

Abstract
Various classes of nitrovasodilators release nitric oxide (NO) through distinct reaction pathways, many of which involve endogenous reductants and/or oxidants. This study examined relaxations of isolated rat coronary arteries induced by spermine NONOate (SPNO), 3-morpholinosydnonimine (SIN-1), nitroprusside (NP), S-nitroso-N-acetylpenicillamine (SNAP) and nitroglycerin (NTG) in order to assess whether their potency was influenced by any of six redox compounds: 1 mM ascorbate, 1 mM dehydroascorbate, 0.1 mM dithiothreitol, 10 μM diamide, 0.1 mM ferrocyanide, and 0.1 mM ferricyanide.
Only SPNO spontaneously generated NO at measurable levels. These levels were decreased by the presence of ascorbate and dithiothreitol, which likewise decreased the potency of SPNO.
The potency of SIN-1 was unaffected by any redox compound except ferricyanide, which increased the potency not only of SIN-1, but also of other nitrovasodilators and NO-independent vasodilators.
The potency of NP was decreased by two structurally similar multivalent anions, ferrocyanide and ferricyanide, suggesting that NP metabolism requires ionic binding to tissue.
SNAP lost its potency in solutions containing ascorbate or dehydroascorbate. SNAP potency was also decreased by the glutathione oxidant, diamide, and by ferrocyanide and ferricyanide, suggesting that glutathione and ionic binding may be required for NO release.
NTG appeared to relax arteries via two pathways. One required only low concentrations of NTG and a labile endogenous factor that was preserved by dithiothreitol and eliminated by ferricyanide. A distinct second pathway required higher concentrations of NTG.
These distinct attributes of nitrovasodilator metabolism may underlie differences in regional specificity or tolerance development, and therefore might eventually be exploited in the development and use of nitrovasodilators.
Keywords: Ascorbate, coronary arteries, diamide, ferricyanide, glutathione, nitric oxide, nitroglycerin, nitroprusside, S-nitroso-N-acetylpenicillamine
Introduction
Nitric oxide is endogenously synthesized as a potent vasodilator, and is likewise generated from a number of clinically important compounds referred to as nitrovasodilators. Nitroglycerin (NTG) is the prototypical example, and for decades has remained a useful therapy against angina. Remarkably, there is not yet agreement on the chemical pathway by which NTG releases NO. Considerable evidence suggests that cellular thiols are critical (Ignarro & Gruetter, 1980; Gruetter & Lemke, 1986; Fung & Bauer, 1994; Marks et al., 1995; Haj-Yehia & Benet, 1996), but there is disagreement about their identity (Feelisch & Noack, 1987a,1987b; Hutter et al., 1988; Wheatley et al., 1994; Haj-Yehia & Benet, 1996), the role of enzymes (Feelisch & Noack, 1987a; Schroder, 1992; Seth & Fung, 1993; Kurz et al., 1993; Chung & Fung, 1993; Salvemini et al., 1993; Singhal et al., 1996), and the nature of any intermediates (Marks et al., 1995). The uncertainty about NTG metabolism has led to multiple explanations for the phenomenon of tolerance (Fung & Bauer, 1994; Laursen et al., 1996), and for the regional specificity of NTG-induced vasodilation (Wheatley et al., 1994; MacAllister et al., 1995; Khan et al., 1998).
In an effort to avoid tolerance and attain regional specificity, other chemical classes of nitrovasodilators have been identified and clinically tested. Yet even in these cases the mechanism of NO release is also not always clear. Spermine NONOate (SPNO) is an example of the NONOate complexes that form between amines and NO. These compounds are thermolabile, and spontaneously release two molecules of NO in aqueous solutions (Morley & Keefer, 1993; Ioannidis et al., 1996). Although redox reactions are not required for NO release from SPNO, it is possible that the rate of NO release may be modulated by redox reactions or other processes in vivo. 3-Morpholinosydnonimine (SIN-1) is a sydnonimine, and is thought to undergo a 1 electron oxidation (via a direct reaction with oxygen, if available), followed by spontaneous release of NO (Feelisch et al., 1989). Other oxidants can also initiate NO release from SIN-1 (Bohn & Schonafinger, 1989; Bozinovski et al., 1994), but it is unclear to what extent NO release in vivo is initiated by oxygen vs endogenous oxidants (Schror et al., 1991). Nitroprusside (NP) is a multivalent anion thought to require a 1 electron reduction in order to initiate NO release, but it is unclear which tissue component(s) mediates the reduction (Lad et al., 1981; Bates et al., 1991; Rochelle et al., 1994; Kleschyov et al., 1994; Rao & Cederbaum, 1995). S-Nitroso-N-acetylpenicillamine (SNAP) is an example of a nitrosothiol. Nitrosothiols are interconvertable through transnitrosation reactions, and may be endogenous intermediates in NO storage and transport (Ignarro & Gruetter, 1980; Arnelle & Stamler, 1995; Singh et al., 1996). It is unlikely that nitrosothiols release NO through spontaneous homolytic cleavage (Kowaluk & Fung, 1990; Mathews & Kerr, 1993), rather, release may be driven by reactions with enzymes (Gordge et al., 1998), with copper (De Man et al., 1996; Singh et al., 1996; Al-Sa'doni et al., 1997), and/or with endogenous reductants such as ascorbate (Henry et al., 1989; Arnelle & Stamler, 1995; Arnelle et al., 1997; Kashiba-Iwatsuki et al., 1997; Scorza et al., 1997).
In light of these uncertainties concerning the nature and importance of tissue-dependent redox reactions in promoting the release of NO from these five classes of nitrovasodilators, a systematic study was designed in which six redox compounds were tested for their influence on the nitrovasodilator-induced relaxations of isolated coronary arteries. The redox compounds were: ascorbate (ASC), an endogenous reductant; dehydroascorbate (DHAA), the oxidized form of ASC; dithiothreitol (DTT), a dithiol reductant; diamide, a compound which oxidizes glutathione to glutathione disulphide; ferricyanide (Fe3+(CN−)6), an impermeable multivalent anionic oxidant; and ferrocyanide (Fe2+(CN−)6), a multivalent anionic reductant. The current study simultaneously assessed the effect of these redox compounds on steady-state levels of NO released by the nitrovasodilators in solution, and also assessed the effect of the redox compounds on general aspects of vasoreactivity, including maximal vasoconstrictions and NO-independent vasorelaxations. The results identified several general effects of redox compounds, and also several interactions between specific redox compounds and nitrovasodilators. The pattern of these interactions gives clues towards the distinct pathways of nitrovasodilator metabolism in this arterial tissue.
Methods
Sources of arteries
Male Sprague-Dawley rats were obtained from Taconic, and housed with free access to water and standard laboratory rat chow. Animals between 300–500 g (10–15 weeks of age) were killed with sodium pentobarbital (35 mg kg−1 i.p.). The heart was removed and placed in 4°C physiological salt solution (PSS) with the composition (in mM): NaCl 118.5; KCl 4.7, KH2PO4 1.2; MgSO4 1.18; NaHCO3 25; Na2EDTA 0.023; CaCl2 1.6; glucose 11; saturated with 95% O2, 5% CO2; and adjusted to pH 7.4. The septal coronary artery was accessed through the right ventricle, isolated and cleaned free of connective tissue. All experiments involving the use of animals were approved by the Institutional Animal Care and Use Committee.
Drugs
Nitroprusside, spermine/nitric oxide complex and S-nitroso-N-acetylpenicillamine were from Research Biochemicals International. 3-morpholinosydnonimine was a gift from Cassella AG. Nitroglycerin was from American Regent Laboratories, Inc. Dehydroascorbic acid was from Aldrich. Diltiazem, forskolin, all other redox compounds and all other reagents were from Sigma.
Preparation of arteries
Segments of the septal artery were mounted in a myograph as previously described (Murphy & Brayden, 1995). Each artery (∼1.5 mm long, ∼300 μm diameter) was mounted using two fine intraluminal wires, and incrementally stretched using a force equivalent to 100 mg per mm artery length. Output data from a force transducer was displayed and stored using PolyView software (Grass Instruments, Inc.). The vessel was superfused with oxygenated PSS at a rate of ∼10 ml min−1, which passed through a water-jacketed heating loop (to maintain 37°C in the chamber). Superfusate was simultaneously removed by a vacuum aspirator at the opposite corner of the chamber. Two criteria were established for acceptance of an artery: (1) an additional constriction of at least 100 mg per mm artery length in the presence of 50 mM KCl; (2) relaxation of at least 50% (of the KCl-induced constriction) in the presence of 10 μM SPNO. Approximately 80% of arteries met these two criteria; other arteries were discarded from the study. Pilot studies showed that arteries retained functional endothelium, but also showed that endothelium removal did not affect responses to nitrovasodilators.
Vasorelaxation protocols
Vasorelaxations represent a reversal of constriction induced by KCl (50 mM, using isosmotic substitution of NaCl), which induced a 2–10 fold increase in force. All superfusate buffers contained both indomethacin (10 μM; to inhibit cyclo-oxygenase) and N-nitro-L-arginine (100 μM; to inhibit NO synthase). Ascorbate, dehydroascorbate and beta-mercaptoethanol were added directly into the superfusate. Other vasodilators and redox compounds were prepared as stock solutions in distilled water (or DMSO in the case of forskolin), and then diluted into the superfusate. When present, redox compounds were added to the PSS 15 min prior to each constriction/dilation protocol, and continued to be present in the PSS throughout the protocol. Four protocols involving one vasodilator were conducted on each artery. The four protocols randomized with respect to the redox compound tested. For each artery, one of the four protocols lacked any redox compound, and served as a control. A 45 min wash-out period was provided between protocols. During each protocol, once a stable KCl-induced pre-constriction was reached, a range of graded nitrovasodilator concentrations were added incrementally to the superfusate. Each dose of nitrovasodilator was maintained for 8–10 min, allowing sufficient time for any relaxation to stabilize. In protocols involving diltiazem and forskolin, 10–12 min were allowed for stable relaxations.
Print-outs of recordings were used to determine the baseline force applied to each artery, as well as the maximal degree of pre-constriction, and the steady-state force that was reached at each concentration of vasodilators (examples in Figure 2A and B). These force values from each artery were used to create a sigmoidal curve using Prism® software (GraphPad Software, Inc.), which calculates the maximal per cent relaxation, the EC50, and the Hill coefficient. Although curves were fitted without regard to the actual maximal pre-constriction that occurred before the addition of the vasodilator, the calculated maximal pre-constriction always fell within 5% of the actual observed value. The only exception occurred with NTG (discussed below), for which the calculated pre-relaxation asymptote value often fell significantly below the observed value of the pre-constriction that developed before the addition of the first dose of NTG.
Figure 2.
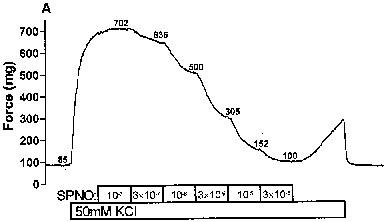
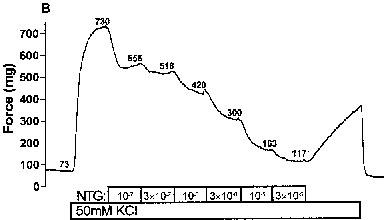
Nitrovasodilator-induced relaxations of rat coronary arteries. The force generated by rat coronary arteries mounted in a wire myograph were recorded as described in the Methods section. Pre-constrictions were induced by 50 mM KCI during the period indicated. During subsequent periods, relaxations were induced by incremental additions of: (A) spermine/nitric oxide complex (SPNO); and (B) nitroglycerin (NTG). The numbers adjacent to each trace denote the force (mg) reached in the presence of the given concentration of nitrovasodilator.
NO measurements
A ISO-NO electrode (2 mm, World Precision Instruments, Inc.) was used to measure the steady-state concentrations of NO. The electrode was calibrated on multiple occasions using anoxic NO stock solutions, with final dilutions (yielding 0.1–1 μM NO) made in 20 ml of superfusion buffer (37°C, 95% O2, 5% CO2). The electrode response (mean of 1.42 pA per nM NO) was not affected by the presence of redox agents, nor was the background current. When measuring the release of NO from nitrovasodilators, assay solutions containing various combinations of nitrovasodilators and redox compounds were prepared and handled exactly as above, but in the absence of arteries. When converting SPNO concentration to NO concentration, a linear regression equation was used to calculate the NO concentration below the electrode's threshold of detection, which was 10–30 nM NO. It should therefore be kept in mind that values for the EC50 of NO (which are <10 nM) should be considered approximate.
Statistics
Results are reported as the mean±s.e.mean. Because data values for the Hill coefficient and for the EC50 are not expected to be normally distributed, these data values were transformed logarithmically prior to the calculation of means and other statistical analyses. In the case of the Hill coefficient, this mean of the transformed values was then re-transformed (exponentially) before being reported as a mean. The symbols in Figure 3 represent the mean dilation achieved at each concentration of the vasodilator. However, the lines in Figure 3 are not fitted to those symbols, rather, the equation of each line is defined by the mean values of the maximal dilation, the Hill coefficient and the pEC50.
Figure 3.
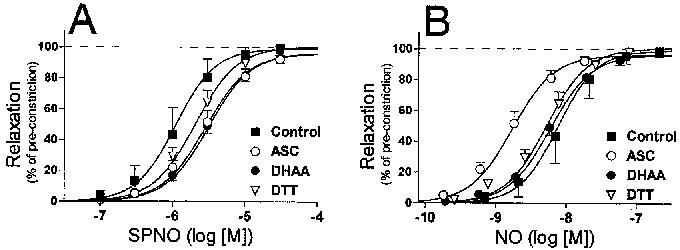
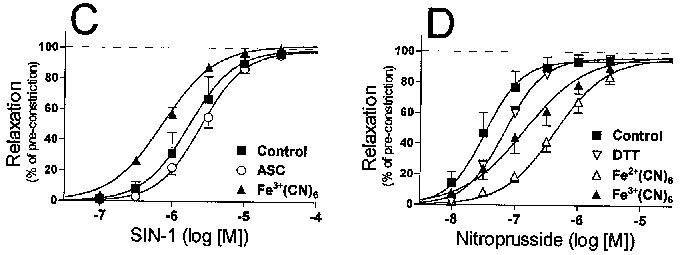
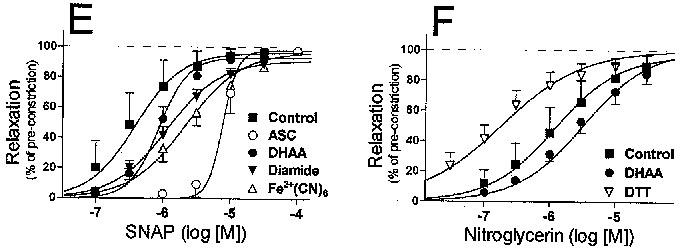
Concentration–response profiles of nitrovasodilator-induced relaxations. Relaxations were calculated as the per cent reversal of the KCI-induced pre-constriction as determined in each individual experiment. Relaxations were induced by: (A) Spermine/nitric oxide complex (SPNO); (B) nitric oxide (NO, released from SPNO); (C) 3-morpholinosydnonimine (SIN-1); (D) nitroprusside; (E) S-nitroso-N-acetylpenicillamine (SNAP); or (F) nitroglycerin (NTG). Symbols denote the mean (±s.e.mean) relaxation at each concentration of nitrovasodilator. The equation of each curve is defined by the mean values of the maximal dilation, the Hill coefficient and the pEC50.
The statistical significance of effects of redox compounds on the responses to a given vasodilator were determined based on Analysis of Variance, with subsequent post hoc analysis based on Fisher's PSLD test, using StatView® software (Abacus Concepts, Inc.). To determine the significance of the general effects of a given redox compound on all vasodilators, a two-way ANOVA was conducted. These statistical analyses were approached in two ways. Firstly, an analysis was conducted on overall mean values. However, this analysis did not account for inherent differences in the baseline responses of individual arteries. So a second analysis was conducted based on paired values, in which the response of each artery in the presence of a redox compound was first compared to a control value obtained on that day, and then these individual changes were statistically examined to determine whether redox compounds had significant effects. The tables report significance differences based on this second approach to analysis, but the significance determined by the first approach was identical with respect to all salient conclusions. Statistical significance was defined as P<0.05 for every type of analysis.
Results
When tested at concentrations of up to 100 mM in PSS at 37°C under 95% O2/5% CO2, SPNO was the only nitrovasodilator that yielded measurable steady-state concentrations of NO. The slope of log-log plots of NO concentration vs SPNO concentration averaged 0.98± 0.04 (s.e.mean, n=17; Figure 1). This slope was not significantly different than 1, indicating that NO concentration was linearly related to SPNO concentration. NO accumulated to a concentration of around 0.7% of each tested SPNO concentration. In the presence of redox compounds, the linearly of SPNO concentrations vs NO concentration was still observed, but the steady-state accumulation of NO was decreased 11.7 fold by ASC (1 mM; Figure 1), 3.6 fold by DHAA (1 mM), and 2.7 fold by DTT (0.1 mM) at any given concentration of SPNO (DHAA and DTT not shown). These decreases were significantly reversed by the addition of superoxide dismutase (SOD; 50 U ml−1), but SOD had no effect on NO levels in the absence of redox compounds. Diamide (10 μM), ferricyanide (100 μM) and ferrocyanide (100 μM) had no significant effect on the steady-state concentration of NO generated by SPNO. Measurable steady-state concentrations of NO did not accumulate in solutions of NTG, NP, or SNAP, and accumulated in solutions of SIN-1 only if SOD was also added. However, SOD was not routinely added to any superfusate solutions in the vasorelaxation experiments.
Figure 1.
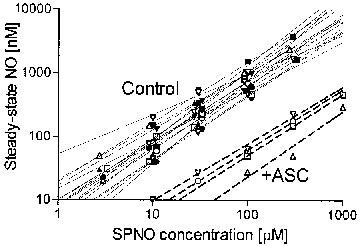
Accumulation of NO from spermine/nitric oxide complex (SPNO). Steady-state NO was measured using a NO electrode as described in the Methods section. Assay solutions contained 3, 10, 30, 100, 300 or 1000 μM SPNO (for clarity, some data points are visually offset from the axis values). Three or four points define each line, which were fit by linear regression. Solid lines (Control) represent data where redox agents were absent from the assay solution. Dashed lines (+ASC) represent data obtained with 1 mM ascorbate in the assay solution.
Isolated rat coronary arteries, pre-constricted with 50 mM KCl, relaxed in a concentration-dependent manner by the addition of nitrovasodilators, and sigmoidal concentration-response curves were created from the results (Figure 2). With the exception of NTG (discussed below), the pre-constrictions and the relaxations were fully reversible and repeatable. The response to a nitrovasodilator did not depend on whether it was tested early or late in the series of four protocols each day, or on whether any redox agents had been present in previous protocols (data not shown). Interestingly, no relaxations at all could be induced by isosorbide dinitrate, which represents a sixth class of nitrovasodilators (data not shown).
SPNO relaxed arteries with a mean EC50 of 1.1±0.1 μM (n=16). ASC (1 mM), DHAA (1 mM) and DTT (0.1 mM) significantly decreased the potency of SPNO, as defined by differences in the mean pEC50 values (Figure 3A and Table 1). However, when SPNO concentrations were converted to NO concentrations, as defined by the linear regression equations mentioned above, NO relaxed arteries with an apparent mean EC50 of 7.6±0.9 nM, and ASC appeared to increase the potency of NO per se (Figure 3B and Table 1).
Table 1.
Influence of redox compounds on pEC50 value of vasodilators in coronary arteries
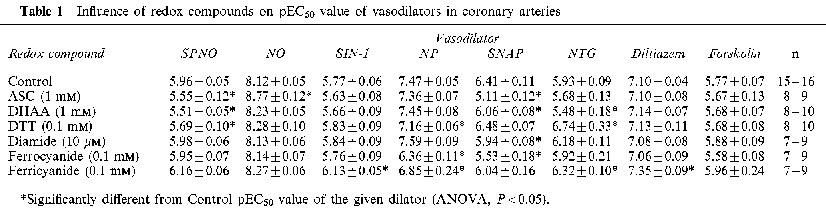
SIN-1 induced relaxations of control arteries with a mean EC50 of 1.7±0.2 μM (n=15). Ferricyanide (0.1 mM) significantly increased its potency (Figure 3C and Table 1). Nitroprusside induced relaxations with a mean EC50 of 3.4±0.4 μM (n=15). The potency of NP was decreased by the presence of DTT, and even more so by ferricyanide and ferrocyanide (0.1 mM) (Figure 3D and Table 1). SNAP induced relaxations with a mean EC50 of 0.39±0.10 μM (n=17). Except for DTT, every redox compound decreased the potency of SNAP, with 1 mM ASC causing a particularly significant 20 fold decrease in the potency (Figure 3E and Table 1). The affects of ASC and DHAA on SNAP potency were accompanied by a significant increase in the Hill coefficient (control, 1.26±.07; ASC, 6.31±1.41; DHAA, 2.00±.24; DTT 1.86±.23).
Relaxations induced by NTG displayed complex profiles that suggested two pathways by which NTG released NO. One pathway appeared to cause an unusually strong initial relaxation to the first time the artery was exposed to even the lowest dose of NTG (example in Figure 2B). This initial relaxation was significantly attenuated during the second dose-response protocol each day, and even more so during the third and fourth protocols (Figure 4; runs 1–4). This loss in sensitivity to the lowest dose of NTG also occurred in the presence of redox compounds, except for two: DTT, which preserved the initial relaxation even until the fourth NTG protocol of the day; and ferricyanide, which significantly attenuated the initial relaxation even during the first NTG protocol (Figure 4). It must be noted that the calculation of concentration-response curves (see Methods section) minimized or eliminated the influence of this initial relaxation on the responses illustrated in Figure 3F and reported in Table 1. Those calculated curves suggest a second pathway of NO release from NTG, which causes a complete relaxation at a higher range of doses of NTG (mean EC50 of 1.2±0.2 μM, n=17). The potency of NTG at these higher doses did not decrease over the course of a day (P>0.05). Also, this relaxation at higher NTG doses was affected by redox compounds in a pattern that differed in two ways from their effects on the initial relaxation. Firstly, DTT and ferricyanide increased the potency of the higher doses of NTG (Table 1), even though they had opposite effects on the inital relaxation (Figure 4); Secondly, DHAA significantly decreased the potency of higher doses of NTG (Table 1), even though DHAA had no effect on the initial relaxation (Figure 4).
Figure 4.
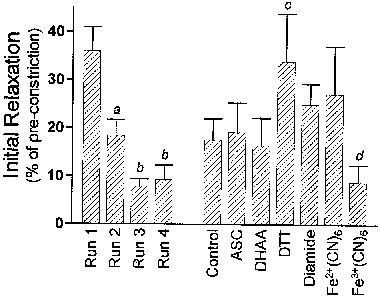
Initial relaxations induced by nitroglycerin. The initial relaxation is defined as the per cent relaxation induced by the first (lowest) concentration of nitroglycerin each time a concentration-response protocol was carried out. The lowest concentration was 3×10−8 M whenever dithiothreitol was present, and was 10−7 M in every other case. The first four columns (runs 1–4) denote the mean initial relaxation (±s.e.mean, n+48) recorded at the beginning of each of the four sequential protocols conducted on each artery, omitting data obtained in the presence of dithiothreitol and ferricyanide. The last seven columns denote the mean initial relaxation (±s.e.mean, n=7–17) recorded in the presence of each redox compound, combining data obtained from all of the four protocols conducted on each artery. aSignificantly less than Run 1; bSignificantly less than Runs 1 and 2; cSignificantly greater than Control; dSignificantly less than Control.
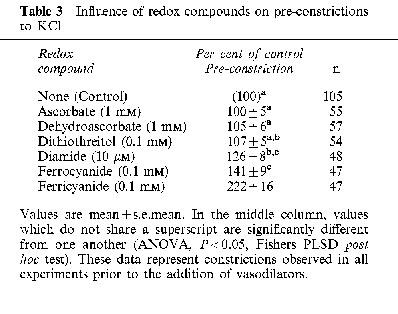
Relaxations were induced by diltiazem, a calcium channel blocker, with a mean EC50 of 79±7 nM (n=17), and this potency was not significantly affected by the redox compounds, except for an increase due to ferricyanide (Table 1). Likewise, relaxations were induced by forskolin, which stimulates adenylyl cyclase and cyclic AMP accumulation, with a mean EC50 of 1.7±0.3 μM (n=9), and this potency was unaffected by all redox compounds (Table 1). The maximal degree of relaxation induced by either diltiazem or forskolin was significantly greater than the maximal relaxation induced by the nitrovasodilators (Table 2). The maximal pre-constriction induced by KCl was significantly potentiated by ferricyanide, was potentiated to a lesser extent by diamide and ferrocyanide, but was not significantly affected by the other redox compounds (Table 3).
Table 2.
Maximal relaxations induced by vasodilators
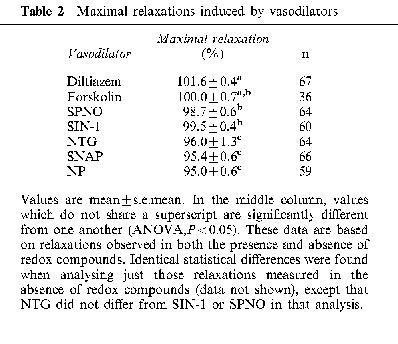
Table 3.
Influence of redox compounds on pre-constrictions to KCl
Discussion
Only SPNO appeared to spontaneously generate NO at levels apparently capable of relaxing arteries. It seems unlikely, or at least unnecessary, that the rate of spontaneous NO release is significantly influenced by arterial tissue, since our value of 7.6 nM for the EC50 of NO (released from SPNO in the absence of redox compounds) is consistent with previous estimates of the potency of NO (Khan et al., 1998). It should be noted, though, that our EC50 value is somewhat approximate, since it is based on extrapolations below the detection limit of the NO electrode. Other reports have also concluded that SPNO does not undergo tissue-dependent metabolism (Maragos et al., 1991; Morley & Keefer, 1993). In contrast, the lack of measurable levels of NO in solutions containing NP, NTG and SNAP confirms expectations that the release of NO from these three nitrovasodilators absolutely requires tissue-dependent metabolism. It is interesting to note that the three nitrovasodilators that require metabolism (NP, NTG and SNAP) consistently led to less maximal relaxation than SPNO or SIN-1, and that all five of these nitrovasodilators led to less maximal relaxation than the calcium channel blocker, diltiazem (Table 2). The reason for these differences is not yet clear.
Even though SIN-1 is known to spontaneously release NO (Feelisch et al., 1989), NO did not accumulate to measurable concentrations in solutions containing SIN-1, even at levels of SIN-1 far greater than needed to relax arteries. One explanation is that peroxynitrite, generated by SIN-1, is the actual vasorelaxant, acting via a glutathione-dependent intracellular generation of NO (Wu et al., 1994; Mayer et al., 1995; Schrammel et al., 1998). However, the lack of inhibition by diamide (Table 1) argues against this possibility. A second explanation is that arterial tissue may catalyze the initial oxidation of SIN-1, and accelerate local NO release, but there is no evidence for or against this possibility. A third explanation is based on the likelihood that the accumulation of NO is limited by superoxide simultaneously generated in solutions of SIN-1. Arterial tissue contains both cytosolic and extracellular SOD, and these particular arteries are thick enough to allow a gradient of NO concentration to exist. Therefore, even if SIN-1 generates NO within the artery merely at the same rate as in bulk solution, it is possible that the NO is locally protected from destruction, and accumulates to active levels.
Redox compounds had significant effects on three general aspects of vasoreactivity. The first is that diamide, ferrocyanide, and particularly ferricyanide, potentiated KCl-induced pre-constrictions (Table 3). The second is that diamide consistently diminished the maximal relaxation elicited by all vasodilators, and the effect was significant in the case of NP, SNAP and forskolin (Figure 3E and other data not shown). A third effect is that ferricyanide consistently increased the potency of vasodilators (Table 1), with the exception of NP and SNAP, which probably had additional specific ionic interactions with ferricyanide (see below). These three general effects of redox compounds on vasoreactivity have not been previously reported, and their mechanism is not yet clear. Nevertheless, these effects need to be kept in mind for the interpretation of the apparent interactions between redox compounds and specific nitrovasodilators.
ASC, DHAA and DTT caused a specific, significant decrease in the potency of SPNO. This likely involves the generation of superoxide, which reacts with NO to form peroxynitrite, and thus decrease the steady-state concentrations of NO in solutions containing redox compounds that generate superoxide (Moncada et al., 1986). However, once this decrease was accounted for by directly measuring NO, the results indicated that ASC significantly increased the potency of NO per se. One possible explanation (discussed above for SIN-1) is that the local concentration of NO within arteries, where it is protected from superoxide, is actually higher than in the bulk PSS, such that the electrode data leads to an overestimation of the potency of NO. An additional explanation is that intra-arterial concentrations of NO are elevated through an ASC-driven intracellular generation of NO from nitrosothiols (Henry et al., 1989; Kashiba-Iwatsuki et al., 1997; Scorza et al., 1997), which form when cellular thiols react with the peroxynitrite generated in the bulk solution (Wu et al., 1994; Mayer et al., 1995). However, the inability of ASC to potentiate SIN-1, which also produces peroxynitrite, argues against this possibility. A third explanation is that ASC enhances tissue responsiveness to NO. Reductants are known to optimize guanylyl cyclase activity (Craven & DeRubertis, 1978; Lad et al., 1981), and ASC can potentiate SPNO-induced cGMP accumulation in monolayer cultures of vascular smooth muscle cells, where gradients of NO are less likely to exist (unpublished observations). However, the inability of ASC to potentiate SIN-1, NP or NTG (Table 1) argues against a general potentiation of guanylyl cyclase. Further investigations examining the effects of superoxide, SOD, peroxynitrite and other compounds are needed to resolve these issues.
It is unclear why DTT decreased the potency of NP. Although one report suggested that DTT decreased responses to NP (Rapoport et al., 1985), it is more generally thought that reductants like DTT accelerate the release of NO from NP (Lad et al., 1981; Bates et al., 1991; Rochelle et al., 1994; Kleschyov et al., 1994; Rao & Cederbaum, 1995). One possibility is that DTT does accelerate NP breakdown, but that any NO is immediately consumed in a reaction with superoxide, so that the net effect is merely a lower concentration of NP in solution. Additional work is needed to resolve this. On the other hand, the significant decrease of NP potency in the presence of ferricyanide and ferrocyanide can be easily explained by competitive antagonism of ionic binding. NP is an impermeable multivalent anion, and some evidence suggests that NP generates NO extracellularly (Kleschyov et al., 1994). Although it is attractive to speculate that a reduction of NP and a release of NO occurs at this proposed extracellular binding site, this idea is not supported by our observation that NP was inhibited by ferrocyanide, a reductant, just as effectively as by ferricyanide, an oxidant which might have consumed reducing equivalents on the cell surfaces. Other experiments would be needed to determine whether the anions, particularly endogenous anions, may also inhibit NP-induced relaxations, and whether regional differences in the density of such binding sites underlies the regional specificity of NP.
Several studies show that SNAP is degraded through reactions involving ASC (Henry et al., 1989; Singh et al., 1996; Kashiba-Iwatsuki et al., 1997; Scorza et al., 1997), and the resulting rapid depletion of SNAP likely accounts for the apparent loss in SNAP potency in the presence of ASC (Figure 3E). The loss in SNAP potency due to DHAA had a similar profile (i.e., a significantly increased Hill coefficient) suggesting that it occurs through the same mechanism as ASC. It is not yet clear, though, whether SNAP is destroyed because a trace contamination of ASC exists in the presence of DHAA, or because ASC and DHAA are both converted to the same critical reactive intermediate. The data also suggest that the tissue-dependent metabolism may require extracellular binding of SNAP (which exists as an anion at pH 7), since its potency was diminished equivalently by both ferricyanide and ferrocyanide, reminiscent of how these two impermeable multivalent anions decreased the potency of NP. The decrease in SNAP potency due to diamide further suggests that the tissue-dependent release of NO from SNAP requires glutathione. This is consistent with a report that diamide diminished nitrosothiol metabolism in fibroblasts (Gordge et al., 1998). It is noteworthy, in contrast, that diamide tended to increase the potency of all other nitrovasodilators (Table 1). This increase not only reinforces the conclusion that thiols like glutathione are not critical to NO-induced relaxations in general (Ignarro & Gruetter, 1980), but is also consistent with the evidence that diamide may actually increase the sensitivity of guanylyl cyclase to NO (Wu et al., 1992).
The data suggest that one pathway of NO release from NTG, which accounts for the initial relaxation to low doses of NTG, requires a limited labile cell component that efficiently promotes NO release from NTG. The effects DTT on initial relaxations suggests that DTT preserves or repletes that component. The likelihood that this cell component is a reductant is also supported by the observation that ferricyanide eliminated this initial relaxation, whereas ferrocyanide partially preserved it. However, even though glutathione has been implicated in NTG metabolism in canine (Wheatley et al., 1994) and bovine (Chung & Fung, 1993) coronary arteries, the lack of inhibition by diamide indicates that glutathione is not required for this first pathway in rat arteries. The data also suggest that a second pathway of NO release from NTG requires a cell component that is either more abundant or less labile, but which is inhibited by DHAA. Although one known effect of DHAA is to oxidize glutathione, the lack of effect of diamide on the potency of higher doses of NTG suggests that glutathione is not involved in this effect of DHAA. It is unclear whether the potentiation of both low and high doses of NTG by DTT occurs through two distinct mechanisms, or only one.
The evidence that rat coronary arteries support two pathways of NTG metabolism clearly highlights the need for caution when interpreting in vitro studies. The half-maximal effective concentration of NTG in most types of arteries is <100 nM, suggesting that the first pathway of NTG metabolism in rat coronary arteries, rather than the second, is the relevant route in vivo. This would diminish the relevance of any conclusions about NTG metabolism that could be drawn from Figure 3F. In contrast, the EC50 values of every other nitrovasodilator examined in this study fall in the approximate range of clinically effective concentrations, supporting the relevance of those data.
In summary, redox compounds interacted in several specific ways with nitrovasodilators, which lead to the following conclusions: (1) the rate of NO degradation in solution may be uniquely relevant to SPNO because it is the only nitrovasodilator which spontaneously generates active concentrations of NO in solution; (2) redox compounds have little influence on relaxations induced by SIN-1 because it does not require metabolism, and its simultaneous generation of superoxide becomes irrelevant to the actions of the NO it generates within tissue; (3) thiols do not drive NO release from NP in tissue, but ionic binding of NP to tissue is required; (4) SNAP is highly susceptible to degradation by ASC, requires glutathione for release of NO, and requires ionic binding for metabolism; and (5) NTG is metabolized by two pathways in tissue, one is a high-affinity and requires a labile reductant, the other is low affinity, and requires a different reductant which is not glutathione. These distinct features of nitrovasodilator metabolism should prove useful in further investigations into the basis of regional specificity and tolerance to nitrovasodilators.
Acknowledgments
This work was supported by a Grant-in-Aid from the American Heart Association (960130). The author thanks Katherine Marciniszyn and Beth Zigmund for excellent technical assistance.
Abbreviations
- ASC
ascorbate
- DHAA
dehydroascorbic acid
- DTT
dithiothreitol
- NP
nitroprusside
- NTG
nitroglycerin
- PSS
physiological saline solution
- SIN-1
3-morpholinosydnonimine
- SNAP
S-nitroso-N-acetylpenicillamine
- SPNO
spermine/nitric oxide complex
References
- AL-SA'DONI H.H., MEGSON I.L., BISLAND S., BUTLER A.R., FLITNEY F.W. Neocuproine, a selective Cu(I) chelator, and the relaxation of rat vascular smooth muscle by S-nitrosothiols. Br. J. Pharmacol. 1997;121:1047–1050. doi: 10.1038/sj.bjp.0701218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNELLE D.R., DAY B.J., STAMLER J.S. Diethyl dithiocarbamate-induced decomposition of S-nitrosothiols. Nitric Oxide. 1997;1:56–64. doi: 10.1006/niox.1996.0107. [DOI] [PubMed] [Google Scholar]
- ARNELLE D.R., STAMLER J.S. NO+, NO, and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch. Biochem. Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- BATES J.N., BAKER M.T., GUERRA R., JR, HARRISON D.G. Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem. Pharmacol. 1991;42 Suppl.:S157–S165. doi: 10.1016/0006-2952(91)90406-u. [DOI] [PubMed] [Google Scholar]
- BOHN H., SCHONAFINGER K. Oxygen and oxidation promote the release of nitric oxide from sydnonimines. J. Cardiovasc. Pharmacol. 1989;14 Suppl. 11:S6–S12. [PubMed] [Google Scholar]
- BOZINOVSKI J., BRIEN J.F., MARKS G.S., NAKATSU K. Inhibition of nitrovasodilators by pyocyanin and methylene blue is dissociated from nitric oxide formation. Can. J. Physiol. Pharmacol. 1994;72:746–752. doi: 10.1139/y94-106. [DOI] [PubMed] [Google Scholar]
- CHUNG S.J., FUNG H.L. Relationship between nitroglycerin-induced vascular relaxation and nitric oxide production. Probes with inhibitors and tolerance development. Biochem. Pharmacol. 1993;45:157–163. doi: 10.1016/0006-2952(93)90388-d. [DOI] [PubMed] [Google Scholar]
- CRAVEN P.A., DERUBERTIS F.R. Restoration of the responsiveness of purified guanylate cyclase to nitrosoguanidine, nitric oxide, and related activators by heme and hemeproteins. Evidence for involvement of the paramagnetic nitrosyl-heme complex in enzyme activation. J. Biol. Chem. 1978;253:8433–8443. [PubMed] [Google Scholar]
- DE MAN J.G., DE WINTER B.Y., BOECKXSTAENS G.E., HERMAN A.G., PELCKMANS P.A. Effect of Cu2+ on relaxations to the nitrergic neurotransmitter, NO and S-nitrosothiols in the rat gastric fundus. Br. J. Pharmacol. 1996;119:990–996. doi: 10.1111/j.1476-5381.1996.tb15769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEELISCH M., NOACK E. Nitric oxide (NO) formation from nitrovasodilators occurs independently of hemoglobin or non-heme iron. Eur. J. Pharmacol. 1987a;142:465–469. doi: 10.1016/0014-2999(87)90090-2. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., NOACK E.A. Correlation between nitric oxide formation during degradation of organic nitrates and activation of guanylate cyclase. Eur. J. Pharmacol. 1987b;139:19–30. doi: 10.1016/0014-2999(87)90493-6. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., OSTROWSKI J., NOACK E. On the mechanism of NO release from sydnonimines. J. Cardiovasc. Pharmacol. 1989;14 Suppl. 11:S13–S22. [PubMed] [Google Scholar]
- FUNG H.L., BAUER J.A. Mechanisms of nitrate tolerance. Cardiovas. Drugs Ther. 1994;8:489–499. doi: 10.1007/BF00877927. [DOI] [PubMed] [Google Scholar]
- GORDGE M.P., ADDIS P., NORONHA-DUTRA A.A., HOTHERSALL J.S. Cell-mediated biotransformation of S-nitrosoglutathione. Biochem. Pharmacol. 1998;55:657–665. doi: 10.1016/s0006-2952(97)00498-x. [DOI] [PubMed] [Google Scholar]
- GRUETTER C.A., LEMKE S.M. Effects of sulfhydryl reagents on nitroglycerin-induced relaxation of bovine coronary artery. Can. J. Physiol. Pharmacol. 1986;64:1395–1401. doi: 10.1139/y86-236. [DOI] [PubMed] [Google Scholar]
- HAJ-YEHIA A.I., BENET L.Z. In vivo depletion of free thiols does not account for nitroglycerin-induced tolerance: a thiol-nitrate interaction hypothesis as an alternative explanation for nitroglycerin activity and tolerance. J. Pharmacol. Exper. Therap. 1996;278:1296–1305. [PubMed] [Google Scholar]
- HENRY P.J., DRUMMER O.H., HOROWITZ J.D. S-nitrosothiols as vasodilators: implications regarding tolerance to nitric oxide-containing vasodilators. Br. J. Pharmacol. 1989;98:757–766. doi: 10.1111/j.1476-5381.1989.tb14603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTTER J., SCHMIDT M, RITTLER J. Effects of sulfhydryl-containing compounds on nitroglycerin-induced coronary dilatation in isolated working rat hearts. Eur. J. Pharmacol. 1988;156:215–222. doi: 10.1016/0014-2999(88)90324-x. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., GRUETTER C.A. Requirement of thiols for activation of coronary arterial guanylate cyclase by glyceryl trinitrate and sodium nitrite: possible involvement of S-nitrosothiols. Biochim. Biophys. Acta. 1980;631:221–231. doi: 10.1016/0304-4165(80)90297-4. [DOI] [PubMed] [Google Scholar]
- IOANNIDIS I., BATZ M., PAUL T., KORTH H.G., SUSTMANN R., DE GROOT H. Enhanced release of nitric oxide causes increased cytotoxicity of S-nitroso-N-acetyl-DL-penicillamine and sodium nitroprusside under hypoxic conditions. Biochem. J. 1996;318:789–795. doi: 10.1042/bj3180789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KASHIBA-IWATSUKI M., KITOH K., KASAHARA E., YU H., NISIKAWA M., MATSUO M., INOUE M. Ascorbic acid and reducing agents regulate the fates and functions of S-nitrosothiols. J. Biochem. 1997;122:1208–1214. doi: 10.1093/oxfordjournals.jbchem.a021883. [DOI] [PubMed] [Google Scholar]
- KHAN S.A., HIGDON N.R., MEISHERI K.D. Coronary vasorelaxation by nitroglycerin: involvement of plasmalemmal calcium-activated K+ channels and intracellular Ca++ stores. J. Pharmacol. Exp. Ther. 1998;284:838–846. [PubMed] [Google Scholar]
- KLESCHYOV A.L., SEDOV K.R., MORDVINTCEV P.I., VANIN A.F. Biotransformation of sodium nitroprusside into dinitrosyl iron complexes in tissue of ascites tumors of mice. Biochem. Biophys. Res. Commun. 1994;202:168–173. doi: 10.1006/bbrc.1994.1908. [DOI] [PubMed] [Google Scholar]
- KOWALUK E.A., FUNG H.L. Spontaneous liberation of nitric oxide cannot account for in vitro vascular relaxation by S-nitrosothiols. J. Pharmacol. Exp. Ther. 1990;255:1256–1264. [PubMed] [Google Scholar]
- KURZ M.A., BOYER T.D., WHALEN R., PETERSON T.E., HARRISON D.G. Nitroglycerin metabolism in vascular tissue: role of glutathione S-transferases and relationship between NO. and NO2-formation. Biochem. J. 1993;292:545–550. doi: 10.1042/bj2920545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAD P.J., LIEBEL M.A., WHITE A.A. Activation of rat lung soluble guanylate cyclase by sodium nitroprusside: effects of hemoglobin and reducing agents. Biochem. Biophys. Res. Commun. 1981;103:629–637. doi: 10.1016/0006-291x(81)90497-6. [DOI] [PubMed] [Google Scholar]
- LAURSEN J.B., MULSCH A., BOESGAARD S., MORDVINTCEV P., TRAUTNER S., GRUHN N., NIELSEN-KUDSK J.E., BUSSE R., ALDERSHVILE J. In vivo nitrate tolerance is not associated with reduced bioconversion of nitroglycerin to nitric oxide. Circulation. 1996;94:2241–2247. doi: 10.1161/01.cir.94.9.2241. [DOI] [PubMed] [Google Scholar]
- MACALLISTER R.J., CALVER A.L., RIEZEBOS J., COLLIER J., VALLANCE P. Relative potency and arteriovenous selectivity of nitrovasodilators on human blood vessels: an insight into the targeting of nitric oxide delivery. J. Pharmacol. Exp. Ther. 1995;273:154–160. [PubMed] [Google Scholar]
- MARAGOS C.M., MORLEY D., WINK D.A., DUNAMS T.M., SAAVEDRA J.E., HOFFMAN A., BOVE A.A., ISAAC L., HRABIE J.A., KEEFER L.K. Complexes of .NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- MARKS G.S., MCLAUGHLIN B.E., JIMMO S.L., POKLEWSKA-KOZIELL M., BRIEN J.F., NAKATSU K. Time-dependent increase in nitric oxide formation concurrent with vasodilation induced by sodium nitroprusside, 3-morpholinosydnonimine, and S-nitroso-N-acetylpenicillamine but not by glyceryl trinitrate. Drug Metab. Dispos. 1995;23:1248–1252. [PubMed] [Google Scholar]
- MATHEWS W.R., KERR S.W. Biological activity of S-nitrosothiols: the role of nitric oxide. J. Pharmacol. Exper. Ther. 1993;267:1529–1537. [PubMed] [Google Scholar]
- MAYER B., SCHRAMMEL A., KLATT P., KOESLING D., SCHMIDT K. Peroxynitrite-induced accumulation of cyclic GMP in endothelial cells and stimulation of purified soluble guanylyl cyclase. Dependence on glutathione and possible role of S-nitrosation. J. Biol. Chem. 1995;270:17355–17360. doi: 10.1074/jbc.270.29.17355. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M., GRYGLEWSKI R.J. Mechanism of action of some inhibitors of endothelium-derived relaxing factor. Proc. Natl. Acad. Sci. U.S.A. 1986;83:9164–9168. doi: 10.1073/pnas.83.23.9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORLEY D., KEEFER L.K. Nitric oxide/nucleophile complexes: a unique class of nitric oxide-based vasodilators. J. Cardiovasc. Pharmacol. 1993;22 Suppl.7:S3–S9. [PubMed] [Google Scholar]
- MURPHY M.E., BRAYDEN J.E. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J. Physiol. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO D.N., CEDERBAUM A.I. Production of nitric oxide and other iron-containing metabolites during the reductive metabolism of nitroprusside by microsomes and by thiols. Arch. Biochem. Biophys. 1995;321:363–371. doi: 10.1006/abbi.1995.1406. [DOI] [PubMed] [Google Scholar]
- RAPOPORT R.M., WALDMAN S.A., SCHWARTZ K., WINQUIST R.J., MURAD F. Effects of atrial natriuretic factor, sodium nitroprusside, and acetylcholine on cyclic GMP levels and relaxation in rat aorta. Eur. J. Pharmacol. 1985;115:212–229. doi: 10.1016/0014-2999(85)90694-6. [DOI] [PubMed] [Google Scholar]
- ROCHELLE L.G., KRUSZYNA H., KRUSZYNA R., BARCHOWSKY A., WILCOX D.E., SMITH R.P. Bioactivation of nitroprusside by porcine endothelial cells. Toxicol. Appl. Pharmacol. 1994;128:123–128. doi: 10.1006/taap.1994.1189. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., PISTELLI A., VANE J. Conversion of glyceryl trinitrate to nitric oxide in tolerant and non-tolerant smooth muscle and endothelial cells. Br. J. Pharmacol. 1993;108:162–169. doi: 10.1111/j.1476-5381.1993.tb13457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRAMMEL A., PFEIFFER S., SCHMIDT K., KOESLING D., MAYER B. Activation of soluble guanylyl cyclase by the nitrovasodilator 3-morpholinosydnonimine involves formation of S-nitrosoglutathione. Mol. Pharmacol. 1998;54:207–212. doi: 10.1124/mol.54.1.207. [DOI] [PubMed] [Google Scholar]
- SCHRODER H. Cytochrome P-450 mediates bioactivation of organic nitrates. J. Pharmacol. Exp. Ther. 1992;262:298–302. [PubMed] [Google Scholar]
- SCHROR K., FORSTER S., WODITSCH I. On-line measurement of nitric oxide release from organic nitrates in the intact coronary circulation. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991;344:240–246. doi: 10.1007/BF00167225. [DOI] [PubMed] [Google Scholar]
- SCORZA G., PIETRAFORTE D., MINETTI M. Role of ascorbate and protein thiols in the release of nitric oxide from S-nitroso-albumin and S-nitroso-glutathione in human plasma. Free Rad. Biol. Med. 1997;22:633–642. doi: 10.1016/s0891-5849(96)00378-4. [DOI] [PubMed] [Google Scholar]
- SETH P., FUNG H.L. Biochemical characterization of a membrane-bound enzyme responsible for generating nitric oxide from nitroglycerin in vascular smooth muscle cells. Biochem. Pharmacol. 1993;46:1481–1486. doi: 10.1016/0006-2952(93)90115-d. [DOI] [PubMed] [Google Scholar]
- SINGH R.J., HOGG N., JOSEPH J., KALYANARAMAN B. Mechanism of nitric oxide release from S-nitrosothiols. J. Biol. Chem. 1996;271:18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- SINGHAL S.S., PIPER J.T., SRIVASTAVA S.K., CHAUBEY M., BANDOROWICZ-PIKULA J., AWASTHI S., AWASTHI Y.C. Rabbit aorta glutathione S-transferases and their role in bioactivation of trinitroglycerin. Toxicol. Appl. Pharmacol. 1996;140:378–386. doi: 10.1006/taap.1996.0234. [DOI] [PubMed] [Google Scholar]
- WHEATLEY R.M., DOCKERY S.P., KURZ M.A., SAYEGH H.S., HARRISON D.G. Interactions of nitroglycerin and sulfhydryl-donating compounds in coronary microvessels. Am J. Physiol. 1994;266:H291–H297. doi: 10.1152/ajpheart.1994.266.1.H291. [DOI] [PubMed] [Google Scholar]
- WU M., PRITCHARD K.A., JR, KAMINSKI P.M., FAYNGERSH R.P., HINTZE T.H., WOLIN M.S. Involvement of nitric oxide and nitrosothiols in relaxation of pulmonary arteries to peroxynitrite. Am. J. Physiol. 1994;266:H2108–H2113. doi: 10.1152/ajpheart.1994.266.5.H2108. [DOI] [PubMed] [Google Scholar]
- WU X.B., BRUNE B., VON APPEN F., ULLRICH V. Reversible activation of soluble guanylate cyclase by oxidizing agents. Arch. Biochem. Biophys. 1992;294:75–82. doi: 10.1016/0003-9861(92)90139-n. [DOI] [PubMed] [Google Scholar]