent vasodilation response to DABK.
Endotoxin treatment (6 h) in vivo promoted a hypotensive response to DABK (ED50=29.7 nmol kg−1, n=10) which was antagonised by DALBK (3–6 nmol kg−1 min−1, P<0.05, n=7). DALBK (3 nmol kg−1 min−1) and des-Arg10HOE140 (B1 receptor antagonist, 30 nmol kg−1 min−1) produced a 5.3% (n=6, P<0.05) and 8.8% (n=5, P<0.05) reversal, respectively, of endotoxin-induced hypotension.
In summary, we have shown that in endotoxemia activation of B1 receptors causes coronary vasodilation via endothelial prostacyclin release. Additionally, B1 receptor antagonists partially reversed endotoxin-induced hypotension. Therefore activation of B1 receptors may have a role to play in the vascular changes associated with endotoxemia.
Keywords: Kinin B1 receptor, endotoxemia, vascular inflammation, rat heart, coronary circulation, nitric oxide, prostanoids
Introduction
Kinins act on two receptor types, B1 and B2, classified according to the relative potency of their endogenous agonists (Regoli & Barabe, 1980; Bhoola et al., 1992). While the B2 receptor is typically activated by bradykinin (BK) and Lys-bradykinin (Lys-BK), the B1 receptor is selectively sensitive to kinin metabolites without the C-terminal arginine residue, e.g. des-Arg9BK (DABK) and Lys-des-Arg9BK (Lys-DABK) which show relative selectivity for the rat and human B1 receptor respectively (Marceau et al., 1998). B2 receptors are constitutively expressed and mediate many kinin-dependent inflammatory responses including increase in blood flow, vascular permeability and accumulation of inflammatory cells (Regoli & Barabe, 1980; Bhoola et al., 1992). B1 receptors are not expressed in normal physiology but are induced during vascular inflammation. Knowledge of the contribution of B1 receptors to the regulation of vascular tone is of importance since B1 receptors might replace B2 receptors during vascular inflammation (Marceau et al., 1998). The selective ligand for B1 receptors, DABK, normally an inactive metabolite of BK, shows pro-inflammatory activity following an inflammatory insult in vitro or in vivo (Marceau et al., 1998). B1 receptor-mediated effects are diverse and include vascular smooth muscle contraction, endothelium dependent vasodilation and inflammatory cell recruitment (for a recent review see, Marceau et al., 1998). However, the mechanisms of B1 receptor induction and function in the heart is unknown.
B1 receptors may be involved in certain cardiovascular pathologies such as atheromatous disease (Raidoo et al., 1997) and myocardial ischemia (Bouchard et al., 1998) however, of interest to the present study is the possibility that B1 receptors are induced and play a role in the vascular inflammatory response to endotoxin. Kinins are elevated during endotoxemia and experimentally endotoxin treatment is associated with activation of the kallikrein-kinin system (DeLa et al., 1993; Nies et al., 1968). Since BK is a potent hypotensive agent in humans and animals (Kontos et al., 1964; Regoli & Barabe, 1980) it has been suggested that BK acting at constitutive B2 receptors contributes to the development of hypotension in sepsis. Reports investigating the effects of B2 receptor selective antagonists in models of endotoxemia nevertheless are conflicting showing both beneficial (Wilson et al., 1989) or no effect (Feletou et al., 1995). However, administration of a B2 receptor antagonist with moderate efficacy as a B1 receptor antagonist (CP-0127) inhibits hypotension and improves survival in rats and rabbits challenged with endotoxin (Whalley et al., 1992). This compound was also shown to marginally improve survival in patients with gram-negative sepsis (Fein et al., 1997). It is possible that this marginal beneficial effect is correlated to the marginal efficacy of this compound at B1 receptors. Moreover, B1 receptor deficient mice display a blunted hypotensive response to endotoxin (Araujo et al., 1998). It remains unclear however whether the B1 receptor-mediated hypotensive effect is due to an action at the level of the vasculature or directly on the heart.
The effects of endotoxin on the cardiovascular system, both in terms of the systemic vascular action and the direct effects on the heart are at least partially dependent on enhanced NO production but there is a significant NO-independent component (Ungureanu-Longrois et al., 1995; Meng et al., 1997; Filep et al., 1997). Our hypothesis is that de novo induction of B1 receptors contributes to the NO-independent regulation of coronary tone and hypotension in endotoxemia. To test this hypothesis we investigated whether endotoxin treatment is associated with cardiac B1 receptor mRNA expression and correlated this with the development of a coronary vasodilation response ex vivo and a hypotensive response in vivo. We also determined the underlying mechanisms mediating the B1 receptor induction and coronary vasodilation.
Methods
Animal preparation for in vitro studies
Experiments were performed according to the ‘Guide on the Operation of Animals (Scientific Procedures) Act 1986'. Male Wistar rats (320–370 g Charles River, U.K.) either untreated or treated (1, 2, 3, 6, 14 or 24 h) with lipopolysaccharide (LPS, E. coli 0.55:B5, 2.5 mg kg−1, i.p.) were heparinized (sodium heparin, 250 iu, i.p.) and killed by cervical dislocation. Hearts were rapidly excised and either frozen in liquid nitrogen for mRNA extraction or placed in ice-cold perfusion solution (constituents below) prior to coronary perfusion pressure (CPP) measurement.
Measurement of coronary hemodynamics
Hearts were cannulated via the aorta and perfused retrogradely under constant flow (10 ml min−1) using a calibrated roller pump (Gilson, Minipuls-2) with a buffer solution of the following composition (mM): D(+)glucose 11.1, CaCl2 1.4, NaCl 118.5, NaHCO3 25.0, MgSO4 1.2, NaH2PO4 1.2 and KCl 4.0; gassed with 95% O2/5% CO2, (pH 7.4) and perfused at 37°C. The CPP in the aortic line was monitored by a Statham Spectramed pressure transducer connected to a chart recorder (Grass 79E). Air temperature was maintained by means of a heated (37°C) water jacket. On establishing a stable CPP (20–30 min following cannulation), bolus doses (10–30 μl) of BK, DABK, sodium nitroprusside (SNP) or acetylcholine were administered via the aorta and changes in CPP recorded. Other substances were infused at 0.1 ml min−1.
Mechanisms of kinin responses
Receptor antagonism studies
Control dose-response curves to DABK (10–3000 pmol) or BK (3–1000 pmol) were constructed. Hearts were then perfused with either the B1 receptor antagonist des-Arg9-[Leu8]-bradykinin (DALBK, 30 nM) or the B2 receptor antagonist HOE140 (0.1–10 nM) for 20 min prior to and during construction of a second dose-response curve. These experiments were repeated in hearts from rats pretreated with LPS (2.5 mg kg−1; 2, 6, 14 and 24 h). In some experiments CPP was increased in control hearts with endothelin-1 (10 nM, 3 min) prior to the administration of DABK or BK.
Initial studies showed that the induction of the vasodilator response to DABK was maximal in hearts from rats 6 h post LPS-treatment. Therefore the 6 h time point was used in subsequent experiments.
Determination of endothelium-dependency
The endothelium was removed by saponin infusion (Ellwood & Curtis, 1997); delivered at 20 μg ml−1 in three cycles of 2 min followed by 2 min buffer, followed by a 20 min control recovery perfusion. Endothelial and vascular smooth muscle function were tested before and after saponin treatment using the endothelium-dependent vasodilator acetylcholine (3 nmol) and the endothelium-independent vasodilator SNP (10 nmol). DABK (1 nmol)-induced responses were determined before and after saponin.
Involvement of endothelial mediators
In order to investigate involvement of the NO-guanylyl cyclase pathway in mediation of the responses to DABK, dose-response curves were constructed in the absence and presence of either NG-nitro-L-arginine methyl ester (L-NAME, 300 μM, 20 min pretreatment) to inhibit NO synthase (NOS) or 1H-[1,2,4]oxadiazolo [4,3-α]quinoxalin-1-one (ODQ, 1 μM, 20 min pretreatment) to inhibit soluble guanylyl cyclase (Garthwaite et al., 1995). The NO-donor SNP was used to verify the activity of ODQ.
To investigate the involvement of prostanoids, dose-response curves to DABK were constructed in the absence and presence of the cyclooxygenase inhibitor indomethacin (5 μM, 20 min pretreatment). Prostacyclin (PGI2) involvement was assessed by measuring the coronary efflux of 6-keto-PGF1α, the stably hydrolysis product of PGI2, released in response to DABK. Fractions (2 min) were collected immediately before and after administration of DABK. Samples were frozen at −70°C until assay of 6-keto-PGF1α by radioimmunoassay using a commercially available kit (detection limit=0.06 ng ml−1, Amersham Ltd, Bucks, U.K.).
In vitro B1 receptor induction
Control hearts were perfused in vitro with either buffer solution alone (4 h), LPS (1 μg ml−1, 4 h) or interleukin-1β (IL-1β, 5 ng ml−1, 2 h) and the effects of DABK (1 nmol) tested every 15 min. The concentrations of LPS and IL-1β used were based upon circulating concentrations measured during endotoxemia (Waage et al., 1989; Hanasaki et al., 1997). Responses to BK were also assessed every 30 min during these experiments. At the end of the perfusion hearts were frozen in liquid nitrogen for mRNA extraction.
Measurement of blood pressure
Control or LPS-treated rats were anaesthetized with thiopentone sodium (120 mg kg−1, i.p.), the trachea cannulated and temperature maintained at 37°C with a homeothermic blanket. For measurement of mean arterial pressure (MAP) the right common carotid artery was cannulated and connected via a Statham Spectramed pressure transducer to a MacLab4E. The left jugular vein was cannulated for the administration of saline or drugs. To elevate MAP of LPS-treated rats to control levels noradrenaline was infused at a rate of 3–8 μg min−1.
Reverse-transcription polymerase chain reaction
Poly(A)+ mRNA was extracted (Micro-FastTrack kit, Invitrogen, San Diego, CA, U.S.A.) from homogenates of left ventricle, stored at −70°C, from untreated rats, from rats 1,3, or 6 h post LPS treatment in vivo or from hearts perfused in vitro with LPS, IL-1β, or buffer solution alone (as described above). Poly(A)+ mRNA was resuspended in 20 μl of Tris pH 7.5 and 2 μl aliquots reverse transcribed in a 20 μl-reaction mixture containing 150 ng of random hexanucleotides, 2 mM dNTPs, 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol and 200 U SuperScript II RNAse H− reverse transcriptase (Life Technologies, Paisley, U.K.). PCR experiments were performed with 2 μl of the first-strand DNA in a 25 μl reaction mixture containing 200 μM dNTPs, 1.5 mM MgCl2, 25 pmol of specific forward and reverse primers and 1 U of recombinant Taq DNA polymerase (Life Technologies, Paisley, U.K.) and amplified for 30 cycles (Biometra-TRIO thermocycler): 95°C for 1 min, 60°C for 2 min and 72°C for 2 min followed by a final extension step at 72°C for 10 min. Rat B1 receptor cDNA was amplified with the sense primer (5′-GCGGAAATCTACCTGGCTAACTTG-3′) at base position 392–415 and antisense primer (5′-CAGTCACGGGGAGGAGGAAACC-3′) at base position 806–827 of rat B1 receptor cDNA (Ni et al., 1998). The quality of RNA samples was evaluated by RT–PCR using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) specific primers (sense primer, 5′-AAGGTGAAGGTCGGAGTCAACG-3′ (position 8–29); antisense primer 5′-GGCAGAGATGATGACCCTTTTGGC-3′ (position 362–339). A plasmid containing the mouse B1 receptor cDNA (Pesquero et al., 1996) was used as the template for the positive PCR control reaction. PCR products were detected by electrophoresis on 2% agarose gels stained with ethidium bromide. The sequence of the amplified cDNA was verified using direct automated DNA sequencing using the TaqDyeDeoxy terminator cycle sequencing method (Thermosequence, Amersham Ltd, U.K.).
Reagents
All reagents were purchased from Sigma Chemical Co. (Dorset, U.K.), unless otherwise indicated below. Lipopolysaccharide (E. coli 055:B5) was purchased from Becton Dickinson (Oxford, U.K.). Des-Arg10HOE140 was purchased from RBI (Dorset, U.K.). HOE140 was a kind gift from Hoechst (Frankfurt, Germany) and rat recombinant IL-1β a generous gift from Dr R.C. Newton (Du Pont-Merck, Wilmington, DE, U.S.A.). ODQ was synthesised in The Wolfson Institute for Biomedical Research (London, U.K.).
Data analysis
The responses to DABK and BK are expressed as a percentage of their individual maxima. ED50 values were calculated from the 50% response level and expressed as geometric mean with 95% confidence limits in parentheses using a non-linear curve fitting analysis. The potency of the antagonists was expressed as a pA2 value. Apparent pA2 values were calculated using single point analysis from pA2=log(DR-1)-log[B], where DR is the dose-ratio of agonist measured in the absence and presence of antagonist (B). The assumption of simple competition (i.e. slope of unity) between antagonist and agonist was checked with the Schild plot. All other data are given as arithmetic means±s.e.mean or geometric mean with 95% confidence limits. The number of observations is indicated by n. The significance of differences between the values was determined by use of Student's paired or unpaired t-test. Dose-response curves were compared using two-way ANOVA (analysis of variance, Graph Pad Prism, Graph Pad Software Inc., San Diego, U.S.A.). The criterion for statistical significance was set at P<0.05.
Results
Effects of endotoxin treatment
Treatment of rats with LPS produced characteristic symptoms: pilo-erection, malaise and loose stools. Hearts from LPS-treated rats displayed significantly (P<0.05) higher basal CPP values than control hearts (CPP (mm Hg)=66±3 (control, n=42); 114±5 (P<0.05, 6 h, n=40); 111±14 (14 h, n=4); 94±8 (P<0.05, 24 h, n=5).
Influence of endotoxin treatment on coronary reactivity to DABK and BK
The B1 receptor selective agonist DABK had no effect on CPP in hearts from control rats (n=6, Figures 1 and 2). Elevation of CPP with endothelin-1 (CPP=112.7±15.2, n=4) failed to reveal a response to DABK. In contrast, BK produced dose-dependent vasodilations (Figure 1, ED50=37.9 pmol (4.4–329.1); Emax=55.4 mm Hg (40.1–70.7), n=13) in control hearts with endothelin-1-elevated CPP. The B2 receptor antagonist, HOE140 had no effect on basal CPP per se but caused significant rightward shifts (P<0.05, 2-way ANOVA) of the dose-response curve to BK in a competitive fashion without affecting the maximum response (pA2=10.1±1.5, Schild slope=0.96±0.2, n=13); the B1 receptor antagonist, DALBK (100 nM) had no significant effect on the dose-response curve to BK (P>0.05, 2-way ANOVA, n=4).
Figure 1.
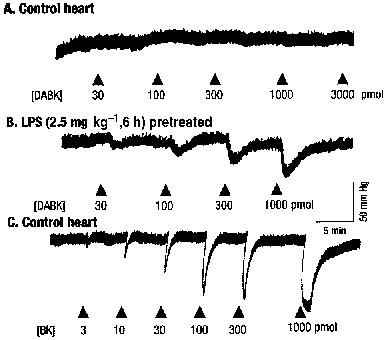
Typical traces showing vasodilator effects of bradykinin (BK) and des-Arg9BK (DABK), measured as decreases in coronary perfusion pressure in the rat isolated heart. (A and C) control heart; (B) heart from LPS (2.5 mg kg−1, 6 h)-pretreated rat.
Figure 2.
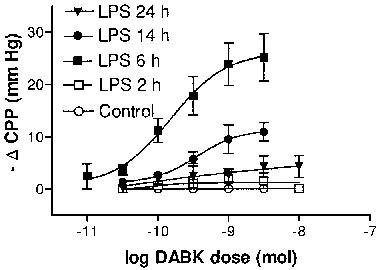
Time-dependent induction of coronary vasodilation responses to des-Arg9BK (DABK), measured as decreases in coronary perfusion pressure (ΔCPP). Hearts were obtained from control or LPS (2.5 mg kg−1)-pretreated rats at 2, 6, 14 and 24 h post treatment. Data are means±s.e.mean (n=4–9).
Pretreatment of rats with LPS promoted a time-dependent induction of coronary vasodilation responses to DABK. Whilst DABK had no effect on CPP in hearts taken from rats 2 h post LPS (n=4), marked dose-dependent reductions in CPP were observed after 6 h LPS treatment (ED50=149.4 pmol (33.0–675.6); Emax=27.0 mm Hg (15.7–36.6), n=9). At the 14 and 24 h time points the Emax of the dose-response curves to DABK were significantly reduced (P>0.05, two-way ANOVA) relative to the 6 h time point; values were reduced by approximately 50 and 80% respectively (Figure 2). DALBK (30 nM) had no effect on basal CPP (6 h post-LPS) but caused a significant (P<0.05, two-way ANOVA) parallel rightward shift of the dose-response curve to DABK (apparent pA2=8.4±0.16, n=5, Figure 3) without affecting the maximum response. In contrast HOE140 (10 nM) did not affect either basal CPP or the dose-response curve to DABK (P>0.05, two-way ANOVA, n=4, Figure 3).
Figure 3.
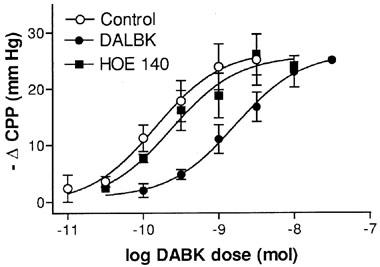
Comparison of the effects of kinin B1 and B2 receptor antagonism on the vasodilator responses to des-Arg9BK (DABK). Vasodilator responses are expressed as decreases in coronary perfusion pressure (ΔCPP) in hearts from LPS (2.5 mg kg−1, 6 h)-pretreated rats. Data obtained in the absence or presence of des-Arg9-[Leu8]-BK (DALBK, B1 receptor antagonist, 30 nM) and HOE140 (B2 receptor antagonist, 10 nM). Each point represents the means±s.e.mean (n=4–9).
Emax values of dose-response curves to BK, in hearts from rats 6 h post LPS (ED50=76.0 pmol (27.2–212.2), Emax=49.6 mm Hg (39.1–60.0), n=5) appeared to be reduced relative to controls, i.e. control hearts with endothelin-1-elevated CPP, but this effect did not reach statistical significance (P>0.05, two-way ANOVA). The BK dose-response curves in hearts from rats 6 h post LPS were unaltered (P>0.05, two-way ANOVA) by DALBK (30 nM) but were significantly inhibited (P<0.05, two-way ANOVA) by HOE140 (10 nM) without reducing the maximum response (apparent pA2=9.5±0.7, n=5).
Mechanisms of kinin responses
Endothelium removal
Endothelium removal produced an increase in CPP in hearts from both untreated rats and rats 6 h post-LPS treatment of 33.7±6.1 mm Hg (n=5) and 23.3±4.2 mm Hg (n=5) respectively (P>0.05). Figure 4 shows that removal of the endothelium caused a significant (P<0.05) reduction in responses to DABK (1 nmol) and acetylcholine (3 nmol, P<0.05) but had no effect on the responses to SNP (10 nmol, P>0.05, n=5).
Figure 4.
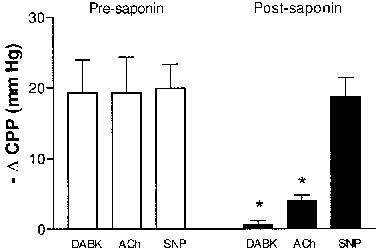
Effects of endothelium removal on des-Arg9bradykinin (DABK)-induced coronary vasodilation. Vasodilator responses are expressed as changes in coronary perfusion pressure (ΔCPP), mm Hg) in response to DABK (1 nmol), acetylcholine (ACh, 3 nmol) and sodium nitroprusside (SNP, 10 nmol) before (open columns) and after (solid columns) saponin treatment in hearts from LPS (2.5 mg kg−1, 6 h)-pretreated rats. Data are means±s.e.mean (n=5). *P<0.05 vs pre-saponin values.
Involvement of endothelial mediators
ODQ, whilst producing an increase in CPP of 12.8±3.7 mm Hg (n=5), had no significant effect on the DABK-induced dose-response curves (P>0.05, two-way ANOVA, Figure 5). ODQ did however completely abolish the dilator response to SNP (10 nmol, n=4, control response=−20±4.2 mm Hg; no detectable response following ODQ treatment). L-NAME (n=5) also increased CPP (34.3±4.3 mm Hg) and reduced the response to acetylcholine (3 nmol) by 75±6.2% (n=4), but did not modify the dose-response curve to DABK (P>0.05, two-way ANOVA, Figure 5).
Figure 5.
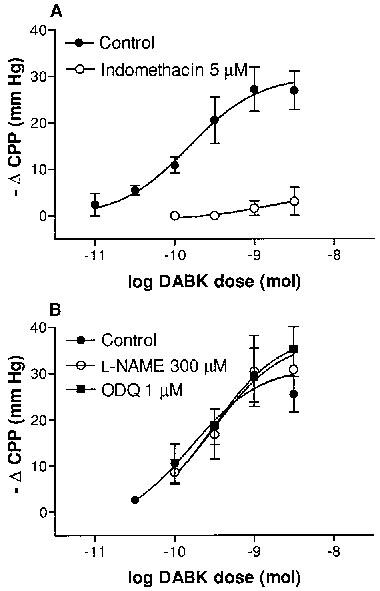
Effects of inhibition of (A) cyclooxygenase and (B) the nitric oxide-guanylyl cyclase pathway on des-Arg9bradykinin (DABK)-induced coronary vasodilation. Vasodilator responses are expressed as changes in coronary perfusion pressure (ΔCPP, mmHg) produced by DABK in hearts from LPS (2.5 mg kg−1, 6 h)-pretreated rats. Data obtained in the absence or presence of (A) indomethacin (5 μM), (B) L-NAME (300 μM) or ODQ (1 μM). Each point represents the means±s.e.mean (n=5–7).
Indomethacin completely abolished the responses to submaximal doses of DABK (<1 nmol) and inhibited the maximal response to DABK by 85% (see Figure 5). Additionally, DABK caused a dose-dependent increase in the concentration of 6-keto-PGF1α measured in the perfusate (Emax=0.3±0.04 ng ml−1) of hearts from rats 6 h post-LPS (Figure 6). No increase in 6-keto-PGF1α levels were observed following DABK administration in hearts from untreated rats (n=3, all doses P<0.06 ng ml−1).
Figure 6.
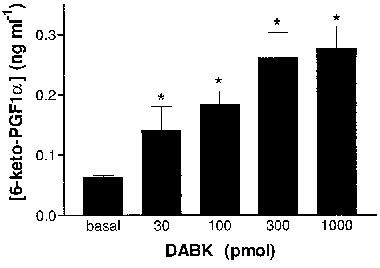
Increases in coronary perfusate concentrations of 6-keto-PGF1α in response to bolus doses of des-Arg9BK (DABK) in hearts from LPS (2.5 mg kg−1, 6 h)-pretreated (n=6) rats. Data shown are the means±s.e.mean of 6-keto-PGF1α concentrations in perfusate collected 2 min before and 2 min after administration of des-Arg9BK. *P<0.05 vs control.
Mechanisms of B1 receptor induction
In vitro perfusion of control hearts with LPS (n=6) produced no effect per se on CPP. Perfusion with the inflammatory cytokine, IL-1β (n=6) caused a gradual, transient (30 min) reduction in CPP of 33.8±4.0 mm Hg (n=6). Figure 7 shows that both treatments induced a time-dependent induction of a vasodilator response to DABK (1 nmol); the responses peaking at 3 h (IL-1β) and >4 h (LPS). These responses to DABK were completely abolished by indomethacin treatment (n=5) and inhibited by 68.5±7.6% (LPS, n=4) and 67.2±3.9% (IL-1β, n=5) in the presence of DALBK (30 nM). No response to DABK was observed in control hearts perfused for 4 h in the absence of LPS or IL-1β. The initial vasodilator response to BK (0.1 nmol, 54.3±4.4 mm Hg, n=6) was not significantly (P>0.05) affected 3 h after the IL-1β perfusion however a 27% reduction was observed after 4 h of LPS treatment (P<0.05). The SNP (10 nmol)-induced vasodilation (control response=25±4.9 mm Hg, n=4) was not significantly affected (P>0.05) by either treatment.
Figure 7.
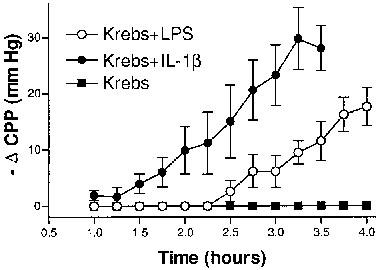
In vitro perfusion of LPS or IL-1β sensitises rat hearts to des-Arg9bradykinin (DABK). Control rat hearts were perfused with Krebs (n=4) or Krebs plus LPS (1 μg ml−1, 4 h, n=6) or IL-1β (5 ng ml−1, 2 h, n=6); vasodilator responses to repeated doses of DABK (1 nmol) were measured as decreases in coronary perfusion pressure (CPP). Data points represent means±s.e.mean.
Molecular expression of B1 receptors
RT–PCR of cardiac mRNA from untreated rats or rats 1 h post-LPS treatment revealed no expression of B1 receptor mRNA. In contrast, B1 receptor mRNA was consistently detected in hearts from rats 3 and 6 h post LPS (Figure 8A).
Figure 8.
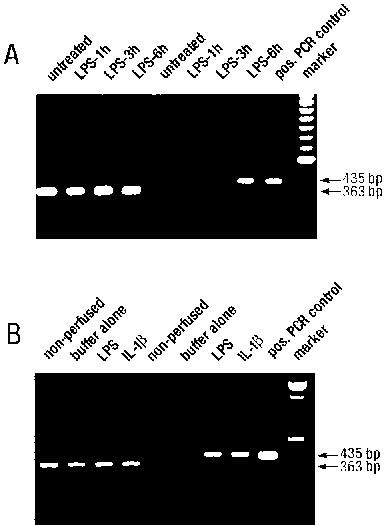
B1 receptor (435 bp) and GAPDH (363 bp) PCR products amplified from poly(A)+ RNA extracted from (A) non-perfused hearts from either untreated or LPS (2.5 mg kg−1, 1, 3 or 6 h)-treated rats and (B) hearts from untreated rats perfused in vitro with either LPS (1 μg ml−1, 4 h), IL-1β (5 ng ml−1, 2 h) or buffer solution alone (4 h). Positive PCR control is a mouse B1 receptor cDNA plasmid.
B1 receptor mRNA was also detected in hearts perfused in vitro with LPS or IL-1β whereas no expression was detected in non-perfused control hearts (Figure 8B). Hearts perfused for 4 h in the absence of LPS or IL-1β also expressed B1 receptor mRNA. GAPDH mRNA was detected in all hearts tested.
The resulting PCR products derived from two separate samples using B1 receptor specific primers were subjected to automated sequencing. The nucleotide sequence of the amplified cDNA revealed identity with the rat B1 receptor cDNA sequence (Ni et al., 1998).
Effects of kinins on blood pressure in vivo
MAP of control untreated rats was 125.9±2.5 mm Hg (n=8). To compare the activity of DABK in control vs LPS-treated rats MAP in LPS-treated rats was raised using noradrenaline. MAP of LPS (2.5 mg kg−1, 6 h)-treated rats infused with noradrenaline was 103.9±3.3 mm Hg (n=10, P>0.05 relative to controls). Bolus doses of DABK (1–100 nmol kg−1) had no effect on MAP in control untreated rats (n=4) but caused dose-dependent decreases in MAP in LPS (2.5 mg kg−1, 6 h)-treated rats with an ED50 of 29.7 (8.3–105) nmol kg−1 and Emax of 45.1 (24–66) mm Hg (n=10, Figure 9). DALBK (3, 6 nmol kg−1 min−1, 5 min) significantly (P<0.05) reduced the hypotensive effect of DABK (Figure 9). DALBK (3 nmol kg−1 min−1, 5 min) produced no effect per se in control animals but in LPS (10 mg kg−1, 6 h)-treated rats it increased MAP by 5.3±1.2% (n=6, P<0.05). The B1 receptor antagonist des-Arg10HOE 140 (30 nmol kg−1 min−1, 5 min) also increased MAP by 8.8±0.34% (n=5, P<0.05) in LPS (10 mg kg−1, 6 h)-treated rats. Captopril (1 mg kg−1) reduced MAP by 11.1±1.9% (n=4, P<0.05) in LPS (10 mg kg−1, 6 h)-treated rats. This effect was reversed by des-Arg10HOE 140 (30 nmol kg−1 min−1) treatment (P<0.05).
Figure 9.
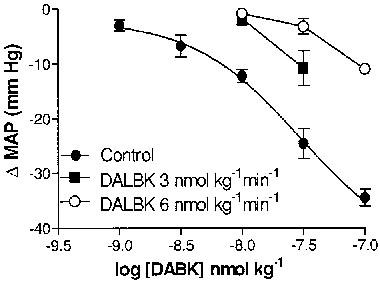
The hypotensive response (ΔMAP) to des-Arg9bradykinin (DABK) and its antagonism by des-Arg9-[Leu8]-BK in LPS (2.5 mg kg−1, 6 h)-pretreated rats. Data obtained in the absence or presence of des-Arg9-[Leu8]-BK (3, 6 nmol kg−1 min−1). Each point represents the means±s.e.mean (n=4–7).
Discussion
The present study has shown that the kinin B1 receptor, whilst not basally expressed, is induced by endotoxin in the rat coronary circulation. This was demonstrated both in terms of mRNA expression as detected by RT–PCR and of a coronary vasodilation response to DABK. Functional responses to DABK increased over time, in parallel with the mRNA expression, peaking at 6 h and almost absent by 24 h post-endotoxin treatment. The coronary vasodilation responses to DABK were B1 receptor-mediated and endothelium-dependent. Furthermore the responses were shown to be mediated by prostanoids but did not involve NO. In vitro treatment with endotoxin revealed that the receptor is induced in the coronary circulation in the absence of circulating cells, possibly by a direct effect of endotoxin within the blood vessel wall.
Induction of B1 receptor-mediated responses and functional relevance in vivo
A single bolus exposure to endotoxin resulted in the development of a functional response to DABK that was maximal at 6 h and almost absent by 24 h. The loss of response to DABK at 24 h may reflect the differences between a continued inflammatory stimulus as opposed to single exposure as in the present study. Whether the functional response is sustained in the presence of a prolonged inflammatory stimulus is unclear and warrants further investigation. The B2 receptor-mediated response to BK was not significantly affected 6 h following endotoxin treatment suggesting that B2 receptor regulation does not accompany B1 receptor induction at this time point in this model. However further studies are required to confirm this.
Whilst it is clear that NO plays a major role in the hypotension associated with endotoxemia there is also a significant NO-independent contribution to this response (Ungureanu-Longrois et al., 1995). Our studies suggest that activation of endotoxin-induced B1 receptors may have a role to play in mediation of the latter component. Indeed the time course of iNOS induction in endotoxemia in the rat is similar to that found for the B1 receptor in the present study (Luss et al., 1995; Kinugawa et al., 1997). This supports our initial hypothesis that the de novo induction of coronary B1 receptors contributes to an NO-independent regulation of coronary tone and this regulation may have relevance in vivo.
Support for the hypothesis that the induction of the B1 receptor occurred at the level of a specific receptor-mediated response was provided by B1 receptor selective agonist and antagonist studies. The B1 receptor selective antagonist DALBK (Marceau et al., 1998) inhibited the DABK-induced coronary vasodilations with an apparent pA2 value of 8.4 and was active against the DABK-induced hypotension in vivo. A partial agonist action of DALBK was also observed both in the isolated heart and on blood pressure which is consistent with its activity at rat B1 receptors (Marceau et al., 1998). The B2 receptor selective antagonist, HOE140, whilst effectively inhibiting the BK-induced coronary vasodilations in both control and endotoxemic hearts did not affect the response to DABK. Whilst these results strongly suggest a specific B1 receptor-mediated response further studies are required to fully confirm induction at the level of receptor protein.
Basal coronary perfusion pressure was elevated in hearts from endotoxin treated rats compared to controls. It has been suggested that this may be due to release of endothelin-1 (Hohlfeld et al., 1995). The possibility that elevation of perfusion pressure alone was responsible for the development of responses to DABK was excluded, since raising coronary perfusion pressure with endothelin-1 in control hearts did not reveal a response to DABK whilst responses to BK remained unchanged. This was further demonstrated in hearts from rats 2 h post-endotoxin in which coronary perfusion pressure was elevated relative to controls yet no response to DABK was observed.
Mechanisms of B1 receptor-mediated coronary vasodilation
DABK-induced vasodilation was endothelium-dependent since removal of the endothelium with saponin abolished the response. This loss of response was not due to damage of the smooth muscle since responses to the endothelium-independent vasodilator, SNP, remained unchanged. These results suggest that the induced B1 receptor expression, in this particular situation, is probably localized to the endothelium. However confirmation of this will require further studies. These results are in keeping with reports showing endothelium-dependency of induced B1 receptor-mediated responses in various other blood vessels (Drummond & Cocks, 1995b; Deblois & Marceau, 1987) including human isolated coronary arteries (Drummond & Cocks, 1995a).
NO did not play a significant role in mediation of these responses since inhibition of NO synthase had no effect on DABK-induced vasodilation. This is in agreement with the study by Nicolau et al. (1996) who show that the DABK-induced hypotension in endotoxin-treated rats was not affected by NO synthase inhibition. However, it is possible that stimulated NO release from preformed stores could be involved in this response. We found that the guanylyl cyclase inhibitor ODQ, at concentrations shown to inhibit NO-mediated effects (this study; Muller et al., 1998), was also inactive suggesting that the activation of guanylyl cyclase downstream is also not involved. Moreover the observation that the cyclooxygenase inhibitor, indomethacin, abolished the response to DABK suggests that prostanoids, most likely PGI2, are the primary if not sole mediator of the response. In support of this, we observed a dose-dependent elevation of 6-keto-PGF1α levels in the effluent following DABK administration. The involvement of endothelial PGI2 in DABK-induced dilator responses is in agreement with data obtained in other isolated vascular beds; including rabbit mesenteric (Churchill & Ward, 1986) and dog renal (Rhaleb et al., 1989) arteries. However, there are also conflicting studies excluding prostanoid involvement in DABK-induced vasorelaxation (Pruneau & Belichard, 1993; Pruneau et al., 1996; Santiago et al., 1995; DeWitt et al., 1994) and hypotension (Nicolau et al., 1996). This difference may be related to species and regional variation or to the absence of specific prostanoid receptor subtypes.
Mechanisms of B1 receptor induction
In vitro LPS perfusion of normal hearts caused a time-dependent induction of functional reactivity to DABK, associated with B1 receptor mRNA expression, in a similar manner to that achieved with in vivo LPS treatment. These effects appear to be selective for LPS treatment as DABK was inactive in hearts not perfused with LPS. Interestingly, unlike the in vivo situation LPS did not raise CPP in vitro suggesting that this is an in vivo phenomenon. However this requires further investigation. These results suggest that induction of B1 receptors in the coronary vasculature in vivo is most likely independent of circulating cells and is probably due to a direct effect of LPS on the blood vessel. Indeed experimental neutropenia has no effect on induction of rabbit aortic B1 receptors by in vivo endotoxin treatment (Bouthillier et al., 1987).
Inflammatory cytokines including IL-1β have also been implicated in the pathogenesis of endotoxemia (Bellomo, 1992; Waage et al., 1991; Martich et al., 1993) and are the endogenous trigger for the induction of kinin B1 receptors in various systems (Marceau et al., 1998). The present study supports this proposal since following perfusion of hearts with IL-1β vasodilator responses to DABK were observed in parallel with B1 receptor expression. Since in vitro IL-1β perfusion caused B1 receptor induction earlier than that achieved by LPS perfusion it is possible that local production of this cytokine, a key mediator in several LPS-induced responses (Glauser, 1996) mediates the in vivo and in vitro effects of LPS, however this requires further investigation for confirmation. Demonstration of B1 receptor mRNA expression in control hearts perfused for 4 h, in the absence of added endotoxin, indicates that the experimental process is sufficient stimulus to cause spontaneous B1 receptor induction as found in other in vitro systems (Deblois & Marceau, 1987; Drummond & Cocks, 1995a,1995b). However, this modest expression of mRNA was not linked to a functional response to DABK at this time point.
Conclusion
We have shown that endotoxin treatment induces de novo functional B1 receptor expression in the rat coronary circulation possibly via a direct action within the vessel wall independent of circulating cells. The B1 receptor-mediated coronary vasodilation is dependent on an intact endothelium and occurs as a result of the secondary release of prostanoids and is independent of NO. The current findings are entirely consistent with the hypothesis that de novo B1 receptor induction plays a role in the regulation of the coronary circulating during endotoxemia and also suggests that B1 receptor induction may contribute to the hypotension associated with this pathological condition.
Acknowledgments
This study and Dr P.G. McLean were supported by the British Heart Foundation (BHF, PG#97013). Dr Ahluwalia is funded by a BHF Intermediate Fellowship and Dr Perretti is a Research Fellow of the Arthritis Research Campaign. The authors kindly thank Dr J.B. Pesquero (Escola Paulista de Medicine, Sao Paulo, Brazil) for the supply of the B1 receptor plasmid.
Abbreviations
- ACh
acetylcholine
- BK
bradykinin
- CPP
coronary perfusion pressure
- DABK
des-Arg9bradykinin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- i.p.
intraperitoneal
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- L-NAME
NG-nitro-L-arginine methyl ester
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- NO
nitric oxide
- PCR
polymerase chain reaction
- PGI2
prostacyclin
- SNP
sodium nitroprusside
References
- ARAUJO R.C., SILVA J.A., WALTHER T., OLIVEIRA S.M., PESQUERO J.B., BADER M.Reduced hypotensive response to endotoxin in kinin B1 receptor deficient mice Abstracts book, The 15th International Conference on Bradykinin and Related Kinins 1998. Kinin '98, Nara, Japan, p.57
- BELLOMO R. The cytokine network in the critically ill. Anaesth. Intensive. Care. 1992;20:288–302. doi: 10.1177/0310057X9202000303. [DOI] [PubMed] [Google Scholar]
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- BOUCHARD J.F., CHOUINARD J., LAMONTAGNE D. Role of kinins in the endothelial protective effect of ischaemic preconditioning. Br. J. Pharmacol. 1998;123:413–420. doi: 10.1038/sj.bjp.0701619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUTHILLIER J., DEBLOIS D., MARCEAU F. Studies on the induction of pharmacological responses to des- Arg9- bradykinin in vitro and in vivo. Br. J. Pharmacol. 1987;92:257–264. doi: 10.1111/j.1476-5381.1987.tb11319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHURCHILL L., WARD P.E. Relaxation of isolated mesenteric arteries by des- Arg9- bradykinin stimulation of B1 receptors. Eur. J. Pharmacol. 1986;130:11–18. doi: 10.1016/0014-2999(86)90178-0. [DOI] [PubMed] [Google Scholar]
- DEBLOIS D., MARCEAU F. The ability of des-Arg9-bradykinin to relax rabbit isolated mesenteric arteries is acquired during in vitro incubation. Eur. J. Pharmacol. 1987;142:141–144. doi: 10.1016/0014-2999(87)90664-9. [DOI] [PubMed] [Google Scholar]
- DELA C.R., SUFFREDINI A.F., PAGE J.D., PIXLEY R.A., KAUFMAN N., PARRILLO J.E., COLMAN R.W. Activation of the kallikrein-kinin system after endotoxin administration to normal human volunteers. Blood. 1993;81:3313–3317. [PubMed] [Google Scholar]
- DEWITT B.J., CHENG D.Y., KADOWITZ P.J. Des-Arg9-bradykinin produces tone-dependent kinin B1 receptor-mediated responses in the pulmonary vascular bed. Circ. Res. 1994;75:1064–1072. doi: 10.1161/01.res.75.6.1064. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Endothelium-dependent relaxation to the B1 kinin receptor agonist des- Arg9-bradykinin in human coronary arteries. Br. J. Pharmacol. 1995a;116:3083–3085. doi: 10.1111/j.1476-5381.1995.tb15108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Endothelium-dependent relaxations mediated by inducible B1 and constitutive B2 kinin receptors in the bovine isolated coronary artery. Br. J. Pharmacol. 1995b;116:2473–2481. doi: 10.1111/j.1476-5381.1995.tb15098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLWOOD A.J., CURTIS M.J. Mechanism of actions of sumatriptan on coronary flow before and after endothelial dysfunction in guinea-pig isolated heart. Br. J. Pharmacol. 1997;120:1039–1048. doi: 10.1038/sj.bjp.0701009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEIN A.M., BERNARD G.R., CRINER G.J., FLETCHER E.C., GOOD J.T.J., KNAUS W.A., LEVY H., MATUSCHAK G.M., SHANIES H.M., TAYLOR R.W., RODELL T.C. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127) JAMA. 1997;277:482–487. [PubMed] [Google Scholar]
- FELETOU M., LONCHAMPT M., ROBINEAU P., JAMONNEAU I., THURIEAU C., FAUCHERE J.L., VILLA P., GHEZZI P., PROST J.F., CANET E. Effects of the bradykinin B2 receptor antagonist S 16118 (p- guanidobenzoyl-[Hyp3,Thi5,D-Tic7,Oic8]bradykinin) in different in vivo animal models of inflammation. J. Pharmacol. Exp. Ther. 1995;273:1078–1084. [PubMed] [Google Scholar]
- FILEP J.G., DELALANDRE A., BEAUCHAMP M. Dual role for nitric oxide in the regulation of plasma volume and albumin escape during endotoxin shock in conscious rats. Circ. Res. 1997;81:840–847. doi: 10.1161/01.res.81.5.840. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- GLAUSER M.P. The inflammatory cytokines. New developments in the pathophysiology and treatment of septic shock. Drugs. 1996;52 Suppl 2:9–17. doi: 10.2165/00003495-199600522-00004. [DOI] [PubMed] [Google Scholar]
- HANASAKI K., YOKOTA Y., ISHIZAKI J., ITOH T., ARITA H. Resistance to endotoxic shock in phospholipase A2 receptor- deficient mice. J. Biol. Chem. 1997;272:32792–32797. doi: 10.1074/jbc.272.52.32792. [DOI] [PubMed] [Google Scholar]
- HOHLFELD T., KLEMM P., THIEMERMANN C., WARNER T.D., SCHROR K., VANE J.R. The contribution of tumour necrosis factor-alpha and endothelin-1 to the increase of coronary resistance in hearts from rats treated with endotoxin. Br. J. Pharmacol. 1995;116:3309–3315. doi: 10.1111/j.1476-5381.1995.tb15140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINUGAWA K., SHIMIZU T., YAO A., KOHMOTO O., SERIZAWA T., TAKAHASHI T. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ. Res. 1997;81:911–921. doi: 10.1161/01.res.81.6.911. [DOI] [PubMed] [Google Scholar]
- KONTOS M.A., MAGEE J.H., SHAPIRO W. General and regional circulation effects of synthetic bradykinin in man. Circ. Res. 1964;14:352. doi: 10.1161/01.res.14.4.351. [DOI] [PubMed] [Google Scholar]
- LUSS H., WATKINS S.C., FREESWICK P.D., IMRO A.K., NUSSLER A.K., BILLIAR T.R., SIMMONS R.L., DEL NIDO P.J., MCGOWAN F.X.J. Characterization of inducible nitric oxide synthase expression in endotoxemic rat cardiac myocytes in vivo and following cytokine exposure in vitro. J. Mol. Cell Cardiol. 1995;27:2015–2029. doi: 10.1016/0022-2828(95)90023-3. [DOI] [PubMed] [Google Scholar]
- MARCEAU F., HESS J.F., BACHVAROV D.R. The B1 receptors for kinins. Pharmacol Rev. 1998;50:357–386. [PubMed] [Google Scholar]
- MARTICH G.D., BOUJOUKOS A.J., SUFFREDINI A.F. Response of man to endotoxin. Immunobiology. 1993;187:403–416. doi: 10.1016/S0171-2985(11)80353-0. [DOI] [PubMed] [Google Scholar]
- MENG X., AO L., BROWN J.M., FULLERTON D.A., BANERJEE A., HARKEN A.H. Nitric oxide synthase is not involved in cardiac contractile dysfunction in a rat model of endotoxemia without shock. Shock. 1997;7:111–118. doi: 10.1097/00024382-199702000-00007. [DOI] [PubMed] [Google Scholar]
- MULLER B., KLESCHYOV A.L., MALBLANC S., STOCLET J.C. Nitric oxide-related cyclic GMP-independent relaxing effect of N- acetylcysteine in lipopolysaccharide-treated rat aorta. Br. J. Pharmacol. 1998;123:1221–1229. doi: 10.1038/sj.bjp.0701737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NI A., CHAO L., CHAO J. Molecular cloning and expression of rat bradykinin B1 receptor. Biochim. Biophys. Acta. 1998;1442:177–185. doi: 10.1016/s0167-4781(98)00163-8. [DOI] [PubMed] [Google Scholar]
- NICOLAU M., FELTRIN M.R., REGOLI D. Induction of bradykinin B1 hypotensive receptors in rats by lipopolysaccharide. Can. J. Physiol. Pharmacol. 1996;74:337–340. [PubMed] [Google Scholar]
- NIES A.S., FORSYTH R.P., WILLIAMS H.E., MELMON K.L. Contribution of kinins to endotoxin shock in unanesthetized Rhesus monkeys. Circ. Res. 1968;22:155–164. doi: 10.1161/01.res.22.2.155. [DOI] [PubMed] [Google Scholar]
- PESQUERO J.B., PESQUERO J.L., OLIVEIRA S.M., ROSCHER A.A., METZGER R., GANTEN D., BADER M. Molecular cloning and functional characterization of a mouse bradykinin B1 receptor gene. Biochem. Biophys. Res. Commun. 1996;220:219–225. doi: 10.1006/bbrc.1996.0384. [DOI] [PubMed] [Google Scholar]
- PRUNEAU D., BELICHARD P. Induction of bradykinin B1 receptor-mediated relaxation in the isolated rabbit carotid artery. Eur. J. Pharmacol. 1993;239:63–67. doi: 10.1016/0014-2999(93)90976-o. [DOI] [PubMed] [Google Scholar]
- PRUNEAU D., LUCCARINI J.M., DEFRENE E., PAQUET J.L., BELICHARD P. Characterisation of bradykinin receptors from juvenile pig coronary artery. Eur. J. Pharmacol. 1996;297:53–60. doi: 10.1016/0014-2999(95)00720-2. [DOI] [PubMed] [Google Scholar]
- RAIDOO D.M., RAMSAROOP R., NAIDOO S., MULLER-ESTERL W., BHOOLA K.D. Kinin receptors in human vascular tissue: their role in atheromatous disease. Immunopharmacology. 1997;36:153–160. doi: 10.1016/s0162-3109(97)00015-5. [DOI] [PubMed] [Google Scholar]
- REGOLI D., BARABE J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- RHALEB N.E., DION S., BARABE J., ROUISSI N., JUKIC D., DRAPEAU G., REGOLI D. Receptors for kinins in isolated arterial vessels of dogs. Eur. J. Pharmacol. 1989;162:419–427. doi: 10.1016/0014-2999(89)90332-4. [DOI] [PubMed] [Google Scholar]
- SANTIAGO J.A., GARRISON E.A., CHAMPION H.C., SMITH R.E., DEL RIO O., KADOWITZ P.J. Analysis of responses to kallidin, DABK, and DAK in feline hindlimb vascular bed. Am. J. Physiol. 1995;269:H2057–H2064. doi: 10.1152/ajpheart.1995.269.6.H2057. [DOI] [PubMed] [Google Scholar]
- UNGUREANU-LONGROIS D., BALLIGAND J.L., SIMMONS W.W., OKADA I., KOBZIK L., LOWENSTEIN C.J., KUNKEL S.L., MICHEL T., KELLY R.A., SMITH T.W. Induction of nitric oxide synthase activity by cytokines in ventricular myocytes is necessary but not sufficient to decrease contractile responsiveness to beta-adrenergic agonists. Circ. Res. 1995;77:494–502. doi: 10.1161/01.res.77.3.494. [DOI] [PubMed] [Google Scholar]
- WAAGE A., BRANDTZAEG P., ESPEVIK T., HALSTENSEN A. Current understanding of the pathogenesis of gram-negative shock. Infect. Dis. Clin. North. Am. 1991;5:781–791. [PubMed] [Google Scholar]
- WAAGE A., BRANDTZAEG P., HALSTENSEN A., KIERULF P., ESPEVIK T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. J. Exp. Med. 1989;169:333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHALLEY E.T., SOLOMON J.A., MODAFFERI D.M., BONHAM K.A., CHERONIS J.C. CP-0127, a novel potent bradykinin antagonist, increases survival in rat and rabbit models of endotoxin shock. Agents. Actions. Suppl. 1992;38:413–420. [PubMed] [Google Scholar]
- WILSON D.D., DE GARAVILLA L., KUHN W., TOGO J., BURCH R.M., STERANKA L.R. D-Arg-[Hyp3-D-Phe7]-bradykinin, a bradykinin antagonist, reduces mortality in a rat model of endotoxic shock. Circ. Shock. 1989;27:93–101. [PubMed] [Google Scholar]