

Abstract
Tricyclic antidepressants (TCAs) are associated with cardiovascular side effects including prolongation of the QT interval of the ECG. In this report we studied the effects of two TCAs (imipramine and amitriptyline) on ionic current mediated by cloned HERG potassium channels.
Voltage clamp measurements of HERG currents were made from CHO cells transiently transfected with HERG cDNA. HERG-encoded potassium channels were inhibited in a reversible manner by both imipramine and amitriptyline. HERG tail currents (IHERG) following test pulses to +20 mV were inhibited by imipramine with an IC50 of 3.4±0.4 μM (mean±s.e.mean) and a Hill coefficient of 1.17±0.03 (n=5). 3 μM amitriptyline inhibited IHERG by 34±6% (n=3). The inhibition showed only weak voltage dependence.
Using an ‘envelope of tails' comprised of pulses to +20 mV of varying durations, the τ of activation was found to be 155±30 ms for control and 132±26 ms for 3 μM imipramine (n=5). Once maximal channel activation was achieved after 320 ms (as demonstrated by maximal tail currents), further prolongation of depolarization did not increase imipramine-mediated HERG channel inhibition.
Taking current measurements every second during a 10 s depolarizing pulse from −80 mV to 0 mV, block was observed during the first pulse in the presence of imipramine and the level of IHERG block was similar throughout the pulse (n=5).
A three pulse protocol (two depolarizing pulses to +20 mV separated by 20 ms at −80 mV) revealed that imipramine did not significantly alter the kinetics of IHERG inactivation. The τ of inactivation was 8±2 ms and 5.6±0.4 ms (n=5) in the absence and presence of 3 μM imipramine, respectively, and currents inactivated to a similar extent.
Our data are consistent with TCAs causing components of block of the HERG channel in both the closed and open states. Any component of open channel block occurs rapidly upon depolarization. Inhibition of IHERG by the prototype TCAs imipramine and amitriptyline may suggest a mechanism for QT prolongation associated with risks of arrhythmia and sudden death that accompany high concentrations of TCAs following overdose.
Keywords: HERG, imipramine, amitriptyline, tricyclic antidepressant, ‘rapid' delayed rectifier, IKr, cardiac arrhythmia, long QT syndrome, potassium channel
Introduction
Imipramine and amitriptyline are agents belonging to the tricyclic class of antidepressants (TCAs), and are used for treatment of depression and other conditions including panic disorder, obsessive compulsive disorder, bulimia, and nocturnal enuresis (Gorman, 1996; Daly & Wilens, 1998). TCAs have a wide spectrum of actions including inhibition of noradrenaline and 5-hydroxytryptamine reuptake at nerve endings, as well as anti-histaminergic, anticholinergic activities, and block of sodium channels (Coupland et al., 1997; Stahl, 1998).
Tricyclic antidepressants such as imipramine and amitriptyline have cardiovascular side effects including orthostatic hypotension, atrioventricular conduction delay, reduced heart rate variability in response to exercise, tachycardia, syncope, and lengthening of the QT interval (which may be associated with arrhythmias), particularly in cases with high dosages and in patients with concurrent cardiovascular disease (Coupland et al., 1997). Overdose of antidepressant agents (and particularly TCAs) is epidemiologically important from the standpoint of mortality (Henry, 1997).
The cardiac actions of TCAs have been suggested to result from ‘quinidine-like' activity (Pentel & Benowitz, 1986). Quinidine, a type I antiarrhythmic, is known to cause slowing of phase 0 depolarization of the action potential resulting in slowing of conduction through the His-Purkinje system and myocardium. It is well-established that, in addition to its antiarrhythmic effects, quinidine can have pro-arrhythmic activity (Grace & Camm, 1998). Recent data suggest that quinidine and mexilitene (another type I antiarrhythmic), can also inhibit delayed rectifier potassium currents (IK; Balser et al., 1991; Mitcheson & Hancox, 1997; Po et al., 1999). The rapid component of the delayed rectifier potassium current (IKr) is widely accepted as being mediated by the HERG gene-product, and block of this current can lead to QT prolongation and its concomitant risk of the polymorphic ventricular tachyarrhythmia, torsade de pointes (Sanguinetti et al., 1995; Sanguinetti & Keating, 1997). Furthermore, a variety of pharmacological agents known to cause QT elongation with its associated risk of arrhythmia have been shown to block IHERG, including type III antiarrhythmics (Trudeau et al., 1995; Spector et al., 1996a; Busch et al., 1998), non-sedating antihistamines (Roy et al., 1996; Suessbrich et al., 1996), and antipsychotics (Suessbrich et al., 1997a).
Imipramine has been shown to inhibit IKr in guinea-pig ventricular myocytes (Valenzuela et al., 1994), but the characteristics and concentration-dependence of the inhibition were not studied. In this study, therefore, we investigated the ability of the TCAs imipramine and amitriptyline to block heterologously expressed IHERG. The concentration-dependence and mechanism underlying inhibition were also studied.
Methods
DNA for the HERG transcript (kindly provided by Mike Sanguinetti and Mark Keating, Salt Lake City, U.S.A.) was subcloned into pGW1H (kindly provided by Graham Roberts, British Biotech), which includes a CMV promoter and a polyadenylation site. GFP in pCMX (kindly provided by Jeremy Tavare, Bristol, U.K.), also driven by a CMV promoter, was co-transfected at a 3 : 1 HERG:GFP ratio into CHO cells using lipofectamine (Life Technologies International, Basingstoke, U.K.). Lipofection followed the manufacturer's recommendations, with 1 μg of DNA and 7 μl of lipofectamine used for each 30 mm culture dish; the first stage of lipofection took place in Optimem medium (Life Technologies, Basingstoke, U.K.) and lasted for 4 h. Low passage CHO cells were maintained in Ham's F-12 medium+glutamax (Life Technologies International, Basingstoke, U.K.) with 100 u ml−1 penicillin and 100 μg ml−1 streptomycin (Sigma, Poole, U.K.), and for experiments the cells were grown on glass coverslips. Cells were used for electrophysiology within 30 h of transfection.
Perforated patch clamp of cells was achieved with borosilicate patch pipettes (Drummond Scientific, Broomall, PA, U.S.A.), with a resistance of 1–2 MΩ. Pipettes were sylgard-coated and fire polished to a final resistance of 2–2.5 MΩ. The pipette filling solution consisted of (in mM) KCl 130, MgCl2 1, EGTA (K+) 5, ATP (Mg2+) 5, HEPES 10, at pH 7.2. Gramicidin (Sigma, Poole, U.K.) freshly dissolved in dimethylsulfoxide (DMSO) was added to an aliquot of the pipette filling solution at a final concentration of 32 μg ml−1, and used to back fill the pipettes; the final concentration of DMSO in the pipette solution was 0.4%. Some experiments were performed in the whole cell mode without gramicidin, using the same pipette filling solution; where appropriate these experiments are noted in the text. Cells were continuously superfused at the rate of 1.5 ml min−1 with an extracellular solution consisting of (in mM:) NaCl 140, KCl 4, CaCl2 2, MgCl2 1, HEPES 5, glucose 10, pH 7.4 at ambient temperature. Both imipramine (Sigma, Poole, U.K.) and amitriptyline (Sigma, Poole, U.K.) were made as 10 mM stock solutions in H2O and were diluted at least 100 fold in extracellular solution. Imipramine and amitriptyline stocks were added to the superfusion solution to give final concentrations between 0.1 and 100 μM. Cells were allowed to equilibrate for five (Figures 1 and 2) or 10 min (Figures 3, 4 and 5) in the appropriate agents before measurements were made; similar effects were noted with either incubation time. Voltage protocols were applied and currents recorded using an Axopatch 200B patch clamp amplifier in combination with a DigiData 1200 A/D converter (Axon Instruments, CA, U.S.A.), employing a PC and software written in Axobasic. GFP positive cells were identified using fluorescence microscopy (excitation at 495 nm, emission>530 nm) and were voltage-clamped at a holding potential of −80 mV. Following membrane perforation with gramicidin, the series resistance was 10–15 MΩ and was compensated by 70–80%. IHERG was measured as the peak tail current at −40 mV following a 400 ms voltage step from −80 mV to the test voltage. During drug equilibration the membrane potential of the cell under study was held at −80 mV. Before measurements were made in the presence of TCAs, protocols were applied until a steady-state response was achieved, with the exception of the long pulse protocol, which was always run after equilibration of the agent at the holding potential of −80 mV. Data were stored on PC and analysed off-line (Axobasic; Excel, Microsoft; Origin, Microcal). Mean values and s.e.means for the effects of each concentration were calculated by pooling data for all cells, while IC50s and Hill coefficients were calculated for each cell, and mean values were then calculated.
Figure 1.
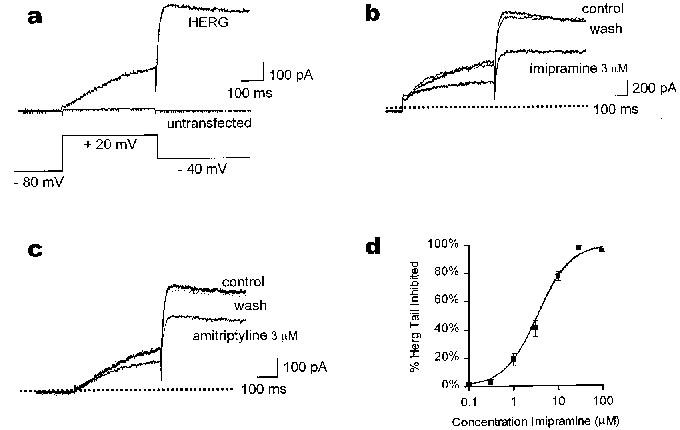
TCAs inhibit current mediated by heterologous HERG. (a) shows the currents elicited from a CHO cell transfected with HERG and from an untransfected cell. Membrane potential was held at −80 mV, depolarized to +20 mV for 400 ms, before the tail was observed at −40 mV. Total current elicited from the untransfected cell during the pulse was under 20 pA, and no tail current could be detected. The HERG current of a typical cell in extracellular solution is shown in (b) in the absence of drug (control), in the presence of 3 μM imipramine, and after washout. The holding potential and voltage steps are identical to those in (a). Interpulse interval was 15 s. (c) shows a typical cell superfused with 3 μM amitriptyline under the same conditions as in (b). The dotted line in (b) and (c) indicates zero current level. In (d), the concentration response curve shows mean (±s.e.mean) inhibition of tail currents measured with the same protocol as above (n=5; at 30 and 100 μM imipramine data using whole cell patch clamp and gramicidin perforated patches were pooled).
Figure 2.
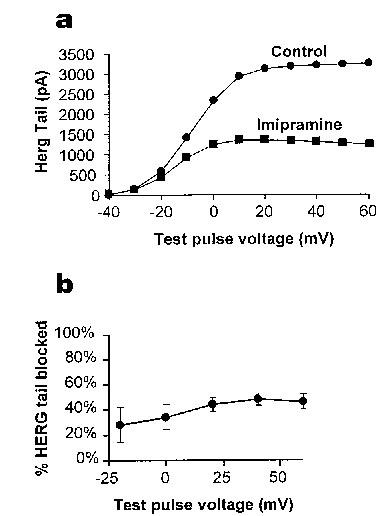
Voltage dependence of the effects of imipramine. (a) demonstrates the effect of voltage on imipramine-mediated block of IHERG. It shows a typical plot of the tail currents (at −40 mV) under these conditions against test pulse potential. In (b) percentage inhibition evoked by 3 μM imipramine at different voltages is shown, with the imipramine-sensitive current being normalized in each cell to the current at the same voltage in the absence of imipramine (n=5).
Figure 3.
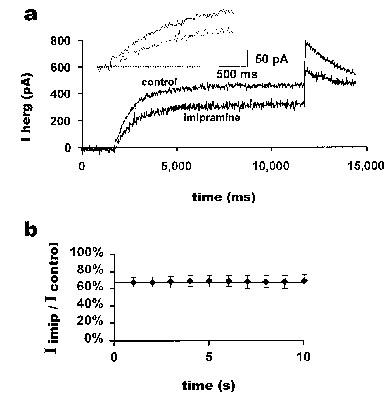
Imipramine inhibition during a long pulse. The currents from a 10 s pulse protocol applied to a typical cell before and after a 10 min application of 3 μM imipramine are shown in (a). The membrane potential was held at −80 mV, and HERG currents were evoked with a 10 s pulse to +20 mV. The cell was held at −80 mV for 10 min while imipramine was applied. The inset shows an expanded portion of the same record at the beginning of the pulse. From the above data, the ratio of current in the presence of imipramine to current before application of imipramine at successive 1 s time points is shown in (b). The horizontal line at the first value of 67% is provided to aid comparison of the data points (n=5).
Figure 4.
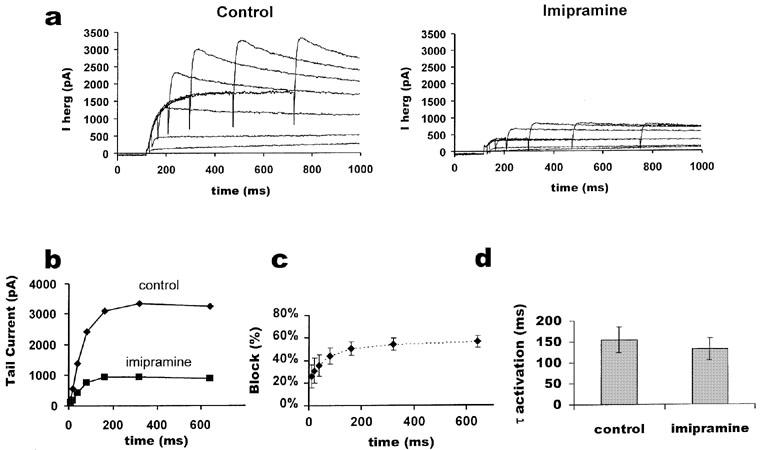
Activation time course of IHERG with and without imipramine. (a) illustrates an ‘envelope of tails' test performed on the same cell before and after block with 3 μM imipramine. Membrane potential was held at −80 mV, depolarized to +20 mV for varying durations of time, and then tail currents were observed at −40 mV. The inter-pulse interval was 30 s. In (b) the peaks of the tail currents were plotted against the duration of the depolarizing pulse. The percentage of current blocked shown in (c) was calculated from the ratio of the matched peak tail currents for imipramine and control at each time point (n=5). (d) shows the mean of the calculated activation time constants (n=5) derived from a fit with a single exponential to the currents as shown in (b) (paired t-test, P>0.05).
Figure 5.
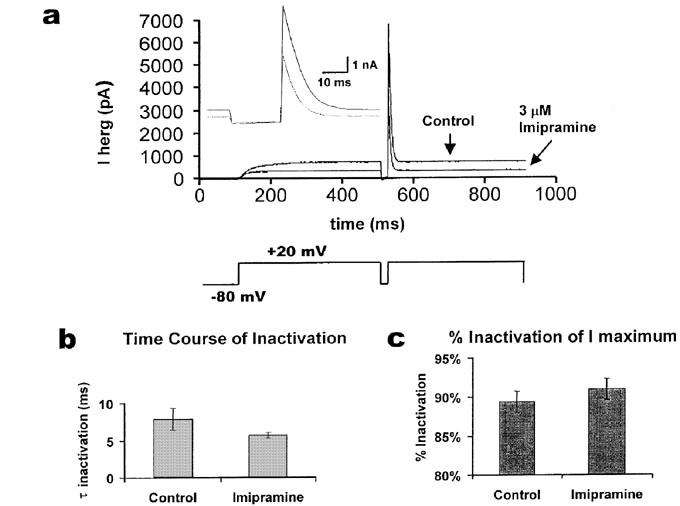
Inactivation of HERG in the presence and absence of imipramine. A three-pulse protocol was used in which the cells were held at −80 mV, depolarized to +20 mV for 400 ms, repolarized for 20 ms to −80 mV, and stepped again to +20 mV for 400 ms as shown in (a) lower panel. Typical responses are shown in the upper panel for control and 3 μM imipramine. During the brief pulse to −80 mV, rapid recovery from inactivation occurred; the time course for current deactivation is long with respect to this period of time. The inset shows the same current records (just before and after the 20 ms step to −80 mV) on an expanded time scale. The time course of inactivation during the second pulse to +20 mV was fitted with a single exponential and compared in (b) (paired t-test, n=5, P=0.23). The extent of inactivation in the two conditions was compared in (c) (paired t-test, n=5, P=0.21).
Results
HERG mediates an outward K+ current with comparatively rapid inactivation and slow deactivation kinetics (Sanguinetti et al., 1995). The rapid inactivation results in inward rectification of the current at positive potentials (Smith et al., 1996; Spector et al., 1996b). These properties result in a distinct current profile with voltage clamp test commands to positive potentials both in transfected cells expressing heterologous HERG and in cardiomyocytes having native IKr (Hancox et al., 1998). Figure 1a shows the currents elicited from a cell transfected with HERG and from an untransfected cell from the holding potential of −80 mV during a test pulse to +20 mV and on repolarization to −40 mV. In the HERG-transfected cell, outward current developed during the pulse and was followed by a large outward tail current upon repolarization to −40 mV. No tail currents were observed in untransfected cells (Figure 1a).
Amitriptyline and imipramine reduced reversibly both IHERG during the test potential and the IHERG tail (Figure 1b and c). Using the same voltage commands as in Figure 1a, imipramine (3 μM) reversibly inhibited 41±4% (mean±s.e.mean) of HERG tail current, and the current at the end of the test pulse was reduced by 42±6% in the same cells (n=5). For each cell the degree of block observed during the pulse and during the tail was similar (paired t-test, P=0.73). Amitriptyline (3 μM) inhibited the IHERG tail by 34±6%, and the current during the test pulse was reduced by 35±9% (n=3). A similar inhibition of tail currents was observed when making measurements in the whole cell configuration (41±5%, 3 μM imipramine, n=5), however, the effects were not fully reversible on wash out.
The concentration-dependence of the imipramine effect is shown in Figure 1d. The data were fit with the Hill equation, which gave an IC50 of 3.4±0.4 μM and a Hill coefficient of 1.17±0.03 (n=5). The observed TCA inhibition of IHERG occurred within the clinically observed range of 1–5 μM for imipramine (Marshall & Forker, 1982) and 0.05–6.9 μM for amitriptyline (Pike & Skuterud, 1982a). Complete block of IHERG could be achieved with 30 μM imipramine or 30 μM amitriptyline (data not shown).
The voltage dependence of the effects of imipramine on IHERG was investigated over a range of test pulses from −40 mV to +60 mV. A representative current voltage plot for the effects of 3 μM imipramine on HERG tail currents is shown in Figure 2a. The degree of imipramine block of IHERG tails is compared at different voltages in Figure 2b. The observed inhibition of HERG appeared weakly voltage sensitive, with more positive test potentials giving a small increase in block, although this difference was not significant. In particular, tail currents elicited following a pulse to −20 mV were blocked by 28±14% by 3 μM imipramine, whereas tail currents elicited following a pulse to +40 mV were blocked by 49±5% (n=5, paired t test, P>0.1).
A ‘long pulse' protocol of 10 s from −80 mV to 0 mV was used to investigate whether the degree of block by imipramine might alter during sustained depolarization. The pulse was first applied in imipramine-free solution. The membrane potential was then held at −80 mV with no stimulation for 10 min in the presence of 3 μM imipramine, after which the long pulse protocol was again applied. In contrast to the methanesulfonanilide class III antiarrhythmic agents E-4031, MK-499 and dofetilide (Sanguinetti et al., 1995; Spector et al., 1996a; Snyders & Chaudhary, 1996), block of IHERG was observed during the first pulse after 10 min application of the drug (Figure 3a, n=5). An expanded view of the early part of the pulse suggests that block may initially increase over time but that some block is present immediately (Figure 3a, inset). Comparing IHERG amplitudes every second, we observed that the current during the pulse in the presence of imipramine did not decrease, and the ratio of current in the presence of imipramine to control showed no significant decrease (Figure 3b, one-way anova, F(9,40)=0.0115, n=5 cells, not significant).
The time course of IHERG activation is typically obtained using an ‘envelope of tails' protocol (Trudeau et al., 1995), in which tail current amplitudes following a series of pulses of increasing duration are measured, because inactivation plays a strong role early in the pulse. To test imipramine's effects on activation, the membrane potential was held at −80 mV, and pulses of increasing duration to +20 mV were applied. Tail currents at −40 mV were measured after each test pulse. Sample traces with and without imipramine (3 μM) are shown in Figure 4a. The peak tail current for each trace is plotted against test pulse duration in Figure 4b. Data from different cells were normalized and pooled in Figure 4c, showing that the inhibition of HERG channels increased over the first 320 ms. A further increase in pulse duration did not further increase imipramine-mediated IHERG inhibition (n=5). The rate of IHERG activation was determined by fitting the peak amplitudes of the tail currents with a single exponential function. Figure 4d shows mean data for five cells. The τ of activation was slightly reduced in the presence of imipramine relative to control (consistent with the increase in fractional block of tail current observed during the early part of the envelope, Figure 4c); however, the observed difference was not statistically significant (P>0.05). Taken collectively, the data in Figures 3 and 4 suggest that part of the block of IHERG by imipramine may result from drug binding to the resting channel, with a further component which occurs rapidly during depolarization, consistent with an open channel block.
To test whether inactivation plays a role in the block, a three pulse protocol (Smith et al., 1996) was used in which the cell was held at −80 mV, depolarized to +20 mV for 400 ms, repolarized briefly to −80 mV for 20 ms, and then depolarized again to +20 mV for 400 ms. Representative traces in control and with imipramine (3 μM) are shown in Figure 5a. The rapid decrease in current after the second pulse to +20 mV (Figure 5a, inset) could be fitted by a single exponential; the inactivation time course was more rapid in the presence of imipramine, but the difference between the inactivation time constants was not significant (Figure 5b). The extent of current inactivation was calculated from the ratio of the final current (at 50 ms after the second depolarization) to the peak current (Figure 5c). The current inactivation was observed to be increased in the presence of imipramine (3 μM), but not significantly.
Discussion
While more than one type of K+ channel can be blocked by TCAs, for example the transient outward current Ito (Casis & Sanchez-Chapula, 1998), it is now well-established that pharmacological block of the HERG-encoded channel can be associated with arrhythmia in an analogous manner to genetic mutations in HERG being responsible for chromosome 7-associated familial ‘Long QT Syndrome' (Curran et al., 1995). Here we show that two TCAs block heterologously expressed HERG at concentrations similar to clinically measured serum levels. Any interpretation regarding clinical relevance must be qualified, however, as the unbound concentration of these drugs may not necessarily reflect drug levels in particular organ systems (Hilberg et al., 1999; Pike et al., 1982b).
Although this is the first direct demonstration of amitriptyline affecting HERG current, there is evidence that imipramine can completely block native cardiac IKr at 1 μM in guinea-pig myocytes (Valenzuela et al., 1994). A reduced sensitivity to drugs of heterologously expressed HERG to that of IKr has been observed previously (Spector et al., 1996a; Suessbrich et al., 1996), particularly for HERG expressed in oocytes, in which either the vitelline membrane, or yolk-sac may limit drug access to the channel and raise the apparent IC50 compared to that observed for HERG in mammalian cell lines (e.g. Po et al., 1999). The apparently higher sensitivity of native IKr than HERG to imipramine may be due to the recording conditions in the two studies, intrinsic differences in the two cell types, to differences in post-translational modifications, or to different auxiliary subunits (e.g. minK and MiRP1) which can be associated with the HERG potassium channel alpha subunit (McDonald et al., 1997; Abbott et al., 1999). However, because guinea-pig IK is a composite current composed of IKr and IKs, Valenzuela et al. (1994) reported that it was not possible to study the detailed effects of imipramine on IKr selectively nor to obtain an IC50 in their experiments. By studying cloned HERG channels, we have been able to investigate the effects of TCAs in more detail than is possible with composite guinea-pig IK.
The imipramine-mediated block is complex in that it occurs while the channel is maintained at −80 mV (at which potential HERG channels are closed; Sanguinetti et al., 1995; Zhou et al., 1998), and the block also increases as activation increases. Within 1 s at +20 mV the block reaches its maximum level. This differs from the methanesulphonanilides dofetilide (Snyders & Chaudhary, 1996) and MK-499 (Spector et al., 1996a) which mediate no significant block on the first pulse after drug addition when the cells are held at −80 mV, and which mediate significant increases in block when channels are held in the open state during a long depolarizing pulse.
Imipramine appears to decrease IHERG amplitude without significant effects on channel inactivation. Both the degree of current inactivation and the time course of inactivation were not significantly changed by imipramine; this suggests that under these conditions the block is not preferentially mediated by binding to the inactivated channel as it is in the presence of terfenadine (Suessbrich et al., 1996), astemizole (Suessbrich et al., 1996), haloperidol (Suessbrich et al., 1997a) and clofilium (Suessbrich et al., 1997b). The block by imipramine is equivalent during the pulse and tail currents. The data are consistent with the drug binding both to channels in the resting state (accounting for block observed during brief pulses and at the start of the ‘long pulse' in Figure 3), and subsequent to this, rapid binding to open channels occurring early during a depolarizing step (Figure 4). This is consistent with the apparent acceleration of IHERG activation observed during the ‘envelope of tails', although the difference in τ activation did not attain statistical significance. The block was only weakly voltage dependent and not significantly voltage dependent for voltages above 0 mV, the range in which full activation occurs. Imipramine and amitriptyline are weak bases (pKAs of 9.5 and 9.4 respectively) and thus at physiological pH they are mostly charged (Jack, 1992). The block by the imidazolidinedione azimilide, another class III antiarrhythmic agent, has been shown to be reverse use-dependent and voltage independent, and its block does not affect inactivation (Busch et al., 1998). The present data show that imipramine has a pharmacological profile which is more similar to azimilide's than to that of the methanesulphonanilides.
In addition to effects on HERG channels in the cardiovascular system, potassium channels in the nervous system also may be affected by TCAs. In rat isolated sympathetic neurones TCAs have been shown to block the voltage gated delayed rectifier potassium current (Wooltorton & Mathie, 1993). Our observations that the block of the heterologous channel does not alter dramatically the kinetics of IHERG, and that the block is relatively voltage independent are in accord with observations of TCA block of the sustained component of IKv made in sympathetic ganglia (Wooltorton & Mathie, 1993). HERG has been demonstrated to be involved in spike-frequency adaptation (Chiesa et al., 1997), and three related members of the erg family have been identified in nervous tissue (Shi et al., 1997). The three erg family members are expressed in peripheral sympathetic ganglia, potentially implicating these transcripts in the mediation of sympathetic outflow to the heart.
Previously, TCAs were known to have type I antiarrhythmic activity and were recommended for patients with ventricular arrhythmias (Bigger et al., 1977). Due to more recent evidence showing that type I antiarrhythmics have significant pro-arrhythmic effects after myocardial infarct, the cardiac actions of TCAs have led to the recommendation that TCAs be avoided under those circumstances (Glassman & Roose, 1994). The present study shows that clinically measured concentrations of imipramine and amitriptyline can inhibit the current mediated by HERG. It is therefore possible that inhibition of HERG-IKr contributes to the observed cardiovascular side-effects of TCAs; this action could be especially marked at elevated drug concentrations which might occur during overdose.
Acknowledgments
This work was supported by the British Heart Foundation and by the Wellcome Trust (grants to E.P. Seward and J.C. Hancox). We gratefully acknowledge David Nutt, Robert Meech, Peter Taberner and Richard Canter for valuable discussion, Mike Sanguinetti and Mark Keating for generous donation of the original HERG clone, and William Orbit for technical support during the study.
Abbreviations
- HERG
Human Ether-a-go-go Related Gene
- IHERG
current mediated by the HERG channel
- TCA
Tricyclic Antidepressant
- IK
delayed rectifier potassium current
- IKr
‘rapid' component of the delayed rectifier current
- GFP
Green Fluorescent Protein
- CMV
Cytomegalovirus
- s.e.mean
standard error of the mean
References
- ABBOTT G.W., SESTI F., SPLAWSKI I., BUCK M., LEHMANN M.H., TIMOTHY K.W., KEATING M.T., GOLDSTEIN S.A.N. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- BALSER J.R., BENNETT P.B., HONDEGHEM L.M., RODEN D.M. Suppression of time-dependent outward current in guinea-pig ventricular myocytes. Actions of quinidine and amiodarone. Circ. Res. 1991;69:519–529. doi: 10.1161/01.res.69.2.519. [DOI] [PubMed] [Google Scholar]
- BIGGER J.T., GIARDINA E.G., PEREL J.M., KANTOR S.J., GLASSMAN A.H. Cardiac antiarrhythmic effect of imipramine hydrochloride. N. Engl. J. Med. 1977;296:206–208. doi: 10.1056/NEJM197701272960407. [DOI] [PubMed] [Google Scholar]
- BUSCH A.E., EIGENBERGER B., JURKIEWICZ N.K., SALATA J.J., PICA A., SUESSBRICH H., LANG F. Blockade of HERG channels by the class III antiarrhythmic azimilide: mode of action. Br. J. Pharmacol. 1998;123:23–30. doi: 10.1038/sj.bjp.0701575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASIS O., SANCHEZ-CHAPULA J.A. Disopyramide, imipramine, and amitriptyline bind to a common site on the transient outward K+ channel. J. Cardiovasc. Pharm. 1998;32:521–526. doi: 10.1097/00005344-199810000-00003. [DOI] [PubMed] [Google Scholar]
- CHIESA N., ROSATI B., ARCANGELI A., OLIVOTTO M., WANKE E. A novel role for HERG K+ channels: spike-frequency adaptation. J. Physiol. (London) 1997;501.2:313–318. doi: 10.1111/j.1469-7793.1997.313bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUPLAND N., WILSON S., NUTT D. Antidepressant drugs and the cardiovascular system: a comparison of tricyclics and selective serotonin reuptake inhibitors and their relevance for the treatment of psychiatric patients with cardiovascular problems. J. Psychopharmacol. 1997;11:83–92. doi: 10.1177/026988119701100118. [DOI] [PubMed] [Google Scholar]
- CURRAN M.E., SPLAWSKI I., TIMOTHY K.W., VINCENT G.M., GREEN E.D., KEATING M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- DALY J.M., WILENS T. The use of tricyclic antidepressants in children and adolescents. Pediatr. Clin. North Am. 1998;45:1123–1135. doi: 10.1016/s0031-3955(05)70065-1. [DOI] [PubMed] [Google Scholar]
- GLASSMAN A.H., ROOSE S.P. Risks of antidepressants in the elderly: tricyclic antidepressants and arrhythmia-revising risks. Gerontology. 1994;40 Suppl 1:S15–S20. doi: 10.1159/000213616. [DOI] [PubMed] [Google Scholar]
- GORMAN J.M. Comorbid depression and anxiety spectrum disorders. Depress. Anxiety. 1996;4:160–168. doi: 10.1002/(SICI)1520-6394(1996)4:4<160::AID-DA2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- GRACE A.A., CAMM A.J. Quinidine. N. Engl. J. Med. 1998;338:35–45. doi: 10.1056/NEJM199801013380107. [DOI] [PubMed] [Google Scholar]
- HANCOX J.C., LEVI A.J., WITCHEL H.J. Time course and voltage dependence of expressed HERG current compared with native ‘rapid' delayed rectifier K current during the cardiac ventricular action potential. Pflügers Arch. 1998;436:843–853. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- HENRY J.A. Epidemiology and relative toxicity of antidepressant drugs in overdose. Drug Safety. 1997;16:374–390. doi: 10.2165/00002018-199716060-00004. [DOI] [PubMed] [Google Scholar]
- HILBERG T., ROGDE S., MORLAND J. Postmortem drug redistribution–human cases related to results in experimental animals. J. Forensic Sci. 1999;44:3–9. [PubMed] [Google Scholar]
- JACK D.B. Handbook of Clinical Pharmacokinetic Data. Macmillan: Basingstoke; 1992. [Google Scholar]
- MARSHALL J.B., FORKER A. Cardiovascular effects of tricyclic antidepressant drugs: therapeutic usage, overdose, and management of complications. Am. Heart J. 1982;103:401–414. doi: 10.1016/0002-8703(82)90281-2. [DOI] [PubMed] [Google Scholar]
- MCDONALD T.V., YU Z., MING Z., PALMA E., MEYERS M.B., WANG K.W., GOLDSTEIN S.A.N., FISHMAN G.I. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- MITCHESON J.S., HANCOX J.C. Modulation by mexiletine of action potentials, L-type Ca current and delayed rectifier K current recorded from isolated rabbit atrioventricular nodal myocytes. Pflügers Arch. 1997;434:855–858. doi: 10.1007/s004240050476. [DOI] [PubMed] [Google Scholar]
- PENTEL P.R., BENOWITZ N.L. Tricyclic antidepressant poisoning. Management of arrhythmias. Med. Toxicol. 1986;1:101–121. doi: 10.1007/BF03259831. [DOI] [PubMed] [Google Scholar]
- PIKE E., SKUTERUD B. Plasma binding variations of amitriptyline and nortriptyline. Clin. Pharmacol. Ther. 1982a;32:228–234. doi: 10.1038/clpt.1982.152. [DOI] [PubMed] [Google Scholar]
- PIKE E., SKUTERUD B., KIERULF P., LUNDE P.K. Significance of lipoproteins in serum binding variations of amitriptyline, nortriptyline, and quinidine. Clin. Pharmacol. Ther. 1982b;32:599–606. doi: 10.1038/clpt.1982.209. [DOI] [PubMed] [Google Scholar]
- PO S.S., WANG D.W., YANG I.C.H., JOHNSON J.P., NIE L., BENNETT P.B. Modulation of HERG potassium channels by extracellular magnesium and quinidine. J. Cardiovasc. Pharmacol. 1999;33:181–185. doi: 10.1097/00005344-199902000-00002. [DOI] [PubMed] [Google Scholar]
- ROY M.L., DUMAINE R., BROWN A.M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., KEATING M.T. Role of delayed rectifier potassium channels in cardiac repolarization and arrhythmias. News Physiol. Sci. 1997;12:152–157. [Google Scholar]
- SHI W., WYMORE R.S., WANG H.-S., PAN Z., COHEN I.S., MCKINNON D., DIXON J.E. Identification of two nervous system-specific members of the erg potassium channel gene family. J. Neurosci. 1997;17:9423–9432. doi: 10.1523/JNEUROSCI.17-24-09423.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH P.L., BAUKROWITZ T., YELLEN G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., CHAUDHARY A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol. Pharm. 1996;49:949–955. [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M.E., KEATING M.T., SANGUINETTI M.C. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulphonanilides. Circ. Res. 1996a;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- SPECTOR P.S., CURRAN M.E., ZOU A., KEATING M.T., SANGUINETTI M.C. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 1996b;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAHL S.M. Basic psychopharmacology of antidepressants, part 1: Antidepressants have seven distinct mechanisms of action. J. Clin. Psychiatry. 1998;59 Suppl 4:S5–S14. [PubMed] [Google Scholar]
- SUESSBRICH H., SCHÖNHERR R., HEINEMANN S.H., ATTALI B., LANG F., BUSCH A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Brit. J. Pharmacol. 1997a;120:968–974. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUESSBRICH H., SCHÖNHERR R., HEINEMANN S.H., LANG F., BUSCH A.E., SUNAMI A., FAN Z., NITTA J., HIRAOKA M. Specific block of cloned Herg channels by clofilium and its tertiary analog LY97241. FEBS Lett. 1997b;414:435–438. doi: 10.1016/s0014-5793(97)01030-2. [DOI] [PubMed] [Google Scholar]
- SUESSBRICH H., WALDEGGER S., LANG F., BUSCH A.E. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- TRUDEAU M.C., WARMKE J.W., GANETZKY B., ROBERTSON G.A. HERG, a human inward rectifier in the voltage gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., SANCHEZ-CHAPULA J., DELPON E., ELIZALDE A., PEREZ O., TAMARGO J. Imipramine blocks rapidly activating and delays slowly activating K+ current activation in guinea-pig ventricular myocytes. Circ. Res. 1994;74:687–699. doi: 10.1161/01.res.74.4.687. [DOI] [PubMed] [Google Scholar]
- WOOLTORTON J., MATHIE A. Block of potassium currents in rat isolated sympathetic neurones by tricyclic antidepressants and structurally related compounds. Brit. J. Pharmacol. 1993;110:1126–1132. doi: 10.1111/j.1476-5381.1993.tb13931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Z., GONG Q., YE B., MAKIELSKI J.C., ROBERTSON G.A., JANUARY C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]