

Abstract
G-protein coupled receptors can exhibit constitutive activity resulting in the formation of active ternary complexes in the absence of an agonist. In this study we have investigated constitutive activity in C6 glioma cells expressing either the cloned δ-(OP1) receptor (C6δ), or the cloned μ-(OP3) opioid receptor (C6μ).
Constitutive activity was measured in the absence of Na+ ions to provide an increased signal. The degree of constitutive activity was defined as the level of [35S]-GTPγS binding that could be inhibited by pre-treatment with pertussis toxin (PTX). In C6δ cells the level of basal [35S]-GTPγS binding was reduced by 51.9±6.1 fmols mg−1 protein, whereas in C6μ and C6 wild-type cells treatment with PTX reduced basal [35S]-GTPγS binding by only 10.0±3.5 and 8.6±3.1 fmols mg−1 protein respectively.
The δ-antagonists N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH (ICI 174,864), 7-benzylidenenaltrexone (BNTX) and naltriben (NTB), in addition to clocinnamox (C-CAM), acted as δ-opioid receptor inverse agonists. Naloxone, buprenorphine, and naltrindole were neutral antagonists. Furthermore, naltrindole blocked the reduction in [35S]-GTPγS binding caused by the inverse agonists. The inverse agonists did not inhibit basal [35S]-GTPγS binding in C6μ or C6 wild-type cell membranes.
Competition binding assays in C6δ cell membranes revealed a leftward shift in the displacement curve of [3H]-naltrindole by ICI 174,864 and C-CAM in the presence of NaCl and the GTP analogue, GppNHp. There was no change in the displacement curve for BNTX or NTB under these conditions.
These data confirm the presence of constitutive activity associated with the δ-opioid receptor and identify three novel, non-peptide, δ-opioid inverse agonists.
Keywords: Constitutive activity, inverse agonism, δ-opioid receptor, μ-opioid receptor, G-protein, pertussis toxin
Introduction
The μ- (OP3), δ- (OP1), and κ- (OP2) opioid receptors belong to the superfamily of seven transmembrane domain, G-protein coupled receptors (Dhawan et al., 1996). In this family of receptors signal transduction is achieved by agonist occupied receptors activating one or more G-proteins. It is now widely accepted that G-protein coupled receptors can exist in equilibrium between an active (R*) and an inactive (R) state of the receptor (for review, see Leff, 1995). Even in the absence of agonist these receptors can maintain a conformation which can activate G-protein and so display constitutive activity (Costa & Herz, 1989, Lefkowitz et al., 1993). Ligands that preferentially stabilize the R form of the receptor abolish this spontaneous, agonist-independent activity and are termed inverse agonists. Neutral antagonists bind to the receptor but do not alter the equilibrium between active and inactive states. The only known opioid receptor inverse agonists are the δ-selective peptide ICI 174,864 (N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH) (Costa & Herz, 1989) and the structurally related ICI 154,129 (N,N-diallyl-Tyr-Gly-Gly-ψ-(CH2S)-Phe-Leu-OH) (Shaw et al., 1982) and diallyl-G (N,N-diallyl-Tyr-D-Leu-Gly-Tyr-Leu-OH) (Georgoussi & Zioudrou, 1993). No non-peptide δ-opioid inverse agonists have been identified to date. Moreover, no ligand has been discovered to have inverse agonist activity at the μ- or κ-opioid receptor, although Wang et al. (1994) have proposed that naloxone exhibits negative intrinsic activity in SH-SY5Y cells under conditions of narcotic tolerance.
In this study constitutive activity and inverse agonism in C6 glioma cells transfected with either the rat cloned δ- (C6δ) or μ- (C6μ) opioid receptor have been investigated. To study constitutive activity the agonist-independent binding of the stable GTP analogue [35S]-GTPγS was employed (Traynor & Nahorski, 1995), in the absence of sodium ions. It has been shown that spontaneous activity can be more easily detected in the absence of Na+ (Costa & Herz, 1989; Szekeres & Traynor, 1997). The findings confirm constitutive activity associated with the cloned δ-receptor and identify the long-lasting μ-antagonist clocinnamox (C-CAM, Comer et al., 1992), and the δ-receptor antagonists 7-benzylidenenaltrexone (BNTX, Portoghese et al., 1992) and naltriben (NTB, Sofuoglu et al., 1991) as novel non-peptide inverse agonists. Under similar conditions the μ-receptor exhibited no significant constitutive activity. A preliminary report of these data has been presented previously (Neilan et al., 1998).
Methods
Cell culture and membrane preparation
C6 glioma cells transfected with either the rat cloned μ- (C6μ; Thompson et al., 1993; Emmerson et al., 1996) or δ- (C6δ; Meng et al., 1995; Clark et al., 1997) receptor were used for this study. Cells were cultured under a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium supplemented with 10% foetal calf serum. For subculture one flask from each passage was grown in the presence of 1 mg ml−1 Geneticin. Cells used for experiments were grown in the absence of Geneticin with no significant reduction in receptor number. Once cells had reached confluency they were harvested in HEPES (N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid], 20 mM, pH 7.4)-buffered saline containing 1 mM EDTA, dispersed by agitation and collected by centrifugation at 500×g. The cell pellet was suspended in 50 mM Tris-HCl buffer, pH 7.4, and homogenized with a tissue tearor (Biospec Products). The resultant homogenate was centrifuged for 15 min at 40,000×g at 4°C and the pellet collected, resuspended and recentrifuged. The final pellet was resuspended in 50 mM Tris-HCl buffer pH 7.4; separated into 0.5 ml aliquots (0.75–1.0 mg protein) and frozen at −80°C. For pertussis toxin (PTX) treatment cells were incubated with 100 ng ml−1 PTX for 24 h prior to harvesting.
[35S]-GTPγS assays
These were performed as previously described (Traynor & Nahorski, 1995). Briefly, cell membranes (60–70 μg protein) prepared as above were incubated for 1 h at 30°C in GTPγS binding buffer (pH 7.4) comprising (in mM): HEPES 20, MgCl2 10 and either KCl 100 or NaCl 100 as appropriate, [35S]-GTPγS (guanosine-5′-O-(3-thio)triphosphate) (0.1 nM) and GDP (guanosine 5′-diphosphate) (30 μM) in a final volume of 1 ml. The reaction was terminated by filtration through GF/C glass fibre filters mounted in a Brandel 24 well harvester. The filters were subsequently washed three times with ice-cold GTPγS binding buffer, pH 7.4, and radioactivity determined by liquid scintillation counting after addition of 3 ml of scintillation fluid. EC50 values were determined using GraphPad Prism, version 2.01 (GraphPad, San Diego, CA, U.S.A.).
Receptor binding assays
For competition binding assays, cell membranes (40–60 μg protein) were incubated at 25°C for 1 h in Tris-HCl, pH 7.4 in the presence or absence of NaCl (100 mM) and the GTP analogue GppNHp (50 μM) with [3H]-naltrindole (0.2 nM) and various concentrations of unlabelled ligand in a final volume of 1 ml. For saturation binding assays, membranes were incubated in Tris-HCl as above with various concentrations of [3H]-diprenorphine as previously described (Traynor & Wood, 1989). Non-specific binding was defined in all experiments with 10 μM naloxone. Again, the reaction was terminated by rapid filtration and radioactivity determined by liquid scintillation counting. Affinity measures (Ki or KD) and Bmax values were obtained using GraphPad Prism.
Materials
[3H]-Naltrindole (33 Ci mmol−1), [3H]-diprenorphine (56 Ci mmol−1) and [35S]-GTPγS (1250 Ci mmol−1) were purchased from DuPont NEN, (Boston, MA, U.S.A.). Clocinnamox (C-CAM) was provided by Dr J.W. Lewis, University of Bristol, U.K. ICI 174,864 was purchased from Tocris Cookson, (Ballwin, MO, U.S.A.). 7-Benzylidenenaltrexone (BNTX), naltriben (NTB) and naltrindole (NTI) were kind gifts from Craig Bertha, NIH (Bethesda, MD, U.S.A.). All tissue culture materials were from Gibco Life Sciences (Grand Island, NY, U.S.A.). All other biochemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.) and were of analytical grade.
Results
Saturation binding
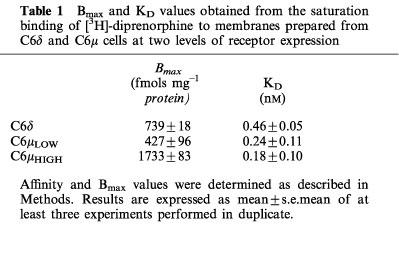
Saturation binding experiments were carried out using [3H]-diprenorphine in order to determine the receptor number in a C6δ clone and two groups of C6μ cells, expressing either a high or a lower receptor number. Bmax values and dissociation constants are given in Table 1. The μ-cells expressed 60 or 220% of the number of receptors in the C6δ cells.
Table 1.
Bmax and KD values obtained from the saturation binding of [3H]-diprenorphine to membranes prepared from C6δ and C6μ cells at two levels of receptor expression
Constitutive activity
In the presence of Na+ ions the basal binding of [35S]-GTPγS to membranes from C6δ cells was 41.3±1.8 fmols mg−1 protein (Figure 1). This increased to 72.3±9.5 fmols mg−1 protein when Na+ ions were replaced with K+ ions. Basal binding of [35S]-GTPγS to C6μ cell membranes was lower than the level in C6δ cell membranes, but a similar percentage increase was also observed, from 22.4±0.1 to 37.1±1.8 fmols mg−1 protein, when K+ ions replaced Na+ ions. The level of basal [35S]-GTPγS binding in C6μ cells in the presence of K+ ions was not significantly different from that of wild-type C6 cells (46.6±8.6 fmols mg−1 protein).
Figure 1.
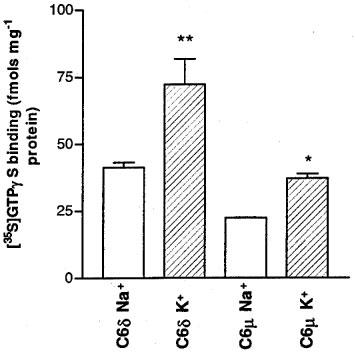
Agonist-independent [35S]-GTPγS binding in C6δ (739 fmols receptor mg−1 protein) and C6μ (1703 fmols receptor mg−1 protein) cell membranes in the presence of either 100 mM NaCl or 100 mM KCl. Values represent mean±s.e.mean for three experiments performed in triplicate. *P<0.05, **P<0.01, significantly different from basal binding in the presence of NaCl, Student's unpaired t-test.
Treatment of C6δ cells with pertussis toxin (PTX) significantly reduced basal levels of [35S]-GTPγS binding by 51.9±6.1 fmols mg−1 protein to 20.4±3.9 fmols mg−1 protein (Figure 2). Since PTX blocks receptor-G protein coupling, but not GTP binding (Katada et al., 1986), this confirmed the presence of constitutively active receptors stimulating Gi/Go proteins. In contrast PTX treatment reduced basal [35S]-GTPγS binding in C6 wild-type cells by only 8.6±3.1 fmols mg−1 protein, to 17.30±1.70 fmols mg−1 protein. A similar low reduction was observed in C6μ cells, regardless of receptor expression level. In the clone expressing a receptor number of 427 fmols mg−1 protein, PTX treatment reduced basal [35S]-GTPγS binding by 10.0±3.5 fmols mg−1 protein, and in the clone expressing the receptor number of 1703 fmols mg−1 protein the reduction in basal binding was 14.6±3.3 fmols mg−1 protein. The reductions in basal binding in both C6μ clones were not statistically significantly different from each other or from the reduction in wild-type C6 cells (Students' unpaired t-test). In contrast, the reduction in basal binding in C6δ cells was significantly different from wild-type (P<0.05).
Figure 2.
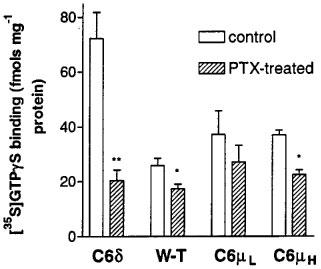
Agonist-independent [35S]-GTPγS binding in membranes prepared from control and pertussis toxin pre-treated (100 ng ml−1, 24 h) C6 glioma cells expressing the δ-opioid receptor at 739 fmols mg−1 protein, untransfected (W-T) C6 cells, or C6 cells expressing the μ-opioid receptor at 427 and 1703 fmols mg−1 protein. Values represent the mean±s.e.mean of at least three experiments performed in duplicate. **P<0.01, *P<0.05, compared to non-PTX treated cells, Student's unpaired t-test.
Identification of non-peptide inverse agonists
The previously reported peptide inverse agonist ICI 174,864 inhibited basal [35S]-GTPγS binding in C6δ cell membranes in a dose-dependent manner, confirming constitutive activity associated with the δ-opioid receptor (Figure 3a). Maximal inhibition was 11.3±0.3 fmols mg−1 protein with an EC50 of 43.6±11.0 nM. Investigation of the antagonist nature of various compounds revealed that the 14-cinnamoylaminomorphinone clocinnamox (C-CAM, Comer et al., 1992), and the δ-receptor antagonists BNTX (Portoghese et al., 1992) and NTB (Sofuoglu et al., 1991) were also able to inhibit the basal binding of [35S]-GTPγS in C6δ cell membranes in a dose-dependent manner, affording EC50 values of 41.0±30.4 nM, 10.7±4.5 nM and 0.90±0.07 nM respectively, and maximal inhibitions of 15.1±2.1, 15.2±1.5, and 22.2±1.5 fmols mg−1 protein respectively (Figure 3b,c,d).
Figure 3.
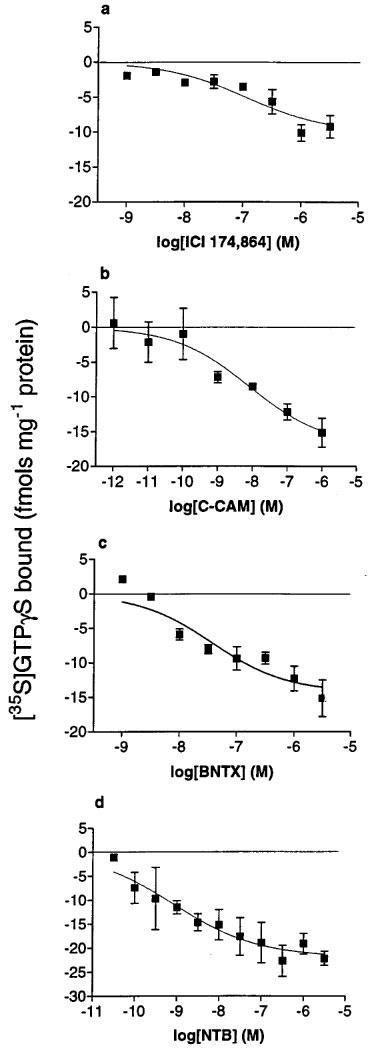
Inhibition of basal [35S]-GTPγS binding to membranes of C6δ cells by (a) ICI 174,864, (b) C-CAM, (c) BNTX and (d) NTB. Values represent mean±s.e.mean for at least three experiments performed in duplicate.
Buprenorphine, naltrindole, and naloxone (all 1 μM) did not alter basal [35S]-GTPγS binding and therefore should be considered neutral antagonists (data not shown). Concurrent addition of the δ-opioid neutral antagonist naltrindole (1 μM) blocked the inverse agonist responses of 1 μM ICI 174,864, C-CAM, or BNTX in C6δ cell membranes (Figure 4). No inhibition of basal [35S]-GTPγS binding by ICI 174,864, C-CAM, or BNTX was observed in membranes prepared from wild-type C6 cells or C6μ cells.
Figure 4.
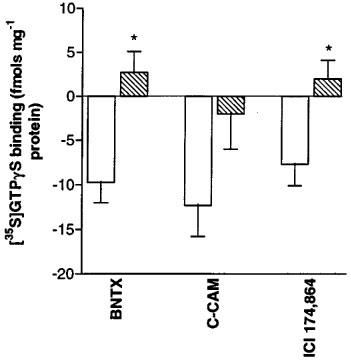
Reversal of inverse agonist activity of 1 μM of BNTX, CCAM or ICI 174864 by the neutral δ-antagonist naltrindole (10 μM) in C6δ cell membranes. Values represent mean±s.e.mean for three experiments performed in duplicate. *P<0.05, Student's unpaired t-test.
It is well documented that the addition of NaCl and guanine nucleotides to the binding assay buffer serves to uncouple the G-protein from the receptor resulting in a low affinity form of the receptor (Pasternak et al., 1975; Childers & Snyder, 1980). Affinity (Ki) values for the inhibition of [3H]-naltrindole binding to C6δ cell membranes were obtained for BNTX, C-CAM, and ICI 174,864 with and without NaCl and GppNHp, a GTP analogue, present in the binding buffer (Table 2). Addition of NaCl and GppNHp afforded a 7 fold leftward shift in the competition curve for ICI 174,864 (Figure 5a). The displacement of [3H]-naltrindole by C-CAM is best fit to a two site model (P=<0.001), a property presumably related to the pseudoirreversible nature of the binding of this ligand (Zernig et al., 1996). Nevertheless a 5 fold leftward shift of the lower affinity site of the competition curve was observed in the presence of NaCl and GppNHp (Figure 5b). No significant shift of the higher affinity site was observed. The inhibition of [3H]-naltrindole binding by BNTX and NTB was unchanged (Figure 5c, d) in the presence of NaCl and GppNHp.
Table 2.
Affinity (Ki) values for the inhibition of [3H]-naltrindole binding to C6δ cell membranes
Figure 5.
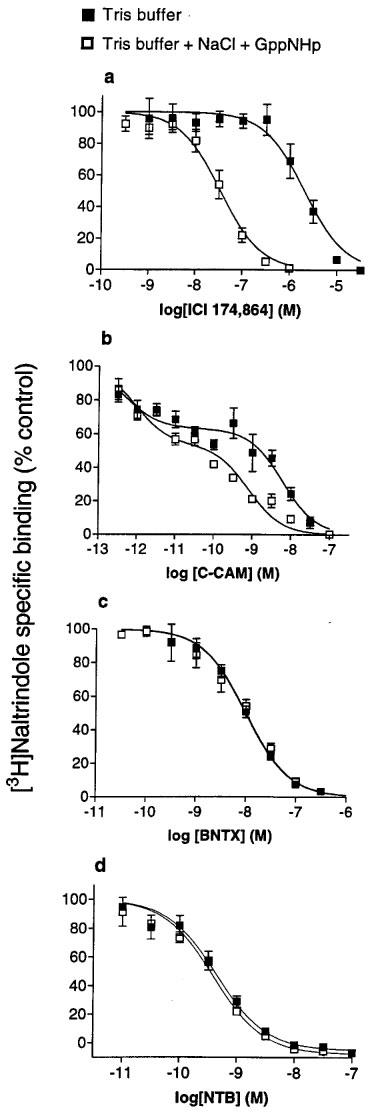
The displacement of [3H]-naltrindole (0.2 nM) binding to membranes of C6δ cells by (a) ICI 174,864, (b) C-CAM, (c) BNTX and (d) NTB in the absence or presence of NaCl (100 mM) and the GTP analogue, GppNHp (50 μM). Values represent mean±s.e.mean for three experiments performed in duplicate.
Discussion
Previous literature suggests that there is a degree of constitutive activity associated with the δ-opioid receptor, due to the ability of the peptide ICI 174,864 to inhibit basal GTPase activity (Costa & Herz, 1989; Mullaney et al., 1996), to inhibit basal binding of [35S]-GTPγS (Szekeres & Traynor, 1997), and to enhance forskolin-stimulated cyclic AMP accumulation (Chiu et al., 1996; Merkouris et al., 1997). However, tools to investigate constitutive activity at δ-opioid receptors are scarce and as yet there have been no reports of similar findings with the μ-opioid receptor. In order to improve the chances of observing constitutive activity, experiments were carried out in GTPγS binding buffer containing K+ ions instead of Na+ ions. It has been shown previously (Costa & Herz, 1989; Szekeres & Traynor, 1997) that the inverse agonist effect of ICI 174,864 is much more pronounced in buffer in which the Na+ ions are replaced by K+ ions. Constitutive activity occurs when receptors exist in the active (R*) state, and therefore can couple to G-proteins in the absence of agonist. Since Na+ ions reduce basal [35S]-GTPγS binding by lowering the affinity of ‘empty' opioid receptors for G-protein, they serve to reduce the constitutive activity of the receptor and as a consequence mask any observable inverse agonist activity. Substitution of Na+ ions with K+ ions provides a higher level of spontaneous G-protein coupling (Costa & Herz, 1989; Szekeres & Traynor, 1997), hence enabling the observation of any reduction in agonist-independent receptor activation.
The level of basal [35S]-GTPγS binding in C6δ cells was much higher than in C6 wild-type cells suggesting the expression of constitutive activity by introduction of the δ-receptor. In contrast, C6μ cells expressing either a high or a low receptor number show the same level of basal [35S]-GTPγS binding as the wild-type cells. Treatment of C6 wild-type cells with pertussis toxin alone reduced basal binding of [35S]-GTPγS showing a tonic level of activation of G-proteins of the Gi/Go class even in the absence of the δ-receptor. However, in C6δ cells the reduction in basal levels by PTX was much more pronounced confirming an increased level of constitutive activity associated with the δ-receptor. C6 cells expressing the μ-opioid receptor showed a similar small reduction of basal [35S]-GTPγS binding by PTX as seen with wild-type cells. A high receptor expression level is thought to lead to increased constitutive activity (Lefkowitz et al., 1993) due to a greater chance of encounter of R* with G, resulting in spontaneous G-protein coupling. However, the lack of constitutive activity observed at the μ-receptor was not changed with increased receptor number, consistent with the μ-opioid receptor being incapable of showing constitutive activity in this cell system. The PTX-induced reduction in basal binding in wild-type and C6μ cells is likely due to the presence of non-opioid, constitutively active, Gi/Go coupled receptors.
In addition to ICI 174,864, the irreversible μ-antagonist C-CAM and the δ-selective antagonists, BNTX and NTB, also acted as inverse agonists in C6δ cell membranes, affording dose-dependent reduction in [35S]-GTPγS binding, with NTB being especially potent. The order of potency was NTB>BNTX>C-CAM=ICI 174,864. The dose-response curve for C-CAM was shallow which may reflect the complex binding at the δ-receptor seen in the competition binding assays. This is likely due to pseudoirreversible binding to the δ-receptor, similar to that which is responsible for the long duration of action of this compound in blocking μ-agonist mediated behaviours (Comer et al., 1992; Zernig et al., 1996). It is interesting to note that Zernig et al., 1995 have reported that C-CAM causes hyperalgesia in the mouse acetic acid-induced writhing assay for nociception. This may be due to the ability of C-CAM to block the action of endogenous opioids that might be released within 5 min of administration of the irritant acetic acid. However, a feasible alternative is that C-CAM causes hyperalgesia by acting as an inverse agonist at the δ-opioid receptor. Activation of δ- receptors provides antinociception in this assay so it is possible that constitutive activity of the δ-receptor could serve to dampen the nociceptive stimulus. Removal of this effect by C-CAM would likely provide increased response to the stimulus.
The inverse agonist activity of ICI 174,864, C-CAM, BNTX or NTB was not seen in wild-type or C6μ cell membranes, suggesting that the response was indeed mediated via the δ-opioid receptor, a finding supported by the prevention of inverse agonist activity by the neutral δ-antagonist naltrindole. In addition to naltrindole, the μ-partial agonist buprenorphine and the non-selective antagonist naloxone behaved as neutral antagonists at the δ-receptor expressed in C6 cells.
None of the inverse agonists were able to inhibit basal [35S]-GTPγS binding in C6δ cells to the level of that seen in non-transfected C6 cells. This suggests that the compounds did not completely prevent δ-receptor mediated constitutive activity in the C6δ cells. The fact that the inverse agonists cannot block all the δ-mediated agonist-independent [35S]-GTPγS binding indicates that the compounds may be partial inverse agonists. Indeed, the compounds do seem to have differential efficacy in the order NTB>C-CAM=BNTX>ICI 174,864. Szekeres & Traynor (1997) obtained similar findings for ICI 174,864 acting on membranes prepared from NG108-15 neuroblastoma×glioma hybrid cells. However, Mullaney et al. (1996), reported that in rat-1 fibroblasts expressing the cloned mouse δ receptor at a level of 6100 fmols mg−1 protein, ICI 174,864 did inhibit basal binding of [35S]-GTPγS to the same extent as pre-treatment of cells with pertussis toxin. The authors concluded that ICI 174,864 was an inverse agonist of high negative intrinsic efficacy. The differences may relate to the 8 fold greater δ-opioid receptor expression level in the rat-1 fibroblasts than in the C6δ cells, and the 12 fold greater expression level than in NG108-15 cells. Collision coupling theory (Stickle & Barber, 1992) suggests that the greater the receptor number in the system, the greater the chance of spontaneous activity occurring.
According to the two state model of receptor activation (Leff, 1995), inverse agonists preferentially stabilize the inactive (R) conformation of the receptor at the expense of the active (R*) form. Therefore, it is predicted that ICI 174,864, C-CAM, BNTX, and NTB would have a higher affinity for the uncoupled (R) state of the δ-receptor. To test for this competition binding assays were performed with the addition of NaCl and the non-hydrolyzable GTP analogue, GppNHp, to the assay buffer in order to drive equilibrium between R and R* greatly in favour of the uncoupled, R form. Under these conditions the affinity of an agonist for the receptor is decreased whereas the affinity of a neutral antagonist is unchanged, and if the hypothesis were correct, the affinity of an inverse agonist would be increased. This held true for both ICI 174,864 and C-CAM, indeed ICI 174,864 exhibited a 7 fold shift consistent with the findings of Appelmans et al. (1986), who showed that the affinity of ICI 174,864 for the δ-opioid binding site is increased 8–16 fold upon addition of 25 mM NaCl to the reaction buffer, depending upon the system studied. In contrast, the affinity of BNTX and NTB for the receptor did not change. This suggests that the abilities of BNTX and NTB to inhibit [35S]-GTPγS binding do not relate to their relative affinity for the δ-receptor under conditions promoting R and R* forms of the receptor.
There are several lines of evidence that differential affinity for receptor states is not a complete explanation for inverse agonism. δ-Opioid receptor constitutive activity has been demonstrated in intact HEK-293 cells and rat-1 fibroblasts expressing the δ-receptor. ICI 174,864 acts as an inverse agonist at the level of forskolin-stimulated adenylyl cyclase in these cells, presumably by relieving the tonic inhibition of cyclic AMP accumulation afforded by the constitutively active δ-receptor (Chiu et al., 1996; Merkouris et al., 1997). This effect is blocked, but not mimicked, by PTX showing that forskolin-stimulated adenylyl cyclase is constitutively suppressed even in the presence of PTX. This suggests that ICI 174,864 in these whole cell preparations is not acting as an inverse agonist by decreasing the amount of receptors in the activated state but rather acts through a G-protein coupled receptor mechanism. However, this action appears to be cell-specific since inverse agonist activity of ICI 174,864 is seen in HEK cells expressing the δ-opioid receptor at 966 fmols mg−1 protein but not in whole NG108-15 cells with a similar level of δ-receptor expression (Chiu et al., 1996). Support for these findings comes from work at the dopamine D3 receptor expressed in CHO cells. In membranes from these cells the D2 antagonist haloperidol is an inverse agonist as determined by its ability to inhibit basal [35S]-GTPγS binding in membranes from these cells (Malmberg et al., 1998). Yet haloperidol binds with equal affinity to D3 receptors in membranes from control and PTX-treated cells, suggesting no difference in its affinity for activated, coupled receptors, and inactive, uncoupled receptors.
Constitutive activity within the opioid receptor family appears to be dependent upon opioid receptor type. The apparent lack of constitutive activity at the μ-opioid receptor may explain why, when so many μ-ligands are known, that no μ-inverse agonists have been discovered to date. The findings are also intriguing considering the similarities between the μ- and δ-receptors. The intracellular loops of these two opioid receptor types, in particular the third intracellular loop which is thought to be responsible for G-protein coupling, are highly conserved in their amino-acid sequence with 90% sequence homology, suggesting a common effector coupling mechanism (for review see Dhawan et al., 1996). However, the C-terminus of the μ- and δ-receptor are very different with only 32% sequence homology. In addition it has been reported that activated μ- and δ-receptors are differentially coupled to PTX-sensitive G proteins in membranes of SH-SY5Y cells (Laugwitz et al., 1993). This ability to differentially couple to specific G proteins may play an important part in the expression of constitutive activity.
Acknowledgments
This work was supported by USPHS grants DA 00254 and DA 02265
Abbreviations
- BNTX
7-benzylidenenaltrexone
- C-CAM
clocinnamox
- GDP
guanosine diphosphate
- G-protein
guanosine triphosphate binding protein
- GTPγS
guanosine 5′-[γ-thio]triphosphate
- ICI 174,864
N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH
- NTB
naltriben
- PTX
pertussis toxin
References
- APPELMANS N., CARROLL J.A., RANCE M.J., SIMON E.J., TRAYNOR J.R. Sodium ions increase the binding of the antagonist peptide ICI 174,864 to the delta opiate receptor. Neuropeptides. 1986;7:139–143. doi: 10.1016/0143-4179(86)90089-2. [DOI] [PubMed] [Google Scholar]
- CHILDERS S.R., SNYDER S.H. Differential regulation by guanine nucleotides of opiate agonist and antagonist receptor interactions. J. Neurochem. 1980;34:583–593. doi: 10.1111/j.1471-4159.1980.tb11184.x. [DOI] [PubMed] [Google Scholar]
- CHIU T.T., YUNG L.Y., WONG Y.H. Inverse agonist effect of ICI 174,864 on the cloned δ-opioid receptor: Role of G-protein and adenylyl cyclase activation. Mol. Pharmacol. 1996;50:1651–1657. [PubMed] [Google Scholar]
- CLARK M.J., EMMERSON P.J., MANSOUR A., AKIL H., WOODS J.H., PORTOGHESE P.S., REMMERS A.E., MEDZIHRADSKY F. Opioid efficacy in a C6 glioma cell line stably expressing the delta opioid receptor. J. Pharmacol. Exp. Ther. 1997;283:501–510. [PubMed] [Google Scholar]
- COMER S.D., BURKE T.F., LEWIS J.W., WOODS J.H. Clocinnamox: a novel, systemically active, irreversible opioid antagonist. J. Pharmacol. Exp. Ther. 1992;262:1051–1056. [PubMed] [Google Scholar]
- COSTA T., HERZ A. Antagonists with negative intrinsic efficacy at δ opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHAWAN B.N., CESSELIN R., RAGHUBIR R., REISINE T., BRADLEY P.B., PORTOGHESE P.S., HAMON M. International union of pharmacology. XII. Classification of opioid receptors. Pharmacol. Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- EMMERSON P.J., CLARK M.J., MANSOUR A., AKIL H., WOODS J.H., MEDZIHRADSKY F. Characterisation of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J. Pharmacol. Exp. Ther. 1996;278:1121–1127. [PubMed] [Google Scholar]
- GEORGOUSSI Z., ZIOUDROU C. Effect of delta-opioid antagonists on the functional coupling between opioid receptors and G-proteins in rat brain membranes. Biochem. Pharmacol. 1993;45:2405–2410. doi: 10.1016/0006-2952(93)90220-q. [DOI] [PubMed] [Google Scholar]
- KATADA T., OINUMA M., UI M. Mechanism for the inhibition of the catalytic activity of the enzyme adenylate cyclase by the guanine nucleotide binding proteins serving as the substrate of the islet activating protein, pertussis toxin. J. Biol. Chem. 1986;261:5215–5221. [PubMed] [Google Scholar]
- LAUGWITZ K.-L., OFFERMANS S., SPICHER K., SCHULTZ G. μ and δ opioid receptors differentially couple to G protein subtypes in membranes of human neuroblastoma SH-SY5Y cells. Neuron. 1993;10:233–242. doi: 10.1016/0896-6273(93)90314-h. [DOI] [PubMed] [Google Scholar]
- LEFF P. The two-state model of receptor activation. Trends Pharmacol. Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- LEFKOWITZ R.J., COTECCHIA S., SAMAMA P., COSTA T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol. Sci. 1993;14:203–207. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- MALMBERG A., MIKAELS A., MOHELL N. Agonist and inverse agonist activity at the dopamine D3 receptor measured by guanosine 5′-[γ-thio]triphosphate[35S] binding. J. Pharmacol. Exp. Ther. 1998;285:119–126. [PubMed] [Google Scholar]
- MENG F., HOVERSTEN M., THOMPSON R.C., TAYLOR L., WATSON S.J., AKIL H. A chimeric study of the molecular basis of affinity and selectivity of the κ and the δ opioid receptors: potential role of extracellular domains. J. Biol. Chem. 1995;270:12730–12736. doi: 10.1074/jbc.270.21.12730. [DOI] [PubMed] [Google Scholar]
- MERKOURIS M., MULLANEY I., GEORGOUSSI Z., MILLIGAN G. Regulation of spontaneous activity of the δ-opioid receptor: Studies of inverse agonism in intact cells. J. Neurochem. 1997;69:2115–2122. doi: 10.1046/j.1471-4159.1997.69052115.x. [DOI] [PubMed] [Google Scholar]
- MULLANEY I., CARR C., MILLIGAN G. Analysis of inverse agonism at the δ opioid receptor after expression in Rat 1 fibroblasts. Biochem. J. 1996;315:227–234. doi: 10.1042/bj3150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEILAN C.L., VIVIAN J.A., WOODS J.H., TRAYNOR J.R. Constitutive activity at opioid receptors expressed in C6 glioma cells. Society for Neuroscience Abstracts. 1998;24:2053. [Google Scholar]
- PASTERNAK G., SNOWMAN A.M., SNYDER S. Selective enhancement of [3H]opiate agonist binding by divalent cations. Mol. Pharm. 1975;11:735–744. [PubMed] [Google Scholar]
- PORTOGHESE P.S., SULTANA M., NAGASE H., TAKEMORI A.E. A highly selective δ1-opioid receptor antagonist: 7-benzylidenenaltrexone. Eur. J. Pharmacol. 1992;218:195–196. doi: 10.1016/0014-2999(92)90167-3. [DOI] [PubMed] [Google Scholar]
- SHAW J.S., MILLER L., TURNBULL M.J., GORMLEY J.J., MORLEY J.S. Selective antagonists at the opiate delta-receptor. Life. Sci. 1982;31:1259–1262. doi: 10.1016/0024-3205(82)90356-3. [DOI] [PubMed] [Google Scholar]
- SOFUOGLU M., PORTOGHESE P.S., TAKEMORI A.E. Differential antagonism of delta opioid agonists by naltrindole and its benzofuran analog (NTB) in mice: Evidence for delta opioid receptor subtypes. J. Pharmacol. Exp. Ther. 1991;257:676–680. [PubMed] [Google Scholar]
- STICKLE D., BARBER R. The encounter coupling model for β-adrenergic receptor/GTP binding protein interaction in the S49 cell. Calculation of the encounter frequency. Biochem. Pharmacol. 1992;43:2015–2028. doi: 10.1016/0006-2952(92)90645-y. [DOI] [PubMed] [Google Scholar]
- SZEKERES P., TRAYNOR J.R. Delta opioid modulation of the binding of guanosine-5′-O-(3-[35S]thio)triphosphate to NG108-15 cell membranes: characterization of agonist and inverse agonist effects. J. Pharmacol. Exp. Ther. 1997;283:1276–1284. [PubMed] [Google Scholar]
- THOMPSON R.C., MANSOUR A., AKIL H., WATSON S.J. Cloning and pharmacological characterization of a rat μ-opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- TRAYNOR J.R., NAHORSKI S.R. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- TRAYNOR J.R., WOOD M.S. [3H]Diprenorphine binding to kappa sites in guinea-pig and rat brain: evidence for apparent heterogeneity. J. Neurochem. 1989;53:173–178. doi: 10.1111/j.1471-4159.1989.tb07310.x. [DOI] [PubMed] [Google Scholar]
- WANG Z., BILSKY E.J., PORRECA F., SADEE W. Constitutive mu opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sci. 1994;50:339–350. doi: 10.1016/0024-3205(94)90022-1. [DOI] [PubMed] [Google Scholar]
- ZERNIG G., BROADBEAR J., BRINE G.A., LEWIS J.W., WOODS J.H. Opioid agonist effects on mouse writhing after irreversible mu receptor blockade with clocinnamox. Exp. Clin. Psychopharm. 1995;3:323–329. [Google Scholar]
- ZERNIG G., BURKE T., LEWIS J.W., WOODS J.H. Mechanism of clocinnamox blockade of opioid receptors: Evidence from in vitro and ex vivo binding and behavioral assays. J. Pharmacol. Exp. Ther. 1996;279:23–31. [PubMed] [Google Scholar]