

Abstract
Cisapride is a prokinetic agent which has been associated with QT prolongation, torsades de pointes and cardiac arrest. The cellular mechanism for these observations is high affinity blockade of IKr (encoded by HERG).
In a chronic transfection model using CHO-K1 cells, cisapride inhibited HERG tail currents after a step to +25 mV with similar potency at room and physiological temperatures (IC50 16.4 nM at 20–22°C and 23.6 nM at 37°C).
Channel inhibition exhibited time-, voltage- and frequency-dependence. In an envelope of tails test, channel blockade increased from 27±8% after a 120 ms depolarizing step to 50±4% after a 1.0 s step. These findings suggested affinity for open and/or inactivated channel states.
Inactivation was significantly accelerated by cisapride in a concentration-dependent manner and there was a small (−7 mV) shift in the voltage dependence of steady state inactivation.
Channel blockade by cisapride was modulated by [K+]o, with a 26% reduction in the potency of channel blockade when [K+]o was increased from 1 to 10 mM.
In conclusion, HERG channel inhibition by cisapride exhibits features consistent with open and inactivated state binding and is sensitive to external potassium concentration. These features may have significant clinical implications with regard to the mechanism and treatment of cisapride-induced proarrhythmia.
Keywords: Cisapride, human ether-a-go-go-related gene (HERG), Chinese hamster ovary (CHO-K1) cell, cardiac arrhythmia, torsades de pointes
Introduction
Cisapride is a gastroprokinetic agent used clinically in the treatment of dyspepsia, gastroparesis, and gastro-oesophageal reflux (Wiseman & Faulds, 1994). Recently, it has been associated with QT prolongation, syncope, serious ventricular arrhythmias (including torsades de pointes) and cardiac arrest (Wysowski & Bacsanyi, 1996; FDA talk paper, 1998). These complications generally occur in patients administered high doses (Bran et al., 1995) or taking concurrent cytochrome p450-inhibiting medications such as ketoconazole or itraconazole (Ahmad & Wolfe, 1995; Hoover et al., 1996). Cisapride causes prolongation of action potential duration (in a reverse use-dependent manner), and early after depolarizations in isolated rabbit Purkinje fibres (Puisieux et al., 1996; Drolet et al., 1998), and action potential prolongation in guinea-pig isolated papillary muscle (Kii & Ito, 1997).
Repolarization of cardiac ventricular myocytes is due mainly to outward potassium currents, the most important of which is the delayed rectifier potassium channel, (IK), which has two components, IKr and IKs (Sanguinetti & Jurkiewicz, 1990). Drugs which are associated with QT prolongation and torsades de pointes commonly act by inhibiting IKr, which is encoded by the human ether-a-go-go-related gene (HERG; Curran et al., 1995; Sanguinetti et al., 1995). Recent studies have shown that cisapride inhibits IKr in rabbit and guinea-pig ventricular myocytes with similar potency (IC50 9 nM, Carlsson et al., 1997; IC50 15 nM, Drolet et al., 1998). Cisapride also causes potent, voltage-dependent inhibition of the channel activity produced by transient transfection with HERG (IC50 6.5 nM, Mohammad et al., 1997; IC50 44.5 nM, Rampe et al., 1997). HERG inhibition by cisapride· required channel activation and the above authors concluded that cisapride has a predominant affinity for the open and/or inactivated states of the HERG channel.
We have recently reported that Chinese hamster ovary (CHO-K1) cells stably transfected with HERG express a delayed rectifier K+ channel with properties very similar to IKr (Walker et al., 1999). The experiments reported below were designed to confirm cisapride blockade in this model and to characterize its voltage- and time-dependence and any modulation of channel kinetics with a view to further elucidating the mechanisms of blockade. Since there is a disparity in the potency of HERG channel blockade at room temperature (20–22°C) and the human therapeutic plasma concentration, we evaluated the effects of temperature and [K+]o on blockade by cisapride.
Methods
Molecular biology
The CHO-K1 cells (American Type Culture Collection, Bethesda, MD, U.S.A.) used in the following experiments were maintained in Dulbecco's modified Eagle's medium-F12 (DMEM-F12, Gibco, BRL, Gaithersburg, MD, U.S.A.), supplemented with 5% foetal calf serum. Eukaryotic expression of HERG was performed by directionally cloning the coding region of the HERG gene (gift from Dr G. Robertson, Department of Physiology, University of Wisconsin Medical School, Madison, WI, U.S.A.) into the expression vector pRc/CMV (Invitrogen, San Diego, CA, U.S.A.), which also carries the G418 resistance gene. This construct was then transfected into CHO-K1 cells. Cell monolayers in 35 mm2 dishes were transfected using 9 μL Lipofectamine Reagent (Gibco, BRL) and 1 μg DNA. Stably transfected cells were then selected with 1000 μg mL−1 G418 (Boehringer, Mannheim). These were subcloned to isolate individual cell clones which expressed substantial HERG-related K+ current. Individual subclones were maintained long-term in tissue culture and used for the patch clamping experiments to be described below.
Electrophysiology
Currents were recorded from CHO-K1 cells at room temperature (20–22°C) and at 37°C, using the whole-cell patch-clamp technique. CHO-K1 cells plated on coverslips were placed at the bottom of a 2 mL perfusion chamber mounted on the stage of an inverted phase contrast microscope (Nikon Diaphot, Nikon Corporation, Tokyo, Japan). Electrodes were positioned using a micromanipulator (Narishige WB 90, Tokyo, Japan). A silver/silver-chloride reference electrode was either placed directly in the perfusion chamber or in a separate chamber connected by a salt bridge. Membrane potentials were adjusted by −15 mV to correct for the junction potential between high K+ pipette and external bath solution (calculated using commercial software, JpCalc, Barry, 1994). Cells were patched using micropipettes fabricated from thin-walled borosilicate glass (Vitrex Microhematocrit Tubes, Modulohm I/S, Denmark) with a vertical pipette puller (Model 720, David Kopf Instruments, CA, U.S.A.). Currents were amplified and filtered at 2 kHz with a 4 pole Bessel filter (−3dB point) using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA, U.S.A.). Stimulation protocols and data acquisition were carried out using a microcomputer (IBM Pentium), running commercial software and hardware (pClamp 6.0/Digidata 1200, Axon Instruments Inc and Scientific Solutions Inc.).
Whole-cell capacitance was determined from capacitative transient decay in current recordings following voltage steps of ±10 mV from the holding potential. At least 80% series resistance compensation was achieved in all reported experiments. Leak subtraction was performed in some experiments by applying three hyperpolarizing pre-pulses before the test pulses (P/3 subtraction protocol).
Solutions and drugs
The intracellular pipette solution contained (mM): K gluconate 120, KCl 20, MgATP 1.5, EGTA 5, N-2-hydroxylethylpiperazine-N1-2-ethanesulphonic acid (HEPES) 10, adjusted to pH 7.3. The superfusion solution contained (mM): NaCl 130, KCl 4.8, MgCl2 1.2, NaH2PO4 1.2, HEPES 10, glucose 12.5, CaCl2 1.0, adjusted to a pH of 7.4. Cisapride was purchased from Janssen Biotech (Olem, Belgium) and was prepared as stock solution in dimethyl sulphoxide (DMSO) and subsequently diluted as required with superfusate (maximum final DMSO concentration=0.1% v v−1). In preliminary experiments, we determined that DMSO 0.1% v v−1 had no effect on the parameters under study.
Statistics
Current analysis was performed using the Clampfit module of the pClamp software. Data are expressed as mean±s.e. for n experiments. Statistical analyses were performed using Prism 2.0 (Graphpad Software, San Diego, CA, U.S.A.). Unpaired t-tests were used for comparisons of two groups and repeated measures ANOVA with post-hoc comparison of means using Bonferroni's test for multiple group comparisons. A P value <0.05 was considered significant. The voltage-dependence of current activation was determined by fitting the values of the normalized tail currents to a Boltzmann function:
where I represents the tail current, V1/2, the voltage at which the current was half activated, Vt, the test potential and k, the slope factor. The relationship between drug concentration and current blockade was determined by fitting values to a Hill equation after normalization of post-drug current to control current:
where I represents the tail current, IC50 the concentration required for 50% channel blockade, D the drug concentration and n the Hill coefficient.
Results
HERG channel inhibition by cisapride
HERG-transfected CHO-K1 cells produce currents (‘HERG currents') with biophysical properties similar to those of HERG-transfected Xenopus oocytes reported by previous investigators (McDonald et al., 1997; Walker et al., 1999). Current-voltage relations and activation kinetics were determined using step depolarizations to potentials between −35 and +25 mV from a holding potential of −75 mV. Currents were recorded in control conditions and during steady state blockade maintained by 0.1 Hz stimulation in the presence of 10 nM cisapride (Figure 1a,b). Cisapride in concentrations of 1 nM to 10 μM, caused, potent, concentration-dependent inhibition of tail currents at +25 mV, with an IC50 of 16.4 nM (95% CI 11.0−24.4 nM) and Hill coefficient of −0.8±0.1 (Figure 1c, n=8) at 20–22°C. Figure 1d demonstrates that channel inhibition was equally potent at 37°C (IC50 23.6, 95% CI 20.7–27.0, Hill coefficient −0.8±0.1, n=4–7). The voltage required to half-maximally activate HERG (V1/2), was shifted from −21.7±1.2 mV in controls to −29.3±1.4 mV by exposure to 10 nM cisapride (P<0.01, n=9), and slope factors were not significantly different (9.2±1.1 in controls and 8.3±1.3 after cisapride, Figure 1e). Tail current inhibition was voltage-dependent, increasing from 12±8% at −25 mV to 44±7% at +35 mV (P<0.05, n=9, Figure 1f).
Figure 1.

HERG channel inhibition by cisapride 10 nM. Currents before (a) and after cisapride 10 nM (b) were elicited by a protocol in which 3.9 s depolarizing steps to between −35 and +25 mV were applied at 10 s intervals from a holding potential of −75 mV. Tail currents were produced by cell repolarization to −55 mV for 5 s. (c) Relative tail currents (Icis/Icon) at +25 mV following 0.1 Hz stimulation were fitted with a Hill equation, yielding an IC50 of 16.4 nM and Hill slope −0.8 (n=8). (d) Using the same protocol, channel inhibition did not vary when temperature was increased to 37°C (IC50 23.6 nM and Hill slope −0.8, n=4–7). Interrupted line indicates dose response relation for tail currents at 22°C. (e) Tail currents in controls and after bath superfusion with cisapride 10 nM were normalized the maximum control current and fit with a Boltzmann function. (V1/2 shifted from −21.7±1.2 mV in controls to −29.3±1.4 mV after cisapride, P<0.05, n=9). Slope factors were not significantly different. (f) Voltage-dependence of current inhibition. In the presence of cisapride 10 nM, tail current inhibition increased as test potential became more positive. *Represents significant difference from current at +35 mV (n=9).
Frequency dependence of channel blockade
We have previously demonstrated that HERG tail current amplitude remains stable at stimulation frequencies of 0.1 and 0.5 Hz in control conditions and that perhexiline maleate exhibits frequency-dependent tail current inhibition (Walker et al., 1999). To examine the effects of pulse frequency on channel binding by cisapride, tail currents were measured at −55 mV, after each of a train of 500 ms voltage steps to +15 mV, delivered at frequencies of 0.5 and 0.1 Hz. In the presence of 10 nM cisapride, channel blockade increased significantly with stimulation frequency from 23±5% after 40 pulses at 0.1 Hz to 47±6% after the same number of pulses at 0.5 Hz, (P<0.01, n=10, Figure 2).
Figure 2.
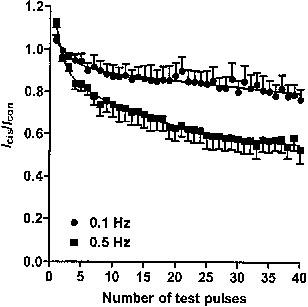
Effect of pulse frequency on current amplitude. Peak tail currents after each pulse in a train of 500 ms voltage steps to +15 mV at frequencies of 0.1 Hz and 0.5 Hz were expressed as relative current and plotted against the number of test pulses. Current amplitude decreased by 47% after 40 pulses at 0.5 Hz, but only 23% with 0.1 Hz stimulation (n=10).
Kinetics of current blockade
Development of channel blockade during depolarization was assessed using an envelope of tails protocol, in which peak tail current amplitude was measured at −55 mV, following depolarizing pulses to +25 mV which varied in duration from 40 ms to 1.0 s (Figure 3). Blockade by cisapride 10 nM increased with pulse duration from 27±8% after a 120 ms depolarizing step to 50±4% after a 1.0 s step (P<0.01, n=7, Figure 3b,c). The time course of block development was well fitted by a single exponential function (mean τ 383 ms, 95% CI 336–444 ms, n=7).
Figure 3.

Development of channel blockade by cisapride 10 nM assessed with envelope of tails test. Depolarizing pulses to +25 mV of variable duration (40 ms to 1.0 s) from a holding potential of −55 mV were applied at 10 s intervals before (a) and after (b) bath exchange with cisapride 10 nM. (c) Peak tail currents corrected for cell capacitance were plotted for controls and after cisapride. Note that current inhibition increased with pulse duration. (d) Relative tail current plotted as a function of the duration of the preceding depolarizing step. Current decreased monoexponentially as test pulse duration increased (τ 383 ms, n=7).
To determine the time course of recovery from channel blockade by cisapride 10 nM, a 500 ms depolarizing prepulse to +25 mV was followed by a step back to −95 mV of variable duration (0.5–240 s). Tail currents were then measured at −55 mV following a second depolarizing pulse of identical duration and amplitude to the first (Figure 4). Recovery from channel inhibition reached 83±3% of the control current after a 4 min hyperpolarizing step. The time course of recovery was fitted with a single exponential function yielding a mean τ of 7.1 s (95% CI 5.9–8.5 s, n=7, Figure 4c,d).
Figure 4.

Recovery from channel blockade following cisapride 10 nM. A 500 ms voltage step to +25 mV was followed by a step back to −95 mV of variable duration (0.5–240 s), then tail currents were elicited by an identical second depolarizing step before (a) and after (b) bath exchange with cisapride 10 nM. (c) Peak tail currents under control conditions and after cisapride were plotted as a function of the duration of the hyperpolarizing pulse. (d) Relative current expressed as a function of the length of the hyperpolarizing interpulse demonstrated a slow recovery, which was fitted with a single exponential function (τ 7.1 s, n=7).
Modulation of channel kinetics by cisapride
HERG channel blockade by cisapride exhibited time, voltage- and frequency-dependence, which suggested greatest affinity for either open or inactivated channel states. We studied the effects of cisapride 10 nM on the time course of channel activation, deactivation and inactivation. Activation time constants were obtained by fitting a single exponential function to activating currents as seen in Figure 1. Time constants of deactivation were obtained by fitting a double exponential function to the deactivating tail currents as seen in Figure 1. Cisapride did not significantly change the time course of channel activation or deactivation at any concentration or potential.
A ‘dual-pulse' protocol (Smith et al., 1996; Spector et al., 1996) was used to assess channel inactivation. Currents were recorded after a 500 ms depolarization to +25 mV from a holding potential of −75 mV, followed by a 20 ms hyperpolarizing step to −125 mV to relieve rapid inactivation, then a second depolarization to potentials between −35 and +25 mV. The time course for the onset of inactivation was determined by fitting a single exponential function to the tail current elicited after the second depolarizing step (Figure 5a, inset). In the presence of cisapride 10 nM, there was a significant acceleration in the time course of current inactivation at most potentials (Figure 5a). At −25 mV, the time constants for inactivation were 14.5±1.1 ms (control), 11.8±0.4 ms (cisapride 10 nM), and 7.2±1.6 ms (cisapride 10 μM) (P<0.05, n=3–8). Recovery from inactivation was determined by fitting a single exponential function to the initial ‘hook' preceding slower deactivation of tail currents elicited by stepping to potentials between −115 and −15 mV following a 500 ms depolarization to +25 mV (Figure 5b, inset). After cisapride, there was no significant change in the time course of recovery from inactivation at any potential (Figure 5b).
Figure 5.

Modulation of the time course of inactivation and recovery from inactivation by cisapride 10 nM. (a) Currents were recorded after a 500 ms depolarization to +25 mV, followed by a 20 ms hyperpolarizing step to −125 mV to relieve rapid inactivation, then a second depolarization to potentials between −35 and +25 mV (inset). Inactivation time constants were determined by fitting a single exponential function to the tail currents elicited by the second depolarizing step. Time constants of inactivation were accelerated in the presence of cisapride at most potentials. (b) Recovery from inactivation was determined by fitting a single exponential function to the initial ‘hook' preceding slower deactivation of tail currents elicited by stepping to potentials between −115 and −15 mV following a 500 ms depolarization to +25 mV (inset). After superfusion with cisapride, there was no change in the time course of recovery from inactivation for potentials in the physiological range (n=8). (c) Voltage-dependence of steady state inactivation. Following a 1 s step to +30 mV, 20 ms pulses were applied to potentials between −120 and +40 mV, followed by a second 1 s step to +30 mV. Current amplitudes elicited by the second step to +30 mV were plotted as a function of the potential of the preceding 20 ms step and were fitted with a Boltzmann function. After cisapride 10 nM, there was a ∼7 mV shift in the voltage-dependence of steady state inactivation (V1/2 −65.6±0.9 mV in controls and −73.4±1.0 mV after cisapride, P<0.05, n=4). Slope factors did not differ significantly between traces.
To determine the effects of cisapride on the voltage dependence of steady state inactivation, we used a protocol similar to that initially described by Smith et al. (1996). Following a 1 s step to +30 mV, 20 ms pulses were applied to potentials between −120 and +40 mV, followed by a second 1 s step to +30 mV. The amplitude of the currents elicited by the second step to +30 mV was plotted as a function of the potential of the preceding 20 ms step and data points were fitted with Boltzmann functions. After cisapride 10 nM, there was a shift in the voltage-dependence of half maximal inactivation from −65.6±0.9 mV in controls to −73.4±1.0 mV after cisapride (P<0.01, n=4), without a significant change to the slope factor (−16.6±0.8 in controls and −15.7±0.9 after cisapride, Figure 5c).
We further studied the state-dependence of channel blockade using a protocol similar to that described by Kiehn et al. (1996). After superfusion with cisapride 100 nM for 2 min, cells were depolarized to 0 mV for 5 s from a holding potential of −80 mV, then to +80 mV for another 5 s, followed by a pulse to 0 mV for 5 s. The representative trace in Figure 6 shows that current decay in the presence of cisapride started during the initial step to 0 mV, at which channels would exist in open and inactivated states. Current inhibition continued to develop during the step to +80 mV, during which virtually all channels would be inactivated, without a significant delay imposed on current decay. Similar results were obtained in another four different cells.
Figure 6.
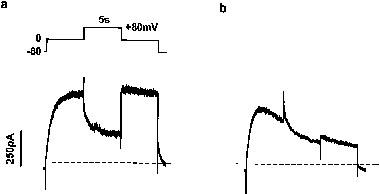
Effect of channel rectification on drug binding. Cells were depolarized to 0 mV for 5 s from a holding potential of −80 mV, then to +80 mV for another 5 s to induce full inactivation, followed by a pulse to 0 mV for 5 s. Current traces under control conditions (a) and after superfusion with cisapride 100 nM (b) are demonstrated. Interrupted lines represent single exponential fits to current traces before and after the step to +80 mV. After cisapride, current decay continued during the intervening step to +80 mV, without significant delay.
Effect of external potassium concentration on block by cisapride
HERG channel inactivation, recovery from inactivation and voltage dependence of inactivation are known to be highly sensitive to [K+]o (Wang et al., 1997a). Several recent reports have shown that blockade of HERG channels and/or IKr by quinidine or dofetilide is enhanced by low [K+] in the superfusate and reduced by high [K+] (Yang & Roden 1996; Wang et al., 1997b; West et al., 1997). We therefore assessed the effect of [K+]o on HERG channel blockade by cisapride 30 nM. At this concentration there was a modest, but statistically significant effect seen, with current inhibition decreasing from 65±3% in 1 mM [K+]o to 51±3% in 10 mM [K+]o (P<0.05, n=7).
Discussion
We have confirmed that cisapride blocks HERG currents in a chronically transfected CHO-K1 cell model with similar potency (IC50 16.4 nM), to that recently reported for inhibition of IKr in mammalian ventricular myocytes (Carlsson et al., 1997; Drolet et al., 1998) as well as HERG channels transfected into HEK 293 and mouse L cell lines (Mohammad et al., 1997; Rampe et al., 1997). The IC50 for HERG channel inhibition by cisapride is an order of magnitude lower than peak plasma concentrations seen in patients receiving therapeutic doses of the drug (150–300 nM; Wiseman & Faulds, 1994; Hofmeyr & Sonnendecker 1986). The drug is however more than 95% bound to plasma proteins which may well explain much of this discrepancy.
An alternative explanation for this difference is that temperature may influence the potency of HERG channel inhibition by cisapride. However, our experiments showed that the potency of HERG channel inhibition by cisapride was not significantly different at room temperature and at 37°C. The potency of channel blockade by an agent may also vary between native currents and those expressed in cultured cell lines. Although the effects of cisapride on human IKr have not been studied, others have reported that cisapride inhibits IKr in rabbit and guinea-pig ventricular myocytes (Carlsson et al., 1997; Drolet et al., 1998) with similar potency to inhibition of HERG channels (Mohammad et al., 1997; Rampe et al., 1997).
State dependence of HERG blockade by cisapride
Previous authors have suggested that cisapride binds to open or inactivated HERG channels on the basis of the observations that blockade required channel activation and was enhanced by prolonged depolarizing steps (Rampe et al., 1997, Mohammad et al., 1997). As well as confirming this, we found that channel inhibition by cisapride was voltage-dependent, increasing at positive potentials, and was associated with a small (∼7 mV) hyperpolarizing shift in the voltage-dependence of activation.
Use- and frequency-dependent blockade indicate greater affinity for an activated or inactivated channel state (Carmeliet & Mubagwa, 1998). We observed that channel inhibition by cisapride was frequency-dependent (degree of blockade increased 2 fold when pulse frequency rose from 0.1 to 0.5 Hz). This frequency-dependence suggests that the drug binds sufficiently slowly so as not to reach equilibrium during the first depolarizing step and that recovery from blockade takes place over a period that allows partial unbinding from the channel (Carmeliet & Mubagwa, 1998). Using an envelope of tails protocol to confirm this hypothesis, we found that the development of channel blockade occurred with a time constant of 383 ms. In Figure 4, recovery from channel blockade is shown to occur with a time constant of 7.1 s, consistent with relatively slow dissociation from the channel during the closed state.
This voltage- and frequency-dependence of HERG channel inhibition suggested that cisapride binds preferentially to open and/or inactivated channel states (Rampe et al., 1997; Toyama et al., 1997; Seussbrich et al., 1997). To further characterize this state dependence, we assessed the effects of cisapride on the kinetics of activation, deactivation and inactivation of the HERG channel. The drug had no effect on activation or deactivation, and we saw no evidence of recovery from channel blockade by cisapride during the course of tail current decay.
We did however, find evidence of cisapride interaction with the inactivated state of the channel. The time course of inactivation was accelerated by cisapride in a concentration-dependent manner at potentials within the range of normal channel activation. In addition, there was a small (∼7 mV) hyperpolarizing shift in the voltage-dependence of inactivation. This reflects a reduction in channel availability and effectively produces enhanced inward rectification (Smith et al., 1996). To further investigate drug binding to the inactivated state, HERG channels were driven into the fully inactivated state by depolarizing to +80 mV in the course of a prolonged step to 0 mV (Figure 6). Our observation that channel decay was not significantly slowed during the step to +80 mV, suggests that some of the current decay seen during these prolonged steps is due to slow development of drug binding to the inactivated channel state. Our findings were quite different from those reported for the same protocol with dofetilide (Kiehn et al., 1996; Snyders & Chaudhary, 1996). Dofetilide preferentially binds to open channels and development of block ceases during the step to +80 mV. We conclude from these observations that cisapride exhibits affinity for both open and inactivated HERG channel states.
Structure function relationships
Cisapride shares structural similarities with a number of other HERG channel blocking agents, which have in common a basic amine group bonded to a substituted phenyl ring by a variable linking group, the length of which relates directly to the potency of channel blockade (Morgan & Sullivan, 1992; Carlsson et al., 1997). Cisapride has a four atom linking group which is almost identical to the analagous group in haloperidol and similar to that of terfenadine (Figure 7), both of which are potent HERG channel blockers (Roy et al., 1996; Seussbrich et al., 1996; 1997). Mosapride, a prokinetic agent with similar pharmacodynamic properties to cisapride, has only a single carbon linking group between the amine and the phenyl ring and is a very weak blocker of the IKr channel (Carlsson et al., 1997). This structural similarity, plus the fact that haloperidol has been shown to bind selectively to the inactivated state of the HERG channel and to bind much less potently to a HERG mutant (S631A), in which the inactivation mechanism is disrupted (Seussbrich et al., 1997), supports our hypothesis that some of the HERG channel blockade due to cisapride results from binding to the inactivated state of the channel.
Figure 7.

Chemical structures of cisapride, haloperidol and terfenadine.
Sensitivity to external potassium
Finally, our observation that the potency of channel blockade by cisapride is modulated by external potassium concentration is consistent with recent reports of a similar phenomenon for dofetilide and quinidine (Yang & Roden 1996; West et al., 1997). Elevation of [K+]o destabilizes C-type inactivation, causing an increase in the time course of inactivation, as well as shifting the voltage-dependence of steady state inactivation to more depolarized potentials (Smith et al., 1996; Wang et al., 1997a). Regardless of the mechanism, our observation is of obvious clinical significance, suggesting increased sensitivity to HERG channel blockade in the presence of hypokalaemia and a possible therapeutic role for intravenous potassium in treating drug-induced QT prolongation and torsades de pointes associated with cisapride as previously reported for quinidine toxicity (Choy et al., 1997).
Conclusions
Cisapride inhibited HERG channels at a potency below the human therapeutic concentration at room temperature and at 37°C. Channel inhibition was time-, voltage- and frequency-dependent, and was accompanied by small hyperpolarizing shifts in the voltage-dependence of channel activation and inactivation. These data suggest that cisapride has affinity for both open and inactivated channel states. We have also shown that channel inhibition is modified by external potassium concentration, an effect which has obvious clinical implications.
Acknowledgments
This work was supported by research grants from the National Health and Medical Research Council of Australia, National Heart Foundation of Australia, St Vincent's Clinic and The Clive and Vera Ramaciotti Foundation.
Abbreviations
- CHO-K1
Chinese hamster ovary
- HEPES
N-2-hydroxylethylpiperazine-N1-2-ethanesulphonic acid
- HERG
human ether-a-go-go-related gene
- IC50
drug concentration producing 50% channel blockade
- IK
delayed rectifier potassium channel
- IKr
rapidly activating component of delayed rectifier potassium channel
- IKs
slowly activating component of delayed rectifier potassium channel
- V1/2
voltage of half maximal activation or inactivation
References
- AHMAD S.R., WOLFE S.M. Cisapride and torsades de pointes. Lancet. 1995;345:508. [Google Scholar]
- BARRY P.H. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neuroscience Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- BRAN S., MURRAY W.A., HIRSCH I.B., PALMER J.P. Long QT syndrome during high dose cisapride. Arch. Intern. Med. 1995;155:756–768. [Google Scholar]
- CARLSSON L., AMOS G.J., ANDERSSON B., DREWS L., DUKER G., WADSTEDT G. Electrophysiological characterization of the prokinetic agents cisapride and mosapride in vivo and in vitro: implications for proarrhythmic potential. J. Pharm. Exp. Ther. 1997;282:220–227. [PubMed] [Google Scholar]
- CARMELIET E., MUBAGWA K. Antiarrhythmic drugs and cardiac ion channels: mechanisms of action. Progress in Physics & Molecular Biology. 1998;70:1–72. doi: 10.1016/s0079-6107(98)00002-9. [DOI] [PubMed] [Google Scholar]
- CHOY A.M., LANG C.C., CHOMSKY D.M., RAYOS G.H., WILSON J.R., RODEN D.M. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149–2154. doi: 10.1161/01.cir.96.7.2149. [DOI] [PubMed] [Google Scholar]
- CURRAN M.E., SPLAWSKI I., TIMOTHY K.W., VINCENT G.M., GREEN E.D., KEATING M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- DROLET B., KHALIFA M., DALEAU P., HAMELIN B.A., TURGEON J. Block of the rapid component of the delayed rectifier potassium current by the prokinetic agent cisapride underlies drug-related lengthening of the QT interval. Circulation. 1998;97:204–210. doi: 10.1161/01.cir.97.2.204. [DOI] [PubMed] [Google Scholar]
- HOFMEYR G.J., SONNENDECKER E.W.W. Secretion of the gastrokinetic agent cisapride in human milk. Eur. J. Clin. Pharmacol. 1986;30:735–736. doi: 10.1007/BF00608226. [DOI] [PubMed] [Google Scholar]
- HOOVER C.A., CARMICHAEL J.K., NOLAN P.E., MARCUS F.I. Cardiac arrest associated with combination cisapride and itraconazole therapy. J. Cardiovasc. Pharmacol. Therapeut. 1996;1:255–258. doi: 10.1177/107424849600100309. [DOI] [PubMed] [Google Scholar]
- KIEHN J., LACERDA A.E., WIBLE B., BROWN A.M. Molecular physiology and pharmacology of HERG. Single channel currents and block by dofetilide. Circulation. 1996;94:2572–2579. doi: 10.1161/01.cir.94.10.2572. [DOI] [PubMed] [Google Scholar]
- KII Y., ITO T. Effects of 5-HT4 receptor agonists, Cisapride, Mosapride citrate, and Zacopride, on cardiac action potentials in guinea pig isolated myocytes. J. Cardiovasc. Pharm. 1997;29:670–675. doi: 10.1097/00005344-199705000-00016. [DOI] [PubMed] [Google Scholar]
- MCDONALD T.V., YU Z., MING Z., PALMA E., MEYERS M.B., WANG K.W., GOLDSTEIN S.A.N., FISHMAN G.I. A minK-HERG complex regulates the cardiac potassium current IKr. Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- MOHAMMAD S., ZHOU Z., GONG Q., JANUARY C.T. Blockade of the HERG human cardiac K+ channel by the gastrointestinal prokinetic agent cisapride. Am. J. Physiol. 1997;273:H2534–H2538. doi: 10.1152/ajpheart.1997.273.5.H2534. [DOI] [PubMed] [Google Scholar]
- MORGAN T.K., JR, SULLIVAN M.E.An overview of class III electrophysiological agents: A new generation of antiarrhythmic therapy Progress in Medicinal Chemistry 1992Amsterdam: Elsevier Science Publishers; 65–108.ed. Ellis, G.P. and Luscomb, D.K. pp [DOI] [PubMed] [Google Scholar]
- PUISIEUX F.L., ADAMANTIDIS M.M., DUMONTIER B.M., DUPUIS B.A. Cisapride-induced prolongation of cardiac action potential and early afterdepolarisations in rabbit Purkinje fibres. Br. J. Pharmacol. 1996;117:1377–1379. doi: 10.1111/j.1476-5381.1996.tb15295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMPE D., ROY M.L., DENNIS A., BROWN A. A mechanism for the proarrhythmic effects of cisapride (Propulsid): high affinity blockade of the human cardiac potassium channel HERG. FEBS Lett. 1997;417:28–32. doi: 10.1016/s0014-5793(97)01249-0. [DOI] [PubMed] [Google Scholar]
- ROY M.L., DUMAINE R., BROWN A.M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of cardiac delayed rectifier K+ current. J. Gen. Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEUSSBRICH H., SCHONHERR R., HEINEMANN S.H., ATTALI B., BUSCH A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in xenopus oocytes. Br. J. Pharmacol. 1997;120:968–974. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEUSSBRICH H., WALDEGGAR S., LANG F., BUSCH A.E. Blockade of HERG channels expressed in xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- SMITH P.L., BAUKROWITZ T., YELLEN G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., CHAUDHARY A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. J. Pharmacol. Exp. Ther. 1996;49:949–955. [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M., ZHOU A., KEATING M.T., SANGUINETTI M.C. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOYAMA J., KAMIYA K., CHENG J., LEE J.K., SUZUKI R., KODAMA I. Vesnarinone prolongs action potential duration without reverse frequency dependence in rabbit ventricular muscle by blocking the delayed rectifier K+ current. Circulation. 1997;96:3696–3703. doi: 10.1161/01.cir.96.10.3696. [DOI] [PubMed] [Google Scholar]
- UNITED STATES FOOD AND DRUG ADMINISTRATION 1998. Talk paper. June 29
- WALKER B.D., VALENZUELA S.M., SINGLETON C.B., TIE H., BURSILL J.A., WYSE K.R., QIU M.R., BREIT S.N., CAMPBELL T.J. Inhibition of HERG channels stably expressed in a mammalian cell line by the antianginal agent perhexiline maleate. Br. J. Pharmacol. 1999;127:243–251. doi: 10.1038/sj.bjp.0702502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S., LIU S., MORALES M., STRAUSS H.C., RASMUSSON R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus Oocytes. J. Physiol. 1997a;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S., MORALES M.J., LIU S., STRAUSS H.C., RASMUSSON R.L. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett. 1997b;417:43–47. doi: 10.1016/s0014-5793(97)01245-3. [DOI] [PubMed] [Google Scholar]
- WEST P.D., MARTIN D.K., BURSILL J.A., WYSE K.R., CAMPBELL T.J. Modulation of the electrophysiologic actions of E-4031 and dofetilide by hyperkalaemia and acidosis in rabbit ventricular myocytes. J. Cardiovasc. Pharmacol. Therapeut. 1997;2:205–212. doi: 10.1177/107424849700200307. [DOI] [PubMed] [Google Scholar]
- WISEMAN L.R., FAULDS D. Cisapride: an update review of its pharmacology and therapeutic efficacy as a prokinetic agent in gastrointestinal motility disorders. Drugs. 1994;47:116–152. doi: 10.2165/00003495-199447010-00008. [DOI] [PubMed] [Google Scholar]
- WYSOWSKI D.K., BACSANYI J. Cisapride and fatal arrhythmias. N. Engl. J. Med. 1996;335:290–291. doi: 10.1056/NEJM199607253350416. [DOI] [PubMed] [Google Scholar]
- YANG T., RODEN D.M. Extracellular potassium modulation of drug block of IKr: implications for torsades de pointes and reverse use-dependence. Circulation. 1996;93:407–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]