

Abstract
The effects of nepalolide A on the expression of inducible nitric oxide synthase (iNOS) caused by incubation with lipopolysaccharide/interferon-γ (LPS/IFN-γ) or tumour necrosis factor-α/interleukin-1β/IFN-γ (TNF-α/IL-1β/IFN-γ, mixed cytokines) in C6 glioma cells and primary astrocytes of rat were investigated. The mechanisms by which nepalolide A confers its effect on iNOS expression were also elucidated.
Treatment with LPS/IFN-γ and mixed cytokines for 24 h elicited the induction of iNOS activity as determined by nitrite accumulation in the culture medium and assay of enzyme activity. Nepalolide A at 10 μM abrogated the LPS/IFN-γ- and mixed cytokines-mediated induction of iNOS by more than 90% in C6 glioma cells, and by 80% for mixed cytokines-induced induction of iNOS in primary astrocytes. The effect of nepalolide A (2–10 μM) was concentration-dependent.
The inhibition of iNOS induction by nepalolide A was attributed to decreases in the content of iNOS protein and the level of iNOS mRNA, as measured by immunoblotting and reverse transcriptase-polymerase chain reaction.
Electrophoretic mobility shift assay was used to evaluate the effect of nepalolide A on the activation of nuclear factor-κB (NF-κB). Results showed that nepalolide A diminished the LPS/IFN-γ-mediated association of NF-κB with consensus oligonucleotide in a concentration-dependent manner. The activation of NF-κB by mixed cytokines was modulated both in the extent of activation and in its time-course by nepalolide A.
The ability of nepalolide A to inhibit NF-κB activation was further confirmed by studies on the degradation of the inhibitor of NF-κB, IκB, as measured by immunoblotting.
The present study demonstrates that the attenuation of NF-κB activation by nepalolide A was mediated by blockade of the degradation of IκB, leading to suppression of the expression of iNOS.
Keywords: Inducible nitric oxide synthase, nepalolide A, C6 glioma, astrocytes, NF-κB, IκB degradation
Introduction
Nitric oxide (NO) is a free radical gas involved in a wide range of physiological and pathophysiological mechanisms, including the control of the cardiovascular, immune, central and peripheral nervous systems, as well as cellular toxicity (Dawson & Snyder, 1994). Nitric oxide is synthesized from L-arginine by the enzyme nitric oxide synthase (NOS), which oxidatively removes the terminal guanidino-nitrogen from L-arginine to form L-citrulline and nitric oxide (Knowles & Moncada, 1994). Three isoforms of NOS have been identified including neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS). nNOS and eNOS are regulated predominantly at the post-transcriptional level by calmodulin in a calcium-dependent manner. In contrast, iNOS is an inducible calmodulin containing but calcium-independent enzyme (Dawson & Dawson, 1996).
In the central nervous system (CNS), iNOS is expressed mainly in activated astrocytes and microglia. High levels of nitric oxide produced via iNOS in CNS are implicated in oligodendrocyte degeneration in demyelinating diseases and neuronal death during trauma (Mitrovic et al., 1994; Merrill et al., 1993; Lipton et al., 1993). iNOS is induced in response to a series of proinflammatory cytokines including interleukin-1β (IL-1β), tumour necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and bacterial lipopolysaccharide (LPS). The recently cloned promoter of murine iNOS contains at least 22 elements homologous to consensus sequences for the binding of transcription factors. Among these are two nuclear factor-κB (NF-κB) binding sites, two activator protein 1 (AP-1) binding sites and one IFN-γ-activated site (GAS). The activation of NF-κB has been shown to be essential for the induction of iNOS. In most cell types, NF-κB is present as a heterodimer composed of p50 (NF-κB1) and p65 (RelA) subunits. NF-κB is sequestered in the cytoplasm by association with a member of the inhibitor family of NF-κB (IκB) (Baeuerle & Henkel, 1994; Thanos & Maniatis, 1995). IκB proteins mask the nuclear localization signal of NF-κB, thereby preventing the NF-κB nuclear translocation. The phosphorylation of serine residues 32 and 36 in IκB-α and residues 19 and 23 in IκB-β by IκB kinase-α (IKK-α) and IKK-β induce degradation of IκB and the dissociation of NF-κB from IκB. Following this NF-κB translocates into the nucleus and interacts with the NF-κB binding motif in the promoters of target genes and so regulates their transcription (Mercurio et al., 1997; DiDonato et al., 1997; Woronicz et al., 1997; Régnier et al., 1997). The activations of protein kinases and other transcription factors are also involved in the regulation of iNOS expression. These include protein kinase C-ε (PKCε), PKCζ, mitogen-activated protein kinase (MAPK), p38, signal transducer and activator of transcription 1 (Stat1), and AP-1 (Díaz-Guerra et al., 1996; Chen et al., 1998; Bhat et al., 1998; Gao et al., 1997; Marks-Konczalik et al., 1998).
Nepalolide A (4β-hydroxy-9-oxo-5β-senecioyloxy-11(13)en-germacran-6α, 12-olide; Figure 1), one kind of a sesquiterpene lactones, has recently been isolated from Carpesium nepalense, a substance used in Chinese traditional medicine for the treatment of hepatitis (Lin et al., 1996). Since our preliminary screening has shown nepalolide A potently inhibits nitrite production. We have investigated the effects of nepalolide A on the expression of iNOS induced by LPS/IFN-γ or TNF-α/IL-1β/IFN-γ (mixed cytokines) in C6 glioma cells and primary astrocytes. The mechanisms by which nepalolide A exerts its effect on iNOS expression have also been examined. Our results show that nepalolide A inhibits the accumulation of nitrite in the culture medium and decreases the enzyme activity of iNOS induced by LPS/IFN-γ and mixed cytokines in C6 glioma cells and by mixed cytokines in primary astrocytes. The inhibitory effects of nepalolide A coincide with a decrease in cellular content of iNOS protein and the mRNA level of iNOS. Nepalolide A abolishes the LPS/IFN-γ-stimulated association of NF-κB with consensus oligonucleotide due to a reduction in the degradation of IκB. Nepalolide A both retards the mixed cytokines-mediated activation of NF-κB and decreases the maximal level of activation by delaying the degradation of IκB.
Figure 1.
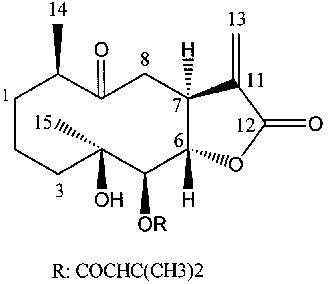
Structure of nepalolide A.
Methods
Cell culture
C6 glioma cells were cultured in F-12K medium supplemented with 15% horse serum and 2.5% foetal bovine serum. Newborn Sprague-Dawley rats were anaesthetized with ether and sacrificed by decapitation. Primary culture of astrocytes were prepared from the cerebral cortices and maintained in DMEM/F12 medium containing 10% foetal bovine serum (Cole & de Vellis, 1992). After confluence, contaminated microglia and oligodendrocytes cells were removed by shaking at 200 r.p.m. with an orbital shaker. Thereafter, the cells were subcultured into culture dishes for experiments.
Measurement of nitrite
Cells were incubated with medium containing 0.2% (w v−1) bovine serum albumin (BSA) plus 5 μg ml−1 LPS/100 units ml−1 IFN-γ or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ to induce expression of iNOS. iNOS activity was assessed by measuring the accumulation of nitrite in the culture medium. The amount of nitrite in medium was determined by using Griess reagent with NaNO2 as standard (Chiou et al., 1997).
Measurement of iNOS activity
Cells were incubated with iNOS inducers or inducers plus nepalolide A for 24 h. After washing twice with ice-cold phosphate buffered saline (PBS), cells were harvested in hypotonic buffer (in mM): HEPES pH 7.5 50, EDTA 1, DTT 1, PMSF 1, aprotinin 5 μg ml−1 and leupeptin 10 μg ml−1 and disrupted by freezing and thawing. Cellular lysates were incubated with iNOS reaction buffer (HEPES pH 7.5 50 mM, NADPH 0.2 mM, FAD 10 μM, FMN 10 μM, tetrahydrobiopterin 100 μM, DTT 1 mM, arginine containing 0.5 μCi [3H]-arginine 0.1 mM, with or without 2 mM Nω-nitro-L-arginine methyl ester) at 37°C for 30 min. The reaction was terminated by addition of 0.8 ml HEPES buffer (in mM): HEPES pH 5.5 20, EDTA 10, citrulline 0.2. [3H]-Arginine and [3H]-citrulline were separated over Dowex AG50W-X8 columns (sodium form). The radioactivity of citrulline was measured by liquid scintillation counting. iNOS activity was presented as pmol of citrulline formed min−1 mg−1 cellular protein.
Assay of cell viability
The reduction of (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) was used to evaluate the cytotoxic effect of nepalolide A on cultured cells. Cells were incubated with medium containing various concentrations of nepalolide A for 24 h before MTT solution was added to the culture medium at a final concentration of 0.5 mg ml−1. After 1 h incubation, the medium was aspirated and the formazan particles dissolved with lysis buffer (10% SDS, 3.3 mM HCl, 50% dimethyl formamide). OD600nm was measured by using a 96-well plate reader.
Immunoprecipitation and immunoblotting
Cells treated with inducers in the presence or absence of nepalolide A were harvested in lysis buffer (in mM): HEPES pH 7.5 50, EDTA 1, NaCl 150, NP-40 1%, PMSF 1, aprotinin 5 μg ml−1 and leupeptin 10 μg ml−1. iNOS was immunoprecipitated from total cellular lysate by using anti-iNOS antibodies. The immunoprecipitates were fractionated by SDS-polyacrylamide gel electrophoresis (SDS–PAGE) and then electrotransferred onto PVDF membrane. Immunoreactive proteins were detected by using anti-rabbit IgG antibodies conjugated with horseradish peroxidase and enhanced chemiluminescence detection reagents. For the immunoblotting of IκB-α and IκB-β, cells were harvested in hypotonic buffer (in mM): HEPES pH 7.5 50, EDTA 1, PMSF 1, aprotinin 5 μg ml−1, leupeptin 10 μg ml−1 and antipain 10 μg ml−1). The total cellular lysates were subjected to SDS–PAGE and immunoblotting.
Estimate of the level of iNOS mRNA
Cells were treated with iNOS inducers plus nepalolide A for 5 h. Total RNA was isolated and reverse transcriptase-polymerase chain reaction (RT–PCR) was employed to determine the mRNA level of iNOS. The detailed experiments were performed according to the manufacturer's protocol. For RT–PCR, two primers, 5′ primer (5′-CCCTTCCGAAGTTTCTGGCAGCAGC-3′) and 3′ primer (5′-GGCTGTCAGAGCCTCGTGGCTTTGG-3′), were used to specifically amplify the cDNA of iNOS. To amplify the cDNA of glyceraldehyde 3-phosphate dehydrogenase (G3PDH), the 5′ primer (5′-TGAAGGTCGGTGTCAACGGATTTGGC-3′) and 3′ primer (5′-CATGTAGGCCATGAGGTCCACCAC-3′) were used. A thermal cycle of 30 s at 94°C, 30 s at 65°C, and 45 s at 68°C for 40 cycles was used for iNOS. For G3PDH, a thermal cycle of 30 s at 94°C, 30 s at 65°C, and 45 s at 68°C for 25 cycles was performed. PCR products were analysed on 2% agarose gel electrophoresis.
Preparation of nuclear extracts and electrophoretic mobility shift assay (EMSA)
Treated cells were washed twice with ice-cold PBS and harvested. The residual PBS buffer was removed and cells used to prepare nuclear extracts (Pahan et al., 1997). The nuclear extracts (10 μg for NF-κB and 5 μg for AP-1) were incubated with 1 ng of 5′-end-labelled consensus (or mutant) oligonucleotide for NF-κB or AP-1 in binding solution (in mM): Tris pH 7.5 10, MgCl2 1, NaCl 140, DTT 0.5, EDTA 0.5, 12% glycerol and 1 μg poly(dl-dC) at room temperature for 30 min. The mixtures were subjected to 5% of nondenatured polyacrylamide gel electrophoresis by using 1×TBE as electrophoresis buffer. The gels were dried and radioactive oligonucleotides were detected by autoradiography.
Materials
C6 glioma cells were from Culture Collection and Research Center (Taiwan, R.O.C.). Human TNF-α and rat IL-1β were from R&D Systems (Minneapolis, MN, U.S.A.). Dowex AG50W-X8 resin and all reagents for electrophoresis were from Bio-Rad Laboratories (Hercules, CA, U.S.A.). Medium for cell culture and rat IFN-γ were from Gibco Laboratories (Gaithersburg, MD, U.S.A.). Radioisotopes, enhanced chemiluminescence detection reagents and conjugated anti-rabbit IgG-horseradish peroxidase were obtained from Amersham Pharmacia Biotech (Buckinghamshire, U.K.). Rabbit polyclonal antibodies for iNOS were obtained from Transduction Laboratories (Lexington, KY, U.S.A.). Rabbit polyclonal antibodies against IκB-α, IκB-β, consensus or mutant oligonucleotide for NF-κB and AP-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Primer sets for iNOS and G3PDH were obtained from CLONTECH Laboratories (Palo Alto, CA, U.S.A.). RNA isolation and RT–PCR kits were obtained from Boehringer Mannheim (Mannheim, Germany). All other reagents were purchased from Sigma (St. Louis, MO, U.S.A.), or Merck (Darmstadt, Germany).
Statistical analysis
Results are expressed as means±s.d., and were analysed by ANOVA with post hoc multiple comparison using a Bonferroni test.
Results
Nepalolide A inhibited the induction of iNOS
Treatment of C6 glioma cells with 5 μg ml−1 LPS/100 units ml−1 IFN-γ or mixed cytokines (5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ) for 24 h caused a significant increase in the accumulation of nitrite in medium from 5.96±3.86 to 127.63±9.62 and 229.98±16.32 nmol (24 h)−1 mg−1 cell protein for LPS/IFN-γ and mixed cytokines treatment, respectively (n=5). These increases were significantly reduced by 1 μM dexamethasone to 35.24±1.97 and 41.99±1.06% of control for LPS/IFN-γ and mixed cytokines, respectively (n=4). Treatment with nepalolide A decreased the accumulation of nitrite in a concentration-dependent manner (Figure 2a). For example, the LPS/IFN-γ- and mixed cytokines-stimulated accumulation of nitrite was decreased to 3.47±2.49 and 14.5±5.9% of control at 10 μM nepalolide A, respectively (n=5). The IC50 of nepalolide A for nitrite accumulation induced by LPS/IFN-γ and mixed cytokines was 1.95 and 4.92 μM, respectively. The effect of nepalolide A on iNOS activity was verified by the formation of citrulline. These experiments showed that 10 μM nepalolide A inhibited the activity of iNOS induced by LPS/IFN-γ and mixed cytokines by 99.08±1.95 and 94.18±4.27%, respectively (n=5) (Figure 2b).
Figure 2.
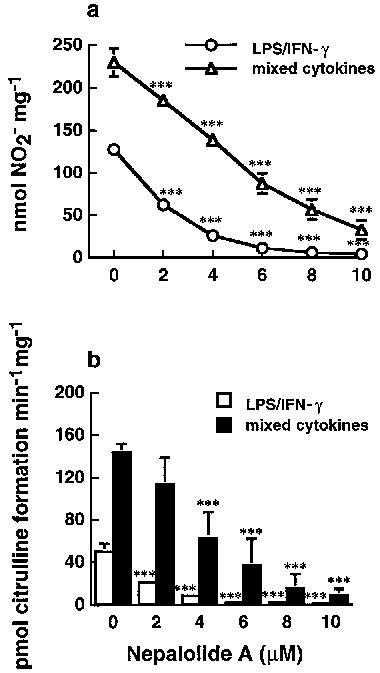
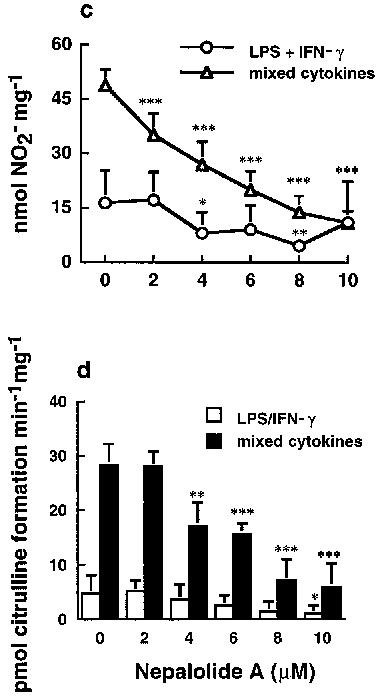
Effects of nepalolide A on the induction of iNOS. C6 glioma cells (a) and primary astrocytes (c) were preincubated with nepalolide A (2–10 μM) for 30 min. After 30 min incubation, cells were challenged with 5 μg ml−1 LPS/100 units ml−1 INF-γ (LPS/IFN-γ) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 INF-γ (mixed cytokines) plus or minus nepalolide A for 24 h. The culture media were collected for nitrite determination. To assay iNOS activity, C6 glioma cells (b) and primary astrocytes (d) were treated with LPS/IFN-γ or mixed cytokines in the presence or absence of nepalolide A as described above. After incubation for 24 h, cells were harvested and the activity of iNOS was determined by citrulline formation. Results are means±s.d. (where large enough to be shown) from five independent experiments. Significant differences between control and nepalolide A-treated cells are indicated by *P<0.05; **P<0.01; and ***P<0.001.
Primary astrocytes were also used to investigate the effect of nepalolide A on the induction of iNOS. Nepalolide A showed a similar effect on the nitrite accumulation and iNOS activity induced by mixed cytokines in primary astrocytes as in C6 glioma cells (Figure 2c and d). Mixed cytokines treatment increased nitrite accumulation from 6.35±3.21 to 48.79±4.44 nmol (24 h)−1 mg−1 cell protein (n=5), and elevated iNOS activity from 5.06±2.1 to 28.40±3.84 pmol citrulline min−1 mg−1. The nitrite accumulation and iNOS activity were reduced to 21.58±6.56 and 22.18±17.67% of control by 10 μM nepalolide A, respectively. However, the effect for LPS/IFN-γ was more variable and not consistent with the results from treatment with mixed cytokines. Therefore, iNOS induction by mixed cytokines was used for further studies in primary astrocytes. Nepalolide A had no effect on the cell viability at concentrations of 2–10 μM (data not shown). Nitrite accumulation increased in a time-dependent manner from 8 to 24 h following exposure to LPS/IFN-γ or mixed cytokines of C6 glioma cells (Figure 3). Ten μM nepalolide A diminished the level of nitrite accumulated in the culture medium during the time periods.
Figure 3.
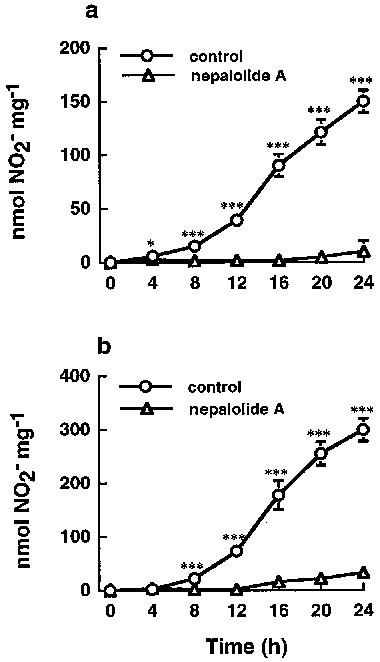
Time-course of the effects of nepalolide A on the accumulation of nitrite in C6 glioma cells. C6 glioma cells were incubated with or without 10 μM nepalolide A for 30 min. After 30 min incubation, cells were challenged with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (a) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (b) in the presence or absence of nepalolide A for the times indicated. After these incubation periods, the culture media were collected for nitrite determination. Results are means±s.d. (where large enough to be shown) from four independent experiments. Significant differences between control and nepalolide A-treated cells are indicated by *P<0.05; and ***P<0.001.
Nepalolide A decreased the level of iNOS protein and mRNA
LPS/IFN-γ significantly increased the content of iNOS in C6 glioma cells (Figure 4a). Similarly, mixed cytokines also significantly elevated the level of iNOS in both C6 glioma cells and primary astrocytes (Figure 4a and b). The protein content of iNOS induced by mixed cytokines was decreased in a concentration-dependent manner by nepalolide A. For example, at 10 μM nepalolide A reduced the iNOS content by 81.86±2.18 and 54.87±4.69% in C6 glioma cells and primary astrocytes, respectively (n=4). Nepalolide A also drastically diminished the level of iNOS induced by LPS/IFN-γ in C6 glioma cells such that at 6 μM nepalolide A reduced the level of iNOS by 87.48±10.51%.
Figure 4.
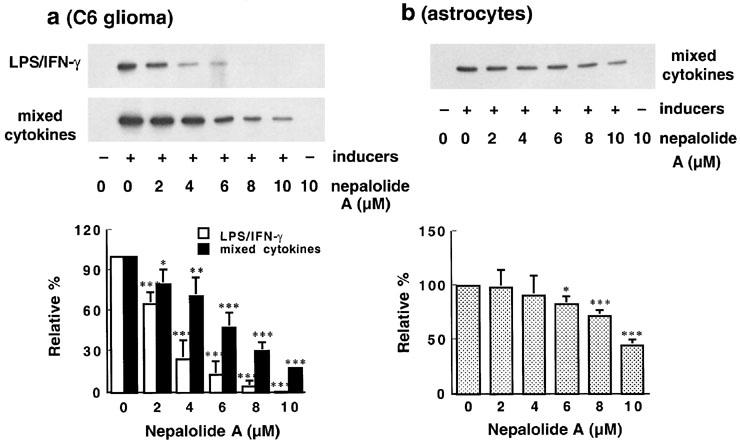
Effect of nepalolide A on the level of iNOS. C6 glioma cells (a) and astrocytes (b) were preincubated with nepalolide A (2–10 μM) for 30 min. After 30 min incubation, C6 glioma cells were challenged with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (LPS/IFN-γ) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (mixed cytokines) plus or minus nepalolide A for 24 h. For astrocytes, cells were challenged with 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ in the presence or absence of nepalolide A for 24 h. After incubation for 24 h, cells were harvested and iNOS was immunoprecipitated and analysed by immunoblotting. The top parts of (a) and (b) are immunoblots of iNOS. The bottom parts are the relative levels of iNOS in untreated cells and cells treated with nepalolide A. Results are means±s.d. (where large enough to be shown) from four independent experiments, and are expressed relative to cells treated with iNOS inducers alone. Significant differences between control and nepalolide A-treated cells are indicates by *P<0.05; **P<0.01; and ***P<0.001.
iNOS mRNA was undetectable in control cells but this was greatly increased by exposure to LPS/IFN-γ and mixed cytokines in C6 glioma cells (Figure 5). Similarly mixed cytokines induced elevation of iNOS mRNA in primary astrocytes. Nepalolide A decreased the level of iNOS mRNA in a concentration-dependent manner. For example, 10 μM nepalolide A inhibited the appearance of iNOS mRNA by 99 and 96% in C6 glioma cells treated with LPS/IFN-γ and mixed cytokines, respectively. In primary astrocytes, 10 μM nepalolide A inhibited the mixed cytokines-induced increase in iNOS mRNA by 97%.
Figure 5.
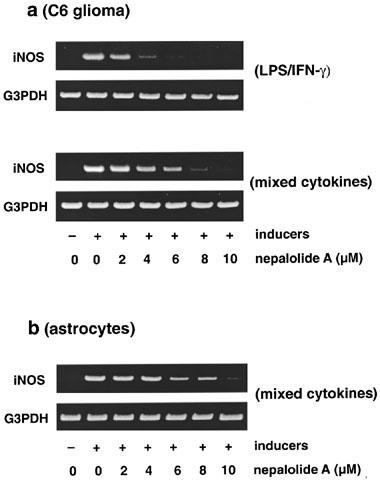
RT–PCR analysis of the level of iNOS mRNA. C6 glioma cells (a) were treated with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (LPS/IFN-γ) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (mixed cytokines) in the presence or absence of various concentrations of nepalolide A for 5 h. For astrocytes (b), cells were treated with mixed cytokines plus or minus nepalolide A for 5 h. After incubation, total RNA was isolated and subjected to RT–PCR analysis. This experiment was repeated three times with similar results. G3PDH, glyceraldehyde-3-phosphate dehydrogenase.
Relationship between the inhibition by nepalolide A of iNOS expression and the activation of NF-κB
Figure 6 showed that the addition of 10 μM nepalolide A from −0.5 to 4 h posterior to the challenge of iNOS inducers completely abolished the accumulation of nitrite in C6 glioma cells, indicating that nepalolide A acts at the early events of iNOS expression.
Figure 6.
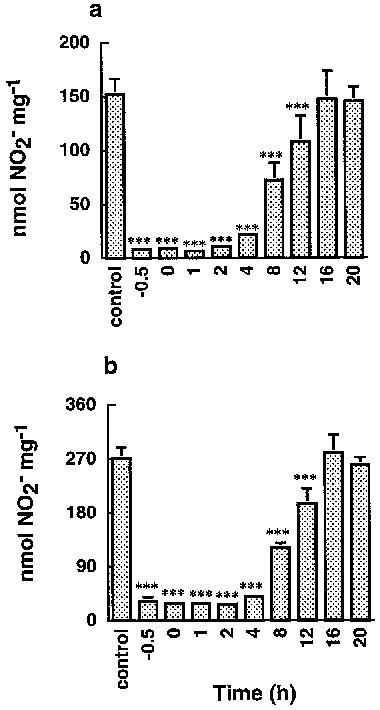
Effects of nepalolide A pretreatment, cotreatment, or post treatment on C6 glioma cells. C6 glioma cells were treated with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (a) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (b) for 24 h. Nepalolide A (10 μM) was added prior to (−0.5 h), during (0 h), or following (1–20 h) exposure to inducers of iNOS. After incubation, the culture media were collected for nitrite determination. Results are means±s.d. (where large enough to be shown) from four independent experiments. Significant differences between control and nepalolide A-treated cells are indicated by ***P<0.001.
Either LPS/IFN-γ or mixed cytokines significantly increased the association of NF-κB with consensus oligonucleotide but not mutant oligonucleotide, demonstrating the specific interaction of NF-κB with the oligonucleotide (Figure 7a). Nepalolide A blocked the association of NF-κB with oligonucleotide triggered by LPS/IFN-γ in a concentration-dependent manner (Figure 7a and c), such that 10 μM nepalolide A decreased the association to 7.45±6.8% of control (n=5). However, nepalolide A failed to affect the association induced by mixed cytokines in both C6 glioma cells and primary astrocytes at 2–10 μM. For AP-1, treatment with mixed cytokines increased the association of AP-1 with consensus oligonucleotide by 37 and 172% in C6 glioma cells and primary astrocytes, respectively (Figure 7b). Neither LPS/IFN-γ nor nepalolide A affected the association of AP-1 with consensus oligonucleotide.
Figure 7.
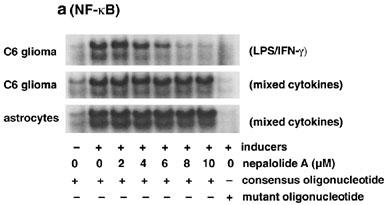
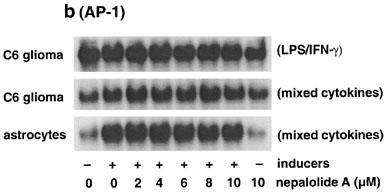
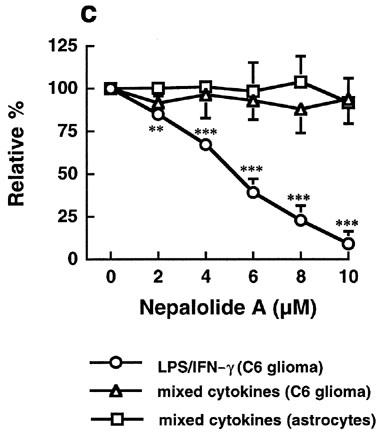
Effect of nepalolide A on the electrophoretic mobility shift assay (EMSA) for NF-κB and AP-1 binding. C6 glioma cells and astrocytes were preincubated with nepalolide A (2–10 μM) for 30 min. After 30 min incubation, cells were challenged with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (LPS/IFN-γ) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (mixed cytokines) plus or minus nepalolide A for 3 h. After incubation, nuclear extracts were isolated. The EMSA of NF-κB and AP-1 binding was performed by using 5′-end-labelled consensus or mutant oligonucleotides. (a and b) are representative autoradiographs of NF-κB and AP-1 binding, respectively. (c) is the relative level of NF-κB binding in untreated cells and cells treated with nepalolide A. Results are means±s.d. (where large enough to be shown) from five independent experiments, and are expressed relative to cells treated with iNOS inducers alone. Significant differences between control and nepalolide A-treated cells are indicated by **P<0.01; and ***P<0.001.
NF-κB activation induced by mixed cytokines was not affected by nepalolide A at 3 h. Thus, the time course effect of nepalolide A on NF-κB activation was studied. Results showed that mixed cytokines significantly elevated the association of NF-κB with consensus oligonucleotide at 10 min in both C6 glioma cells and primary astrocytes (Figure 8a and b). The association reached a maximum at 10–30 min. Thereafter, the activation of NF-κB was decreased from 1 to 3 h. Treatment with 10 μM nepalolide A attenuated the association of NF-κB with oligonucleotide at 10 min by 54.9±12 and 51.3±5.1% in C6 glioma cells and primary astrocytes, respectively (n=4). The association increased with time, reaching a plateau at 1 h in cells treated with 10 μM nepalolide A. Consequently, there was no significant difference in the mixed cytokines-induced activation of NF-κB between control and nepalolide A-treated cells at 3 h. These results established that nepalolide A suppressed NF-κB activation and modulated the time-course profile of activation.
Figure 8.
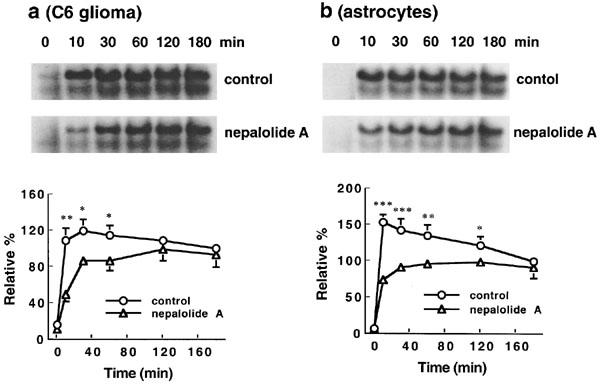
Time-course effect of nepalolide A on the electrophoretic mobility shift assay (EMSA) for NF-κB binding. C6 glioma cells (a) and astrocytes (b) were incubated with 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ in the presence or absence of 10 μM nepalolide A for 0–180 min. After these times, nuclear extracts were isolated and EMSA for NF-κB binding was performed by using 5′-end-labelled consensus oligonucleotide. The top parts of (a and b) are representative autoradiographs of NF-κB binding. The bottom parts are the relative levels of NF-κB binding in untreated cells or cells treated with nepalolide A. Results are means±s.d. (where large enough to be shown) from four independent experiments, and expressed relative to the control cells treated with mixed cytokines for 3 h. Significant differences between control and nepalolide A-treated cells are indicated by *P<0.05; **P<0.01; and ***P<0.001.
Nepalolide A modulated the degradation of IκB-α and IκB-β
LPS/IFN-γ induced the degradation of IκB-α by 43.67±14.14% at 60 min in C6 glioma cells an effect that was significantly suppressed by treatment with 10 μM nepalolide A (n=3) (Figure 9a). Mixed cytokines decreased IκB-α by 74.45±18.5% at 10 min and 76.1±6.6% at 5 min in C6 glioma cells and primary astrocytes, respectively (n=3) (Figure 9b and c). Nepalolide A retarded the mixed cytokines-induced degradation and suppressed the resynthesis of IκB-α. Nevertheless, nepalolide A did not alter the maximum extent of degradation.
Figure 9.
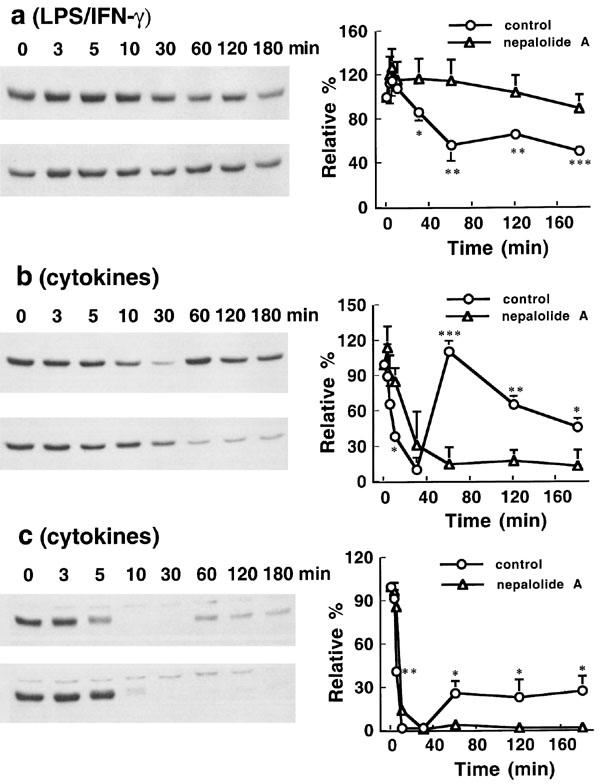
Time-course effect of nepalolide A on the degradation of IκB-α. C6 glioma cells were treated with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (a) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (b) in the presence or absence of 10 μM nepalolide A for 0–3 h. Astrocytes were treated with 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 INF-γ (c) plus or minus 10 μM nepalolide A for 0–3 h. After incubation periods, cellular lysates were prepared and immunoblotting performed. The left parts of (a, b and c) are representative immunoblots of intracellular IκB-α. The right parts are the relative levels of intracellular IκB-α in untreated cells or cells treated with nepalolide A. In each, the top left parts are cells treated with iNOS inducers in the absence of nepalolide A, the bottom left parts are cells treated with iNOS inducers in the presence of nepalolide A. Results are means± s.d. (where large enough to be shown) from three independent experiments, and expressed relative to the cells at zero time. Significant differences between control and nepalolide A-treated cells are indicated by *P<0.05; **P<0.01; and ***P<0.001.
Nepalolide A blocked the degradation of IκB-α in a concentration-dependent manner in C6 glioma cells and primary astrocytes (Figure 10). In C6 glioma cells treated with LPS/IFN-γ for 60 min, the intracellular level of IκB-α was increased from 53.1±10.7 to 73.2±9.6 and 92.1±2.9% by 2 and 4 μM nepalolide A, respectively (n=3) (Figure 10a). Nepalolide A at 4 and 6 μM elevated the level of IκB-α retained in cells treated with mixed cytokines for 10 min from 32.7±9.3 to 55.2±3.0 and 67.5±4.5%, respectively. For primary astrocytes, the intracellular level of IκB-α in cells incubated with mixed cytokines for 5 min was increased from 54.6±12.3 to 83.8±2.2 and 93.0±13.5% by 2 and 4 μM nepalolide A, respectively (n=3) (Figure 10b).
Figure 10.
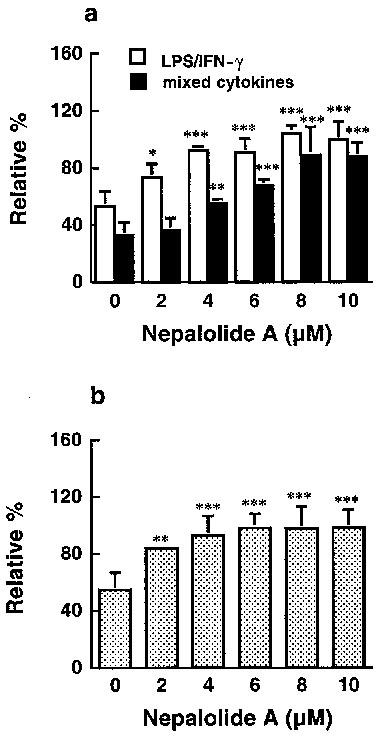
The effect of nepalolide A on the degradation of IκB-α. C6 glioma cells were treated with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (LPS/IFN-γ) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (mixed cytokines) in the presence or absence of nepalolide A (2–10 μM) for 60 and 10 min, respectively (a). Astrocytes were treated with mixed cytokines plus or minus nepalolide A (2–10 μM) for 5 min (b). After treatment, cellular lysates were prepared and immunoblotting of intracellular IκB-α was performed. Results are means±s.d. (where large enough to be shown) from three independent experiments, and expressed relative to the cells at zero time. Significant differences between control and nepalolide A-treated cells are indicated by *P<0.05; **P<0.01; and ***P<0.001.
LPS/IFN-γ and mixed cytokines had similar effects on the degradation of IκB-β as they did on IκB-α (Figure 11). However, there was no obvious resynthesis of IκB-β following treatment with mixed cytokines. Nepalolide A at 10 μM completely inhibited the degradation of IκB-β induced by LPS/INF-γ (Figure 11a). The extent of degradation of IκB-β induced by mixed cytokines was significantly decreased by nepalolide A at 60–120 min and 30–120 min in C6 glioma cells and astrocytes, respectively (Figure 11b and c). These results demonstrate that the attenuation of NF-κB activation by nepalolide A was mediated by blockade of the degradation of IκB, thereby suppressing the expression of iNOS.
Figure 11.
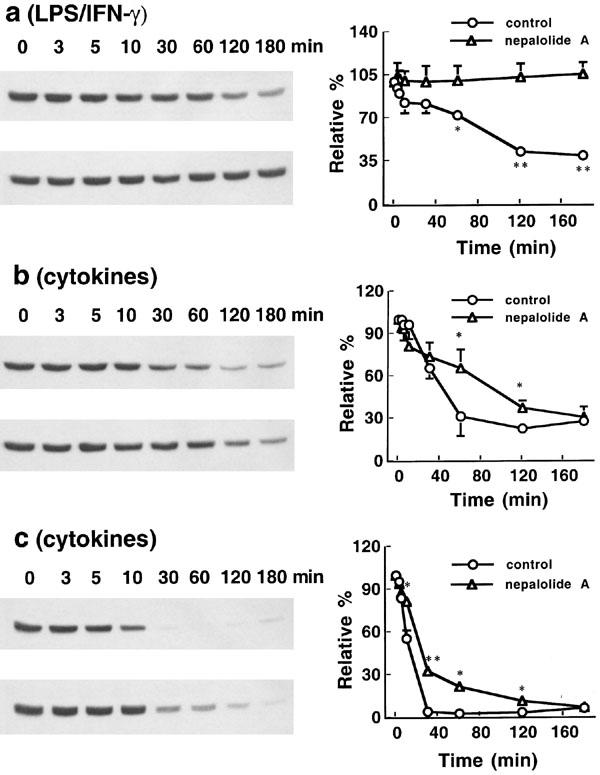
Time-course effect of nepalolide A on the degradation of IκB-β. C6 glioma cells were treated with 5 μg ml−1 LPS/100 units ml−1 IFN-γ (a) or 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (b) in the presence or absence of 10 μM nepalolide A for 0–3 h. Astrocytes were treated with 5 ng ml−1 TNF-α/5 ng ml−1 IL-1β/100 units ml−1 IFN-γ (c) plus or minus 10 μM nepalolide A for 0–3 h. After the incubation periods, cellular lysates were prepared and immunoblotting was performed. The left parts of (a, b and c) are representative immunoblots of intracellular IκB-β. The right parts are the relative levels of intracellular IκB-β in untreated cells or cells treated with nepalolide A. In each, the top left parts are cells treated with iNOS inducers in the absence of nepalolide A, the bottom left parts are cells treated with iNOS inducer in the presence of nepalolide A. Results are means±s.d. (where large enough to be shown) from three independent experiments, and expressed relative to the cells at zero time. Significant differences between control and nepalolide A-treated cells are indicated by * P<0.05; and **P<0.01.
Discussion
Nepalolide A inhibited the LPS/IFN-γ- and mixed cytokines-stimulated accumulation of nitrite in culture medium and the elevation in iNOS activity. The inhibition was in parallel to the decreases in iNOS protein content, the level of iNOS mRNA, the assocation of NF-κB with consensus oligonucleotide and the degradation of IκB.
The structure of nepalolide A consists of an isoprenoide ring system and a lactone ring containing a conjugated exomethylene group (Lin et al., 1996). Both the lactone ring and exomethylene group form a reactive Michael system, which may account for the inhibitory effect of nepalolide A on the induction of iNOS. The present study aimed to investigate the mechanisms by which nepalolide A abrogated the induction of iNOS. The data from Figure 6 showed that the addition of 10 μM nepalolide A following exposure to iNOS inducers for 4 h completely abolished the induction of iNOS. This result excluded the possibility that the inhibitory effect of nepalolide A was due to it interfering with the binding of inducers to their receptors. We could not obtain consistent results for the accumulation of nitrite in primary astrocytes treated with LPS/IFN-γ. The signalling of LPS is mediated by its receptor, membrane-bound CD14 or soluble CD14. Furthermore, recent studies have demonstrated that activation of CD14-negative cells including endothelial cells or primary astrocytes require the presence of soluble CD14 (Galea et al., 1996). The induction of iNOS in astrocytes was performed in medium without serum in this study. Therefore, LPS may well not have been able to exert its effect through soluble CD14, explaining the variable results observed in these studies.
NF-κB activation has been shown to be essential for the expression of iNOS (Xie et al., 1994; Feinstein et al., 1996; Muller et al., 1993). The phosphorylation of serine residues 32 and 36 in IκB-α and residues 19 and 23 in IκB-β by IKK-α and IKK-β trigger the degradation of IκB and the dissociation of NF-κB from IκB. This causes NF-κB to translocate into the nucleus and interact with the NF-κB binding motif in the promoters of target genes (Mercurio et al., 1997; DiDonato et al., 1997; Woronicz et al., 1997; Régnier et al., 1997). The present study demonstrated that the block of NF-κB activation caused by nepalolide A accounted for the inhibition of the induction of iNOS. These results are consistent with the reports that NF-κB plays a critical role in the expression of iNOS. Nepalolide A blocked the LPS/IFN-γ-induced but not mixed cytokines-mediated association of NF-κB with consensus oligonucleotide in a concentration-dependent manner after 3 h incubation. However, nepalolide A did modify the time course profile of mixed cytokines-mediated NF-κB activation, delaying the interaction of NF-κB with consensus oligonucleotide, and reduce the maximal level of association. These results indicate that the time-course of NF-κB activation may be as important as the level of activation. Nevertheless, 10 μM nepalolide A only suppressed the association of NF-κB with consensus oligonucleotide induced by mixed cytokines by about 50% after 10 min induction. If the activation of NF-κB is sufficient for the expression of iNOS, these results imply that there is a threshold concentration for NF-κB associating with the promoter to trigger the transcription of iNOS. Recently, studies by Bergmann et al. (1998) have shown that IκB-α degradation and NF-κB DNA binding are insufficient for IL-1β and TNF-α-induced NF-κB-dependent transcription. From these results they have proposed the existence of a second pathway triggered by TNF-α and IL-1β in parallel to IκB-α degradation, which confers NF-κB transcriptional competency. This hypothesis may provide an explanation for the results obtained in our study. Nepalolide A may interfere with the unknown pathway, in combination with the suppression of NF-κB activation, thereby completely abrogating the mixed cytokines-induced expression of iNOS. Moreover, p38 has been suggested to regulate the induction of iNOS (Bhat et al., 1998). Inhibition of p38 does not affect the localization of NF-κB to nucleus or its DNA binding (Bergmann et al., 1998). It is also possible, therefore, that interference with p38 activation by nepalolide A may also contribute partially to the inhibition of NF-κB-mediated transcription of iNOS.
LPS/IFN-γ and mixed cytokines had differential effects on the degradation of IκB-α and IκB-β, indicating that these iNOS inducers activate NF-κB by different signalling pathways. LPS associates with CD14 thereby eliciting tyrosine phosphorylation, activation of PKCζ, c-Raf, extracellular signal-regulated kinase 1 and 2 (Erk1/Erk2), p38, c-jun N-terminal kinase (JNK) and IκB kinase (Chen et al., 1998; Hambleton et al., 1996; Reimann et al., 1994; Schumann et al., 1998). IFN-γ induces the localization of Stat1 into nucleus and binding to GAS, through tyrosine phosphorylation, by janus kinase (JAK) (Kovarik et al., 1998; Gao et al., 1997). Therefore, LPS is the major factor accounting for the activation of NF-κB in LPS/IFN-γ treatment. PKCζ and p38 have been implicated in the regulation of iNOS induction (Chen et al., 1998; Bhat et al., 1998). However, the activation of Erk1/Erk2 has been shown to be dissociated from the expression of iNOS following LPS/IFN-γ treatment of C6 glioma cells (Nishiya et al., 1997). Multiple lines of evidence have demonstrated that Erk kinase kinase 1 (MEKK1), an activator of JNK, is involved in the activation of IKK-α and IKK-β (Karin & Delhase, 1998; Lee et al., 1998; Hirano et al., 1996; Nakano et al., 1998). It is conceivable, therefore, that LPS may induce the activation of NF-κB by MEKK1 pathway; the activation of IKK-α/IKK-β by TNF-α and IL-1β are mediated by the Rip/TNF receptor-associated factor (TRAF2, TRAF6)→NF-κB-inducing kinase (NIK)→IKK-α/IKKβ pathway (Cao et al., 1996; Hsu et al., 1995; 1996; Woronicz et al., 1997). Since TNF-α and IL-1β also elicit the activation of JNK, we cannot completely rule out the possibility that MEKK1 may also play a marginal role in the activation of NF-κB by mixed cytokines treatment. Two different signalling pathways of LPS/IFN-γ and mixed cytokines converge in the activation of NF-κB. Nepalolide A may act on different (or same) targets within these signalling pathways and so exert differential effects on the degradation of IκB and activation of NF-κB.
Studies into IκB degradation showed a resynthesis of IκB-α following the degradation promoted by LPS/IFN-γ or mixed cytokines. It has been reported that both IKK-α and IKK-β phosphorylate IκB bound to NF-κB more efficiently than they phosphorylate free IκB. This result explains how free IκB can accumulate in cells in which IKK is still active and thus can contribute to the termination of NF-κB activation (Zandi et al., 1998). Studies on the promoter analysis of the gene encoding IκB-α have demonstrated that there are three NF-κB binding sites located in promoter region (Ito et al., 1994). Therefore, the delayed resynthesis of IκB-α observed in the present study further supports the idea of nepalolide A having inhibitory effects on NF-κB transcriptional competency.
In summary, nepalolide A inhibited the LPS/IFN-γ- and mixed cytokines-stimulated accumulation of nitrite and increased activity of iNOS in a concentration-dependent manner in C6 glioma cells. Similar results were obtained in studies employing primary astrocytes treated with mixed cytokines. The inhibitory effect of nepalolide A on the activity of iNOS occurred as a result of suppression of iNOS expression. Nepalolide A suppressed the activation of NF-κB by modulating the degradation of IκB-α and IκB-β triggered by iNOS inducers, thereby attenuating the expression of iNOS.
Acknowledgments
This study was supported by National Science Council Grant NSC87-2811-B-077-0001, NSC87-2314-B-077-017 and NSC88-2314-B-077-011, Taiwan, Republic of China.
Abbreviations
- AP-1
activator protein 1
- Erk
extracellular signal-regulated kinase
- GAS
IFN-γ-activated site
- G3PDH
glyceraldehyde-3-phosphate dehydrogenase
- IFN-γ
interferon-γ
- IκB
inhibitor of nuclear factor-κB
- IKK
IκB kinase
- IL
interleukin
- JAK
janus kinase
- JNK
c-jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MEKK
Erk kinase kinase
- NF-κB
nuclear factor-κB
- NOS
nitric oxide synthase
- PCR
polymerase chain reaction
- PKC
protein kinase C
- RT
reverse transcriptase
- Stat1
signal transducer and activator of transcription 1
- TNF-α
tumor necrosis factor-α
- TRAF
TNF receptor-associated factor
References
- BAEUERLE P.A., HENKEL T. Function and activation of NF-κB in the immune system. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- BERGMANN M., HART L., LINDSAY M., BARNES P.J., NEWTON R. IκBα degradation and nuclear factor-κB DNA binding are insufficient for interleukin-1β and tumor necrosis factor-α-induced κB-dependent transcription. J. Biol. Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- BHAT N.R., ZHANG P., LEE J.C., HOGAN E.L. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J. Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO Z., XIONG J., TAKEUCHI M., KURAMA T., GOEDDEL D.V. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- CHEN C.C., WANG J.K., CHEN W.C., LIN S.B. Protein kinase Cζ mediates lipopolysaccharide-induced nitric-oxide synthase expression in primary astrocytes. J. Biol. Chem. 1998;273:19424–19430. doi: 10.1074/jbc.273.31.19424. [DOI] [PubMed] [Google Scholar]
- CHIOU W.F., SUNG Y.J., LIAO J.F., SHUM A.Y.C., CHEN C.F. Inhibitory effect of dehydroevodiamine and evodiamine on nitric oxide production in cultured murine macrophage. J. Nat. Prod. 1997;60:708–711. doi: 10.1021/np960495z. [DOI] [PubMed] [Google Scholar]
- COLE R., DE VELLIS J.Astrocyte and oligodendrocyte cultures Protocols for Neural Cell Culture 1992The Humana Press Inc: Totowa; 65–78.In: Fedoroff, S. & Richardson, A (eds) [Google Scholar]
- DAWSON V.L., DAWSON T.M. Nitric oxide in neuronal degeneration. Proc. Soc. Exp. Biol. Med. 1996;221:33–40. doi: 10.3181/00379727-211-43950e. [DOI] [PubMed] [Google Scholar]
- DAWSON T.M., SNYDER S.H. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J. Neurosci. 1994;14:5147–5159. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DÍAZ-GUERRA M.J.M., BODELÓN O.G., VELASCO M., WHELAN R., PARKER P.J., BOSCÁ L. Up-regulation of protein kinase C-ε promotes the expression of cytokine-inducible nitric oxide synthase in RAW 264.7 cells. J. Biol. Chem. 1996;271:32028–32033. doi: 10.1074/jbc.271.50.32028. [DOI] [PubMed] [Google Scholar]
- DIDONATO J.A., HAYAKAWA M., ROTHWARF D.M., ZANDI E., KARIN M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- FEINSTEIN D.L., GALEA E., AQUINO D.A., LI G.C., XU H., REIS D.J. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NF-κB activation. J. Biol. Chem. 1996;271:17724–17732. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- GALEA E., XU H., FOX E.S., REIS D.J., FEINSTEIN D.L. CD14 mediate endotoxin induction of nitric oxide synthase in cultured brain glial cells. J. Neuroimmunol. 1996;64:19–28. doi: 10.1016/0165-5728(95)00143-3. [DOI] [PubMed] [Google Scholar]
- GAO J., MORRISON D.C., PARMELY T.J., RUSSELL S.W., MURPHY W.J. An interferon-γ-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-γ and lipopolysaccharide. J. Biol. Chem. 1997;272:1226–1230. doi: 10.1074/jbc.272.2.1226. [DOI] [PubMed] [Google Scholar]
- HAMBLETON J., WEINSTEIN S.L., LEM L., DEFRANCO A.L. Activation of c-jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophage. Proc. Natl. Acad. Sci. U.S.A. 1996;93:2774–2778. doi: 10.1073/pnas.93.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRANO M., OSADA S.I., AOKI T., HIRAI S.I., HOSAKA M., INOUE J.I., OHNO S. MEK kinase is involved in tumor necrosis factor α-induced NF-κB activation and degradation of IκB-α. J. Biol. Chem. 1996;271:13234–13238. doi: 10.1074/jbc.271.22.13234. [DOI] [PubMed] [Google Scholar]
- HSU H., HUANG J., SHU H.B., BAICHWAL V., GOEDDEL D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- HSU H., XIONG J., GOEDDEL D.V. The TNF receptor-1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;82:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- ITO C.Y., KAZANTSEV A.G., BALDWIN A.S., JR Three NF-κB sites in the IκB-α promoter are required for induction of gene expression by TNFα. Nucl. Acids Res. 1994;22:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARIN M., DELHASE M. JNK or IKK, AP-1 or NF-κB, which are the targets for MEK kinase 1 action. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9067–9069. doi: 10.1073/pnas.95.16.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOWLES R.G., MONCADA S. Nitric oxide synthase in mammals. Biochem. J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOVARIK P., STOIBER D., NOVY M., DECKER T. Stat1 combines signals derived from IFNγ and LPS receptors during macrophage activation. EMBO J. 1998;17:3660–3668. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE F.S., PETERS R.T., DANG L.C., MANIATIS T. MEKK1 activates both IκB kinase α and IκB kinase β. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN Y.L., OU J.C., KUO Y.H., LIN J.K., LEE K.H. Nepalolide A-D, four new sesquiterpene lactones from Carpesium nepalense. J. Nat. Prod. 1996;59:991–993. [Google Scholar]
- LIPTON S.A., CHOI Y.B., PAN Z.H., LEI S.Z., CHEN H.S., SUCHER N.J., LOSCALZO J., SINGEL D.J., STAMLER J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compound. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- MARKS-KONCZALIK J., CHU S.C., MOSS J. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor κB-binding sites. J. Biol. Chem. 1998;273:22201–22208. doi: 10.1074/jbc.273.35.22201. [DOI] [PubMed] [Google Scholar]
- MERCURIO F., ZHU H., MURRAY B.W., SHEVCHENKO A., BENNETT B.L., LI J.W., YOUNG D.B., BARBOSA M., MANN M. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- MERRILL J.E., IGNARRO L.J., SHERMAN M.P., MELINEK J., LANE T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- MITROVIC B., IGNARRO L.J., MONTESTRUQUE S., SMOLL A., MERRILL J.E. Nitric oxide as a potential pathological mechanism in demyelination: its differential effects on primary glial cells in vitro. Neurosci. 1994;61:575–585. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- MULLER J.M., LOMS ZIEGLER-HEITBROCKL H.W., BAEUERLE P.A. Nuclear factor kappa B, a mediator of lipopolysaccharide effects. Immunobiology. 1993;187:233–256. doi: 10.1016/S0171-2985(11)80342-6. [DOI] [PubMed] [Google Scholar]
- NAKANO H., SHINDO M., SAKON S., NISHINAKA S., MIHARA M., YAGITA H., OKUMURA K. Differential regulation of IκB kinase α and β by two upstream kinases, NF-κB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3537–3542. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHIYA T., UEHARA T., EDAMATSU H., KAZIRO Y., ITOH H., NOMURA Y. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is independent of interferon-γ-induced MAPK activation that is mediated by p21ras. FEBS Lett. 1997;408:33–38. doi: 10.1016/s0014-5793(97)00383-9. [DOI] [PubMed] [Google Scholar]
- PAHAN K., NAMBOODIRI A.M.S., SHEIKH F.G., SMITH B.T., SINGH I. Increasing cAMP attenuates induction of inducible nitric-oxide synthase in rat primary astrocytes. J. Biol. Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- RÉGNIER C.H., SONG H.Y., GAO X., GOEDDEL D.V., CAO Z., ROTHE M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- REIMANN T., BUSCHER D., HIPSKIND R.A., KRAUTWALD S., LOHMANN MATTHES M.L., BACCARINI M. Lipopolysaccharide induces activation of the Raf-1/MAP kinase pathway. A putative role for Raf-1 in the induction of the IL-1 β and the TNF-α genes. J. Immunol. 1994;153:5740–5749. [PubMed] [Google Scholar]
- SCHUMANN R.R., PFEIL D., FREYER D., BUERGER W., LAMPING N., KIRSCHNING C.J., GOEBEL U.B., WEBER J.R. Lipopolysaccharide and pneumococcal cell wall components activate the mitogen activated protein kinases (MAPK) erk-1, erk-2, and p38 in astrocytes. Glia. 1998;22:295–305. doi: 10.1002/(sici)1098-1136(199803)22:3<295::aid-glia8>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- THANOS D., MANIATIS T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- WORONICZ J.D., GAO X., CAO Z., ROTHE M., GOEDDEL D.V. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- XIE Q.W., KASHIWABARA Y., NATHAN C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- ZANDI E., CHEN Y., KARIN M. Direct phosphorylation of IκB by IKKα and IKKβ: Discrimination between free and NF-κB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]