

Abstract
To elucidate the structural features ensuring action of [D-Ala2, Leu5]-enkephalyl-Arg (dalargin), a series of dalargin analogues were tested for their effectiveness in depressing electrically-evoked contractions of the guinea-pig myenteric plexus-longitudinal muscle preparations (μ- and κ-opioid receptors) and the vasa deferentia of the hamster (δ-opioid receptors), mouse (μ-, δ- and κ-opioid receptors), rat (similar to μ-opioid receptors) and rabbit (κ-opioid receptors). The naloxone KB values in the myenteric plexus were also obtained.
[L-Ala2]-dalargin was 19 times less potent than dalargin, and its pharmacological activity was peptidase-sensitive. The ratio of δ-activity to μ-activity for [L-Ala2]-dalargin was 6.78, and KB was 7.9 nM. This emphasizes the role that D-configuration of Ala2 plays in determining the active folding of dalargin molecule as well as in conferring resistance to peptidases.
[Met5]-dalargin was equipotent to dalargin in the myenteric plexus, but was more potent in the vasa deferentia of hamster and mouse (KB=5.5 nM). Leu5 and the interdependence of Leu5 and D-Ala2 are of importance for the selectivity of dalargin for μ-opioid receptors.
Dalarginamide was more potent and selective for μ-opioid receptors than dalargin, whilst dalarginethylamide, though equipotent to dalarginamide in the myenteric plexus, was more potent at δ-opioid receptors (KB=5.0 nM). [D-Phe4]-dalarginamide and N-Me-[D-Phe4]-dalarginamide were inactive indicating the contribution of L-configuration of Phe4 to the pharmacological potency of dalargin.
N-Me-[L-Phe4]-dalarginamide possessed the highest potency and selectivity for μ-opioid receptors (the ratio of δ-activity to μ-activity was 0.00053; KB=2.6 nM). The CONH2 terminus combined with the N-methylation of L-Phe4 increased the potency and selectivity of dalargin for μ-opioid receptors.
Keywords: Opioid peptides, dalargin, peptide analogues, μ-receptor, δ-receptor, guinea-pig ileum, myenteric plexus, hamster vas deferens, rat vas deferens, mouse vas deferens
Introduction
Thousands of enkephalin analogues have been synthesized to explore the ill-defined physiological and pathophysiological role of μ-, δ- and κ-opioid receptors and to design selective and potent ligands (Hruby & Gehrig, 1989). [D-Ala2,Leu5]-enkephalyl-Arg (dalargin), a synthetic analogue of endogenous [Leu5]-enkephalin, has been found to possess various activities such as: analgesic effects (Alyautdin et al., 1995; Schroëder & Sabel, 1996; Schroëder et al., 1998), cytoprotection (Titov et al., 1985), cardioprotection (Pashutin, 1991) and anti-arrhythmic action (Maslov & Lishmanov, 1993; Pokrovsky & Osadchiy, 1995), antiulcerogenic activity (Timoshin et al., 1991), antistress activity (Dvortsin & Shatalov, 1991) and a therapeutic action during alcohol withdrawal (Aliyev, 1992). However, data concerning the selectivity of this hexapeptide for opioid receptors are limited and rather contradictory. Some authors have reported that dalargin is selective for δ-opioid receptors (Titov et al., 1985), whilst others have reported mixed μ-/δ-activity (Korobov, 1988; Likhvantsev & Smirnova, 1991). Our earlier studies have shown a selectivity of dalargin for μ-opioid receptors (Pencheva et al., 1995, 1996). To further elucidate the structural features important for selective action of dalargin, a series of dalargin analogues, endogenous [Leu5]- and [Met5]-enkephalin and μ-selective agonist [D-Ala2,N-Me-L-Phe4,Gly-ol5]-enkephalin (DAMGO) were tested for their effectiveness in depressing electrically-evoked contractions in five bioassay tissues. Since it is well known that many opioid peptides are rapidly degraded by tissue peptidases, the potencies of peptides were also determined in the presence and in the absence of appropriate peptidase inhibitors (McKnight et al., 1983). The assay preparations used were: the myenteric plexus-longitudinal muscle of the guinea-pig ileum, which has μ- and κ-opioid receptors, but not δ-receptors (Lord et al., 1977), the hamster vas deferens, which contains δ-receptors but not μ- or k-receptors (McKnight et al., 1985), the mouse vas deferens which contains μ-, δ- and κ-receptors (Schultz & Wüster, 1981), but is considered to be a standard model for studying the δ-properties of opioid ligands (Lord et al., 1977; Leslie, 1987), the rat vas deferens, containing mainly opioid receptors whose properties are similar to those of μ-receptors in other peripheral tissues (Miller et al., 1986; Leslie, 1987), and the rabbit vas deferens which is a specific bioassay for κ-opioid ligands (Oka et al., 1980; Hayes & Kelly, 1985). The antagonist potency of naloxone (KB) in the myenteric plexus was obtained for all peptides and was further considered when determining their selectivity.
Methods
Preparations of myenteric plexus-longitudinal muscle
Male guinea-pigs (Dunkin-Hartley, 250–350 g) were fasted overnight before the experiments and were sacrificed by a blow on the neck. A segment of the ileum (excluding the portion 15 cm proximal to the ileocaecal junction) 10–15 cm long, was isolated and intraluminal contents were removed from Krebs solution. Segments of the myenteric plexus-longitudinal muscle (hereafter referred to as myenteric plexus), approximately 1.5 cm long, were set up for field stimulation in a 3 or 5 ml siliconized organ bath, containing Krebs solution, continuously aerated with 95% O2 and 5% CO2 at 36.5°C. Electrically-evoked responses of the longitudinal layer were measured under isometric conditions after standard calibration of a mechanoelectrical transducer (Experimetria Ltd, Hungary) connected to a recording device TZ 4620 (Laboratorni Pristroje, Praha) at a tension equivalent to a load of 0.5 g. Electrical field stimulation (Paton & Zar, 1968) with single, bipolar, rectangular pulses of supramaximal voltage, 75 mA current, 0.5 ms duration and 0.1 Hz frequency, was applied between platinum ring electrodes, situation at the upper and lower ends of the bath.
Preparation of vasa deferentia
Vasa deferentia were removed from Golden Syrian hamsters (MB strain; 200–250 g), from Wistar rats (200–250 g), from TO albino mice (23–29 g), and from Bulgarian White rabbits (2.5–3.5 kg). The connective tissue-free preparations were mounted vertically in 3 or 5 ml siliconized organ bath with warm (36°C), oxygenated (95% O2–5% CO2), modified Krebs solution (according to Stjärne & Astrand, 1985, with lower content of Mg2+). The resting tension was 0.1 g for hamster and mouse vasa deferentia and 0.5 g for rat and rabbit vasa deferentia. Field stimulation was applied as described for myenteric plexus, but instead of single pulses we used trains consisting of three pulses of supramaximal voltage, 25 mA current and 1 ms duration at intervals of 250 ms (4 Hz); the trains were repeated at 0.1 Hz.
Experimental procedure
After preparation, all tissues were allowed to equilibrate for 60 min before the start of experiments. Control agonist dose-response curves were obtained by the cumulative addition of opioid peptides at concentrations increasing from 1 pM to 1 mM at 2–3 min intervals. After a second control curve had been constructed, the tissues were exposed to a cocktail of the peptidase inhibitors, bestatin 10 μM (or 30 μM when the rat or rabbit vas deferens was used), captopril 10 μM, thiorphan 3 μM and Leu-Leu 2 mM (McKnight et al., 1983), for at least 20–25 min and dose-response curves for the same compounds were constructed again.
Potencies of agonists were evaluated by measurement of IC50 values; the data are presented as means±s.e.mean. Differences between means were assessed for statistical significance using t-test for grouped data; P<0.05 or lower was taken to be significant. The maximum inhibition produced by opioid peptides was expressed as percentage of the control electrically-evoked responses before opioid treatment. The values of the maximum inhibition reached, at the respective concentration, were recalculated in mN. The data are presented as means±s.e.mean.
The effects of a single administration of the opioid peptides (10 or 40 nM) were followed only in the myenteric plexus without and in the presence of low (17 nM) and high (100 nM) concentration of naloxone.
The dissociation constant for naloxone in the myenteric plexus was determined by constructing dose-response curves for each opioid with and without three or four concentrations of the antagonist in the presence of the cocktail of peptidase inhibitors. Schild plots were generated according to the method of Arunlakshana & Shild (1959). Computer analysis of the plot provided the pA2 and the slope function. Equilibrium dissociation constant for naloxone (KB value) was calculated from the expression pA2=−Log KB. The KB values are expressed in nM concentrations. The 95% confidence limits of the KB and slope function were calculated.
The statistical procedures and the plotting of the graph were carried out using computer programs (Tallarida & Murray, 1981).
Peptides, drugs and solutions
Dalargin (Titov et al., 1985) and its novel analogues (Table 1) were prepared by solid phase synthesis using the Boc strategy. The guanidyl group of arginine was protected by a tosyl group and the hydroxyl of tyrosine by a 2,6-dichlorbenzyl group. The peptide was cleaved from the resin by HF/anisol treatment. HF was then distilled off the reaction mixture. Crude peptide was solubilized, the resin was filtered off and the filtrate lyophilised. Pure peptides were isolated by preparative RP-HPLS and characterized by amino acid analysis, analytical RP-HPLC, capillary electrophoresis and mass spectrometry. Stock solutions of the peptides were prepared in distilled water and stored at −25°C. Dilutions were made in Krebs or modified Krebs solution. Drugs were added to the bath from siliconized glass microsyringes (Hamilton) in volumes not exceeding 0.5–1% of the bath volume.
Table 1.
Amino acid sequence of opioid peptides and analogues used in this investigation

Inhibitors of peptidases: L-Leu-L-Leu, bestatin ([(2S,3R)-3-amino-2-hydroxy-4-phenylbutanoyl]-l-leucine, thiorphan (DL-3-mercapto-2-benzyl-propanoyl-glycine and captopril ([2S]-1-(3-mercapto-2-methyl-propionyl])-L-proline) were from Sigma Chemical Co. Other peptides and drugs: [Leu5]-enkephalin, [Met5]-enkephalin, DAMGO and naloxone were from Sigma Chemical Co.
The Krebs solution contained (mM): NaCl 120, KCl 5.9, NaHCO3 14.4, NaH2PO4 1.2, MgCl2 1.2, CaCl2 2.5, glucose 11.5. The composition of modified Krebs solution was (mM): NaCl 136.9, KCl 2.7, CaCl2 1.8, MgCl2 0.6, NaHCO3 11.9, KH2PO4 0.5, glucose 11.5, pH 7.3–7.4.
Results
Opioid agonist activity in guinea-pig myenteric plexus and vasa deferentia from hamster and rabbit
Dalargin had a low agonist activity in the hamster vas deferens, but was more active in the guinea-pig myenteric plexus preparation (Figure 1). In the myenteric plexus [D-Ala2,Leu5]-enkephalyl-Arg-NH2 (dalarginamide) and [D-Ala2, N-Me-L-Phe4,Leu5]-enkephalyl-Arg (N-Me-[L Phe4]-dalarginamide) were about twice and 20 times respectively, more potent than dalargin, whereas in the δ-selective hamster vas deferens they maintained a low opioid activity, similar to the μ-selective agonist DAMGO (Table 2). The inhibitory effects of the three opioid peptides in the myenteric plexus were antagonized by a low concentration (17 nM) of naloxone (Figure 2). [D-Ala2,D-Phe4,Leu5]-enkephalyl-Arg-NH2-([D-Phe4]-dalarginamide) and [D-Ala2, N-Me-D-Phe4,Leu5]-enkephalyl-Arg (N-Me-[D-Phe4]-dalarginamide) revealed a poor activity in both myenteric plexus and hamster vas deferens. [D-Ala2,Met5]-enkephalyl-Arg ([Met5]-dalargin) and dalargin were equiactive in the myenteric plexus, whereas in the hamster vas deferens [Met5]-dalargin showed a high activity. The same was the case of dalarginamide and [D-Ala2,Leu5]-enkephalyl-Arg-NH-C2H5 (dalarginethylamide) in the two tissues. In the myenteric plexus [L-Ala2,Leu5]-enkephalyl-Arg ([L-Ala2]-dalargin) was 19 times less potent than dalargin, while its activity in the hamster vas deferens was about 30 times higher. The inhibitory effects of dalarginethylamide, [L-Ala2]-dalargin and [Met5]-dalargin were blocked by 100 nM naloxone, while 17 nM naloxone was ineffective (Figure 3).
Figure 1.
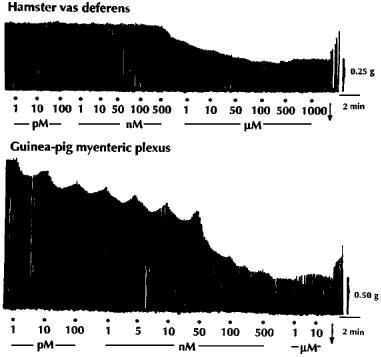
Typical tracings showing the concentration-response inhibitory effects of dalargin, applied cumulatively on the electrically-evoked contractions of the vas deferens of the hamster and the myenteric plexus-longitudinal muscle of the guinea-pig. The Krebs solution contained the following peptidase inhibitors: bestatin 10 μM, thiorphan 3 μM, captopril 10 μM and Leu-Leu 2 mM; •, drug application; ↓, washing out of the drug.
Table 2.
The inhibitory effects (IC50, nM) of dalargin, its analogues, [Leu5]-enkephalin, [Met5]-enkephalin and DAMGO on electrically-evoked contractions of the myenteric plexus-longitudinal muscle of the guinea-pig and the vas deferens of the hamster
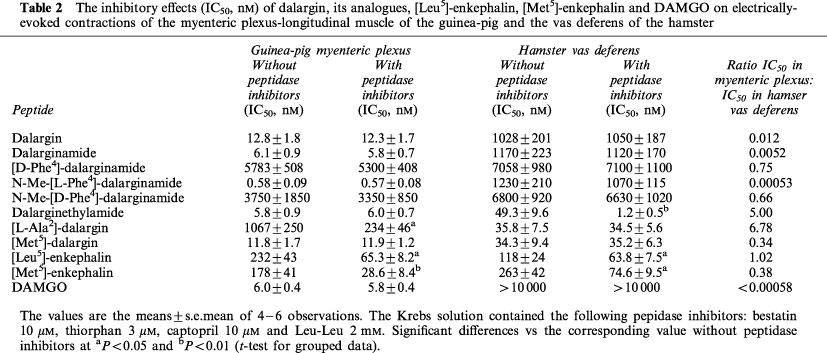
Figure 2.
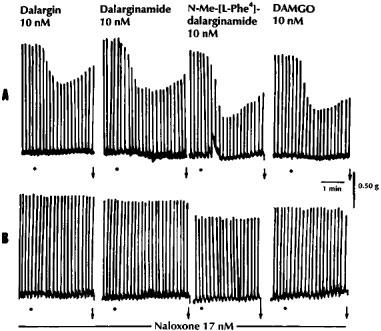
The effects of dalargin, dalarginamide, N-Me-[L-Phe4]-dalarginamide and DAMGO on the electrically-evoked contractions of the myenteric plexus-longitudinal muscle of the guinea-pig in untreated preparations (A) and in the presence of low concentration (17 nM) of naloxone (B). The Krebs solution contained the following peptidase inhibitors: bestatin 10 μM, thiorphan 3 μM, captopril 10 μM and Leu-Leu 2 mM; •, drug application; ↓, washing out of the drug.
Figure 3.
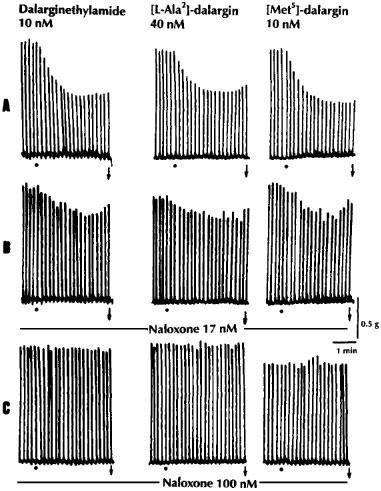
The effects of dalarginethylamide, [L-Ala2]-dalargin and [Met5]-dalargin on the electrically-evoked contractions of the myenteric plexus-longitudinal muscle of the guinea-pig in untreated preparations (A) and in the presence of low (17 nM; B) and high (100 nM; C) concentrations of naloxone. The Krebs solution contained the following peptidase inhibitors: bestatin 10 μM, thiophan 3 μM, captopril 10 μM and Leu-Leu 2 mM; •, drug application; ↓, washing out of the drug.
Combination of the peptidase inhibitors bestatin (10 μM), thiorphan (3 μM), captopril (10 μM) and Leu-Leu (2 mM), did not increase the potency of dalargin and the majority analogues in guinea-pig myenteric plexus nor in the hamster vas deferens. In contrast, similar treatment increased the potency of [L-Ala2]-dalargin in the myenteric plexus 4.5 fold and the potency of dalarginethylamide in the hamster vas deferens 41 fold (Table 2).
Dalargin, dalargin analogues and endogenous enkephalins at concentrations ranging from 10 nM to 10 μM did not significantly inhibit the electrically-evoked contractions of the rabbit vas deferens. In the presence of the peptidase inhibitors neither dalargin, nor its analogues, nor endogenous enkephalins showed changes in their activity except for [L-Ala2]-dalargin, whose IC50 was 3800±420 nM (n=6). The opioid peptides showed no antagonist activity in the bioassay tissues. All these findings indicate that the peptides investigated have no significant affinity for the κ-opioid receptor. Therefore, the ratio of the IC50 value in the guinea-pig myenteric plexus to the IC50 value in the hamster vas deferens could be considered a measure of the degree of selectivity of the peptide for the μ- or δ-opioid receptor. When the ratio was very low, e.g. 0.00053 for N-Me-[Phe4]-dalarginamide, 0.0052 for dalarginamide and 0.012 for dalargin, the degree of μ-selectivity was high. A very low ratio, similar to that of N-Me-[L-Phe4]-dalarginamide, was obtained for μ-receptor selective agonist DAMGO. A relatively high ratio was calculated for [L-Ala2]-dalargin and dalarginethylamide.
The opioid properties of the compounds were further compared according to the maximum inhibition (corresponding to 100% effects of the respective peptide), they exerted on the electrically-evoked contractile activity in guinea-pig myenteric plexus and hamster vas deferens. The data summarized in Figure 4 show: (i) dalargin, dalarginamide and especially N-Me-[L-Phe4]-dalarginamide (9.6±1.3 mN) reached the highest values for maximum inhibition in the guinea-pig myenteric plexus at low concentration; these data were comparable to the μ-agonist DAMGO, though the value for its maximum inhibition was lowest (4.1±0.7 mN); (ii) in the same tissue dalarginethylamide, [L-Ala2]-dalargin and [Met5]-dalargin were less effective, whilst the [D-Phe4-containing analogues reached relatively high values for maximum inhibition, though at very high concentrations; and (iii) in the hamster vas deferens high values for maximum inhibition were obtained for dalarginethylamide, [L-Ala2]-dalargin, [Met5]-dalargin and endogenous enkephalins.
Figure 4.
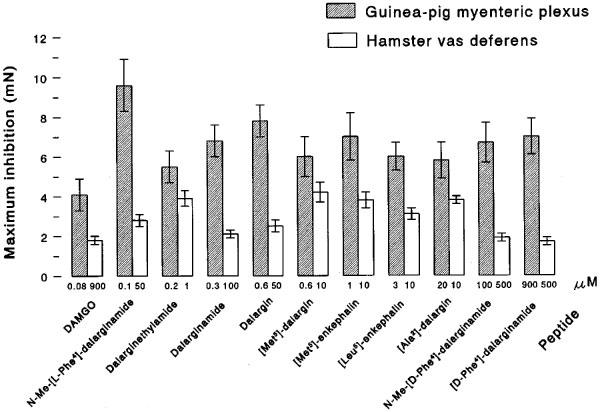
Maximum inhibition (mN), produced at different concentrations (μM) of dalargin, dalargin analogues, [Met5]-enkephalin, [Leu5]-enkephalin and DAMGO on the electrically-evoked contractions of the myenteric plexus-longitudinal muscle of the guinea-pig and the hamster vas deferens. The Krebs solution contained the following peptidase inhibitors: bestatin 10 μM, thiorphan 3 μM, captopril 10 mM and Leu-Leu 2 mM.
Opioid agonist activity in vasa deferentia of rat and mouse
With the exception for D-Phe4-containing analogues the combination of peptidase inhibitors increased the potency of dalargin, its analogues and endogenous enkephalins in the rat vas deferens (Table 3). In the presence of the peptidase inhibitors (only the concentration of bestatin was increased from 10 to 30 μM) dalargin, dalarginamide and N-Me-[L-Phe4]-dalarginamide showed a relatively high agonist activity as compared to the activity of other opioid peptides. No significant differences were found between their IC50 values. In the same preparation [D-Phe4]-dalarginamide and N-Me-[D-Phe4]-dalarginamide were inactive. Dalarginethylamide, [L-Ala2]-dalargin and [Met5]-dalargin exhibited an opioid activity similar to or slightly decreased than that of endogenous [Met5]- and [Leu5]-enkephalin. The more potent analogues were less peptidase-sensitive and the inhibitors enhanced their activity about 4 fold.
Table 3.
The inhibitory effects (IC50, nM) of dalargin, its analogues, [Leu5]-enkephalin, [Met5]-enkephalin and DAMGO on electrically-evoked contractions of the vasa deferentia of the rat and mouse
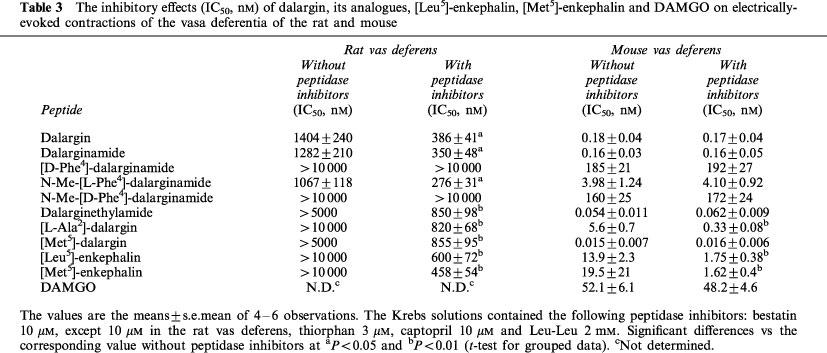
In contrast, in the mouse vas deferens the potencies of dalargin and its analogues were unchanged by the peptidase inhibitors except for [L-Ala2]-dalargin and endogenous enkephalins (Table 3). The opioid agonist potency of all the peptides investigated was high except for that of [D-Phe4]-dalarginamide and N-Me-[D-Phe4]-dalarginamide. The most potent analogues in this tissue were [Met5]-dalargin and dalarginethylamide. Dalargin and dalarginamide were equiactive in the mouse vas deferens with IC50 values <0.2 nM. The potency of N-Me-[L-Phe4]-dalarginamide was decreased about 24 fold compared to that of dalargin and dalarginamide.
Antagonist potency of naloxone (KB) in the guinea-pig myenteric plexus
The potency of the competitive opioid antagonist naloxone in the guinea-pig myenteric plexus was considered indicative of the selectivity of the compounds to opioid receptors. The KB value for naloxone antagonism of μ-opioid effects is 2–3 nM, while of δ- or κ-effects is 20–30 nM (Leslie, 1987; Illes, 1989). In the present study, naloxone was more potent as an antagonist of dalargin and of the potent dalargin analogues with unsubstituted amide terminal group as compared to the other analogues or endogenous [Met5]- and [Leu5]-enkephalins (Table 4). The naloxone KB value for dalargin, dalarginamide and N-Me-[L-Phe4]-dalarginamide was between 2.6 and 3.0 nM and no significant differences were found between them. This KB value was similar to that obtained with the μ-receptor agonist DAMGO suggesting that the action of these peptides was mediated by the μ-receptor. The low potency of [D-Phe4]-dalarginamide and N-Me-[D-Phe4]-dalarginamide prevented the determination of a naloxone KB. The value (ranging from 5.0–7.9 nM), obtained for dalarginethylamide, [L-Ala2]-dalarginamide and [Met5]-dalargin and for endogenous enkephalins were intermediate. The slope of the Schild plots was not significantly different from 1.0, which is indicative of competitive antagonism.
Table 4.
Interaction of the opioid antagonist naloxone with dalargin its analogues, [Leu5]-enkephalin, [Met5]-enkephalin and DAMGO in the myenteric plexus-longitudinal muscle of the guines-pig. KB (nM) and slope (95%) confidence limits)

Discussion
The present investigation provided more detailed and convincing data concerning the high potency and selectivity of dalargin for μ-opioid receptors (Pencheva et al., 1995, 1996). Comparison of the effects of dalargin, its analogues and endogenous enkephalins in models possessing different receptor populations in the presence and in the absence of peptidase inhibitors, allows us to characterize: (i) the tissues, in terms of their reliability for drug-receptor classification; (ii) the opioid peptides in terms of their potency and selectivity; and (iii) the structure-activity relationships, which are important for potency and selectivity of dalargin for μ-opioid receptors.
To assess the activity of dalargin at δ-opioid receptors, we used hamster vas deferens, which contains only δ-receptors, but no μ- and κ-receptors, and the mouse vas deferens, which contains μ-, δ- and κ-receptors. Comparison of the data using these δ-containing tissues indicated that compounds with different ratios of δ- to μ-activity were equiactive or with similar potency in the mouse vas deferens. This finding confirms the result of Sagan et al. (1991), indicating that the mouse vas deferens is not a reliable tissue to assess the agonist potency and receptor selectivity of opioids. The low potency of dalargin and some of its analogues in the hamster vas deferens indicated poor affinity at δ-opioid receptors, despite their high activity in the mouse vas deferens. On the other hand, to further evaluate the activity of dalargin and its analogues at μ-receptors, we used guinea-pig myenteric plexus, containing μ- and κ-receptors and rat vas deferens, containing mainly receptors similar to μ-opioid receptors. Since the majority of peptides investigated were degradated by peptidases in the rat vas deferens it could be assumed that the capacity of the rat vas deferens to inactivation enkephalins was higher than that of other tissues (McKnight et al., 1983). In the enzyme-inhibited rat vas deferens, the activity of dalargin and its analogues were relatively low, but showed some correlation with higher activities in the guinea-pig myenteric plexus. These results could be explained by the less efficient stimulus-response mechanism of the rat vas deferens (Lemaire et al., 1978; Kenakin, 1984). Obviously, the short opioid peptides, selective for δ-receptor, have poor activity in the rat vas deferens, while the μ-dependent peptides only exert pronounced inhibitory effects. However, this tissue was not reliable to determine precise structure-activity relationships.
In the presence and in the absence of peptidase inhibitors dalargin and its analogues failed to produce significant inhibition of electrically-evoked contractions of the rabbit vas deferens containing only κ-receptors. These analogues and dalargin are devoid of κ-agonist activity and thus comparison of the data from myenteric plexus with those from the hamster vas deferens allows the direct correlation of μ-potency with δ-potency. The low value of the ratio, obtained for dalargin and the naloxone KB value for dalargin (3.0 nM) in the myenteric plexus indicate that dalargin is selective for μ-opioid receptors.
We determined the opioid activity of several dalargin analogues using different isolated tissue bioassays in order to elucidate the structural features required for the selective and potent action of dalargin at μ-receptors. The relationship between opioid receptor selectivity and the stereochemistry of the second amino acid of dalargin was assessed by replacing D-amino acid by an L-amino acid. Whether the D-configuration was important for determining the folding of dalargin molecule and/or conferring resistance to proteolysis, was determined by measuring agonist activity in the presence of peptidase inhibitors. Comparison of all parameters of the activity of dalargin and [L-Ala2]-dalargin suggested that the D-configuration of Ala2 was of great importance for both: (i) determining the active folding of dalargin molecule (the L-Ala2 analogue was 20 times less active than dalargin in the myenteric plexus and 30 times more active in the hamster vas deferens; the KB value was 7.9 nM); and (ii) conferring resistance to peptidases as the potency of the [L-Ala2]-containing analogue was affected by peptidase inhibitors in the majority of the tissues examined. It should be noted however, that in the hamster vas deferens, [L-Ala2]-dalargin, which was not protected from the cleavage of amino- and/or carboxypeptidases, was peptidase-resistant similar to protected analogues as dalargin and dalarginamide. Sagan et al. (1991) established that in the same tissue the activity of [L-Ala2]-dermorphin was insensitive to peptidase inhibitors, whilst the activities of [D-Ala2]-containing dermorphin and dermenkephalin analogues were elevated by such treatment. It could be assumed that in hamster vas deferens the D-configuration of Ala2 is of importance for the recognition of δ-opioid receptors rather that for the decreased metabolism of the analogues. The intermediate KB value obtained for naloxone antagonism of [L-Ala2]-dalargin indicates an affinity for more than one receptor type. This explains the high agonist activity of [L-Ala2]-dalargin in the mouse vas deferens and moderate activity in the rat vas deferens. These data also reveal the unreliability of these tissues. Further shortening of the peptide [L-Ala2]-dalargin to [Leu5]-enkephalin resulted in a loss of δ-activity and a slight increase of μ-activity.
The importance of the C-terminal region of dalargin for opioid receptor selectivity was further demonstrated by comparing the activities of dalargin to those of [Met5]-dalargin. Substitution of the Leu5 of dalargin by Met5 decreased the μ-receptor preference of the respective analogue, whose naloxone KB became 5.5 nM, thus changing the selectivity of dalargin from μ-activity to mixed δ-/μ-activity. Obviously, the role of Leu5 in the selectivity of dalargin for μ-receptors was important but an interdependence between Leu5 and the D-configuration of Ala2 in the molecule of the hexapeptide dalargin should be considered because [L-Ala2]-dalargin, which contains Leu5, but L-configuration of the same residue has also mixed affinity for δ- and μ-receptors.
The influence of the COOH terminus on opioid receptor selectivity and on pharmacological potency of dalargin was demonstrated by comparing the activities of dalargin to those of dalarginamide and dalarginethylamide. Replacement of the COOH terminus by CONH2, caused no change in δ-activity, but increased two times the μ-activity of dalarginamide. However, replacement by a CONHC2H5 group maintained the higher potency of dalarginamide in the myenteric plexus, but greatly (>800 times) increased the δ-activity of dalarginethylamide in the hamster vas deferens and changed the selectivity of dalargin to mixed μ-/δ-activity. Similar mixed sensitivity at both μ- and δ-opioid receptors was demonstrated by Fujioka et al. (1984) for another enkephalin derivative containing D-Ala2, Leu5 and an ethylamide terminus. These results are in accordance with the suggestion of Schwyzer (1986) that the different C-terminal ‘address' segments are responsible for the receptor type preference of opioid peptides. However, it should also be noted that the combination of peptidase inhibitors increased the potency of dalarginethylamide in the hamster vas deferens about 41 fold. This finding was surprising as modifications to the structure of this analogue preclude cleavage by amino- and carboxypeptidases and by carboxyamidases. One could speculate that this tissue contains a specific enzyme system which does not participate in the inactivation of dalargin or dalarginamide. It remains of interest to employ enzymological approaches in hamster vas deferens and to obtain data with other ethylated substrates containing different aminoacids.
It was also established that the configuration of the Phe residue in position 4 made a major contribution to the pharmacological potency of dalargin, since [D-Phe4]-dalarginamide and N-Me-[D-Phe4]-dalarginamide were inactive in all tissues examined. These data are in accordance with the results of Shimohigashi et al. (1984) on the role of the stereo-orientation of aromatic groups in the opiate receptor recognition. It is well known that the enkephalin analogues, including the μ-selective agonist DAMGO, containing N-Me-Phe4 possess a high opioid activity (Pless et al., 1979; Kosterlitz & Paterson, 1981; Hruby & Gehrig, 1989). The role of methylation of L-Phe4 in the improvement of the opioid profile of dalargin was evaluated by comparing the agonist activity of dalargin or dalarginamide with that of N-Me-[L-Phe4]-dalarginamide. This structural modification caused no change in δ-activity, which remained low, but increased (ten times as compared with that of dalarginamide; 20 times as compared with that of dalargin) the potency of this analogue in the myenteric plexus. Hence, the ratio of δ-activity to μ-activity was 0.00053. A similar ratio was calculated for DAMGO. However the increased potency of N-Me-[L-Phe4]-dalarginamide was accompanied by a high efficacy, while the maximum inhibitory response of DAMGO was lowest, indicating its less efficient opioid action. The low naloxone KB value (2.6 nM) of N-Me-[L-Phe4]-dalarginamide provided further evidence that methylation of L-Phe4 in the hexapeptide structure of dalargin increases the selectivity for μ-opioid receptors. Obviously, N-Me-[L-Phe4]-dalarginamide fulfils all the structural requirements important for the active, selective and efficient action of dalargin for μ-receptors. We failed to obtain data with the putative endogenous ligands for μ-opioid receptors, endomorphin-1 and endomorphin-2 (Zadina et al., 1997), because in guinea-pig ileum they have opiate agonist and antagonist activity (Zadina et al., 1992), while in human μ-opioid receptor transfected B82 fibroblasts, they act as partial agonists (Hosohata et al., 1998). The relative merits of dalargin and its analogues and of DAMGO and endomorphins as selective and potentially therapeutically useful agonists of μ-receptors remain ill resolved. Moreover, a caution is required when extrapolating data on the activity of the compounds in various bioassays to absolute selectivities for different opioid receptors. Tissues express several receptor subtypes and their responses depend not only upon drug affinity and receptor selectivity but also on the intrinsic activity and receptor reserve of the assay system. Hence, the pharmacological data obtained in our studies should permit further assessment of these novel compounds using appropriate transfected cell lines, expressing a single receptor population. Such studies would more rigorously characterize the sensitivity, selectivity and intrinsic activity of our compounds.
In conclusion, these findings are consistent with a high potency and selectivity of the hexapeptide dalargin for μ-opioid receptors. They characterize some of the structural features that determine its opioid profile and strongly suggest that: (i) D-configuration of Ala2 is of importance for the active folding of dalargin molecule and for conferring resistance to peptidases: (ii) Leu5 and the interdependence between Leu5 and D-Ala2 contribute to the selectivity of dalargin to μ-opioid receptors; (iii) L-configuration of Phe4 makes a major contribution to the pharmacological potency of dalargin; and (iv) CONH2 terminus and especially CONH2 terminus associated with N-methylation of L-Phe4 increases the potency, efficacy and selectivity of dalargin for μ-opioid receptors. These data allow for: (i) the better understanding of the various activities of dalargin and of its therapeutic value in clinical practice; and (ii) the improved design of selective and potent analogues of dalargin and enkephalins.
Acknowledgments
This work was supported by Grant ‘L-637' from the Ministry of Education and Science of Bulgaria, and Grant ‘505/94/0704' from the Grant Agency of the Czech Republic. The expert technical assistance of Velika Deneva is acknowledged.
Abbreviations
- dalargin
[D-Ala2,Leu5]-enkephalyl-Arg
- dalarginamide
[D-Ala2,Leu5]-enkephalyl-Arg-NH2
- dalarginethylamide
[D-Ala2,Leu5]-enkephalyl-Arg-NH-C2H5
- DAMGO
[D-Ala2,N-Me-L-Phe4, Gly-ol5]-enkephalin
- [D-Phe4]-dalarginamide
[D-Ala2,D-Phe4,Leu5]-enkephalyl-Arg-NH2
- [L-Ala2]-dalargin
[L-Ala2,Leu5]-enkephalyl-Arg
- [Met5]-dalargin
[D-Ala2,Met5]-enkephalyl-Arg
- N-Me-[D-Phe4]-dalarginamide
[D-Ala2,N-Me-D-Phe4,Leu5]-enkephalyl-Arg-NH2
- N-Me-[L-Phe4]-dalarginamide
[D-Ala2,N-Me-L-Phe4,Leu5]-enkephalyl-Arg-NH2
References
- ALIYEV N.A. Application of a synthetic analogue of Leu-enkephalin (dalargin) in alcohol withdrawal. Drug Alcohol Depend. 1992;30:209–213. doi: 10.1016/0376-8716(92)90054-g. [DOI] [PubMed] [Google Scholar]
- ALYAUTDIN R., GOTHIER D., PETROV V., KHARKEVICH D., KREUTER J. Analgesic activity of the hexapeptide dalargin absorbed on the surface of polysorbate 80-coated poly(butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 1995;41:44–48. [Google Scholar]
- ARUNLAKSHANA D., SHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DVORTSIN G.F., SHATALOV V.N. Antistressor effect of dalargin in rats with immobilization stress. Biull. Exsp. Biol. Med. (Russia) 1991;111:811–815. [PubMed] [Google Scholar]
- FUJIOKA T., MATSUNAGA T., NAKAYAMA H., KANAOKA Y., HAYASHI Y., KANAGAVA K., MATSUO H. A photoaffinity reagent to label the opiate receptors of guinea-pig ileum and mouse vas deferens. J. Med. Chem. 1984;27:836–840. doi: 10.1021/jm00373a004. [DOI] [PubMed] [Google Scholar]
- HAYES A., KELLY A. Profile of activity of κ receptor agonists in the rabbit vas deferens. Eur. J. Pharmacol. 1985;110:317–322. doi: 10.1016/0014-2999(85)90558-8. [DOI] [PubMed] [Google Scholar]
- HOSOHATA K., BURKEY T.H., ALFARO-LOPEZ J., VARGA E., HRUBY V.J., ROESKE W.R., JAMAMURA H.T. Endomorphin-1 and endomorphin-2 are partial agonists at the human mu-opioid receptors. Eur. J. Pharmacol. 1998;346:111–114. doi: 10.1016/s0014-2999(98)00117-4. [DOI] [PubMed] [Google Scholar]
- HRUBY V.J., GEHRIG C.A. Recent developments in the design of receptor specific opioid peptides. Med. Res. Rev. 1989;9:343–401. doi: 10.1002/med.2610090306. [DOI] [PubMed] [Google Scholar]
- ILLES P. Modulation of transmitter and hormone release by multiple neuronal opioid receptors. Rev. Physiol. Biochem. Pharmacol. 1989;112:139–233. doi: 10.1007/BFb0027497. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.P. The classification of drugs and drug receptors in isolated tissues. Pharmacol. Rev. 1984;36:165–222. [PubMed] [Google Scholar]
- KOROBOV N.V. Dalargin – an opioid peptide with peripheric action. Farmakol. Toksikol. (Russia) 1988;4:35–38. [PubMed] [Google Scholar]
- KOSTERLITZ H.W., PATERSON S.J. Tyr-D-Ala-Gly-MePhe-NH(CH2)2OH is a selective ligand for the μ-opiate binding site. Br. J. Pharmacol. 1981;73:299P. [Google Scholar]
- LEMAIRE S., MAGNAN J., REGOLI D. Rat vas deferens: a specific bioassay for endogenous opioid peptides. Br. J. Pharmacol. 1978;64:327–329. doi: 10.1111/j.1476-5381.1978.tb08653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESLIE F.N. Methods used for study of opioid receptors. Pharmacol. Rev. 1987;39:197–249. [PubMed] [Google Scholar]
- LIKHVANTSEV V.V., SMIRNOVA V.I. Some aspects of using small regulatory peptides in anaesthesiology. Vestn. Ross. Akad. Med. Nauk. (Russia) 1991;7:17–21. [PubMed] [Google Scholar]
- LORD J.A.H., WATERFIELD A.A., HUGHES J., KOSTERLITZ H.W. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- MASLOV L.N., LISHMANOV Y.B. The anti-arrhythmic effect of D-Ala2,Leu5,Arg6 enkephalin and its possible mechanism. Int. J. Cardiol. 1993;40:89–94. doi: 10.1016/0167-5273(93)90269-m. [DOI] [PubMed] [Google Scholar]
- MCKNIGHT A.T., CORBETT A.D., KOSTERLITZ H.W. Increase in potencies of opioid peptides after peptidase inhibition. Eur. J. Pharmacol. 1983;86:393–402. doi: 10.1016/0014-2999(83)90189-9. [DOI] [PubMed] [Google Scholar]
- MCKNIGHT A.T., CORBETT A.D., MARCOLI M., KOSTERLITZ H.W. The opioid receptors in the hamster vas deferens are of the δ-type. Neuropharmacol. 1985;24:1011–1017. doi: 10.1016/0028-3908(85)90184-4. [DOI] [PubMed] [Google Scholar]
- MILLER L., SHAW J.S., WHITING E.M. The contribution of intrinsic activity to the action of opioids in vitro. Br. J. Pharmacol. 1986;87:595–601. doi: 10.1111/j.1476-5381.1986.tb10202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKA T., NEGISHI K., SUDA M., MATSUMIYA T., INAZU T., UEKI M. Rabbit vas deferens: a specific biosassay for opioid κ-receptor agonists. Eur. J. Pharmacol. 1980;73:235–236. doi: 10.1016/0014-2999(81)90098-4. [DOI] [PubMed] [Google Scholar]
- PASHUTIN S.B. Limitation of hemodynamic disturbances in acute hypoxia and reoxygenation in situ by the synthetic peptide bioregulator dalargin. Biull. Exsp. Biol. Med. (Russia) 1991;112:1609–1614. [PubMed] [Google Scholar]
- PATON W.D.M., ZAR A. The origin of acetylcholine released from guinea-pig intestine and longitudinal muscle strip. J. Physiol. 1968;194:13–33. doi: 10.1113/jphysiol.1968.sp008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENCHEVA N., BOCHEVA A., DIMITROV E., IVANCHEVA CHR., RADOMIROV R. [Cys(O2NH2)2]enkephalin analogues and dalargin: selectivity for δ-opioid receptors. Eur. J. Pharmacol. 1996;304:99–108. doi: 10.1016/0014-2999(96)00083-0. [DOI] [PubMed] [Google Scholar]
- PENCHEVA N., IVANCHEVA CHR., DIMITROV E., BOCHEVA A., RADOMIROV R. Dalargin and [Cys-(O2NH2)]2 analogues of enkephalins and their selectivity for μ opioid receptors. Gen. Pharmacol. 1995;26:799–808. doi: 10.1016/0306-3623(94)00244-h. [DOI] [PubMed] [Google Scholar]
- PLESS J., BAUER W., CARDINAUX F., CLOSSE A., HAUSER D., HUGUENIN R., ROEMER D., BUESCHER H.H., HILL R.C. Synthesis, opiate receptor binding and analgetic activity of enkephalin analogues. Helvetica Chimica Acta. 1979;62:398–411. [Google Scholar]
- POKROVSKY V.M., OSADCHIY O.E. Regulatory peptides as modulators of vagal influence on cardiac rhythm. Can. J. Physiol. Pharmacol. 1995;73:1235–1245. doi: 10.1139/y95-175. [DOI] [PubMed] [Google Scholar]
- SAGAN S., CORBETT A.D., AMICHE M., DELFOUR A., NICOLAS P., KOSTERLITZ H.W. Opioid activity of dermenkephalin analogues in the guinea-pig myenteric plexus and the hamster vas deferens. Br. J. Pharmarcol. 1991;104:428–432. doi: 10.1111/j.1476-5381.1991.tb12446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRÖEDER U., SABEL B.A. Nanoparticles, a drug carrier system to pass the blood-brain barrier, permit central analgesic effects of i.v. dalargin injections. Brain Res. 1996;710:121–124. doi: 10.1016/0006-8993(95)01375-x. [DOI] [PubMed] [Google Scholar]
- SCHRÖEDER U., SOMMERFELD P., SABEL B.A. Efficacy of oral dalargin-loaded nanoparticle delivery across the blood-brain barrier. Peptides. 1998;19:777–780. doi: 10.1016/s0196-9781(97)00474-9. [DOI] [PubMed] [Google Scholar]
- SCHULTZ R., WÜSTER M. Are there subtypes (isoreceptors) of multiple opiate receptors in the mouse vas deferens. Eur. J. Pharmacol. 1981;76:61–66. doi: 10.1016/0014-2999(81)90009-1. [DOI] [PubMed] [Google Scholar]
- SCHWYZER R. Molecular mechanism of opioid receptor selection. Biochemistry. 1986;25:6335–6342. doi: 10.1021/bi00368a075. [DOI] [PubMed] [Google Scholar]
- SHIMOHIGASHI Y., COSTA T., NITZ TH, J., CHEN H.-CH., STAMMER CH.H. Importance of the stereo-orientation of aromatic groups in enkephalins to opiate receptor recognition. Biochem. Biophys. Res. Comm. 1984;121:966–972. doi: 10.1016/0006-291x(84)90771-x. [DOI] [PubMed] [Google Scholar]
- STJÄRNE L., ASTRAND P. Relative pre- and postjunctional role of noradrenaline and adenosine 5′-triphosphate as neurotransmitter of the sympathetic nerves of guinea-pig and mouse vas deferens. Neuroscience. 1985;14:929–935. doi: 10.1016/0306-4522(85)90155-1. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of pharmacological calculations with computer programs 1981Springer-Verlag: New York; 1–150.(eds) [Google Scholar]
- TIMOSHIN S.S., ALEKSEENKO S.A., SHTUKA A.A. Effect of dalargin on repair capacity of gastroduodenal mucosa in patients with duodenal ulcer. Klin Med. 1991;69:75–79. [PubMed] [Google Scholar]
- TITOV M.I., VINOGRADOV V.A., BESPALOVA ZH.D. Dalargin – a peptide preparation with cytoprotective action. Biull. Vsesouzn. Kardiolog. Nauk. Tsentr. AMN (Russia) 1985;8:72–76. [PubMed] [Google Scholar]
- ZADINA J.E., HACKLER L., GE L.-J., KASTIN A.J. A potent and selective endogenous agonist for the μ-opiate receptors. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- ZADINA J.E., KASTIN A.J., KERSH D., WYATT A. Tyr-MIF-1 and hemorphin can act as opiate agonists as well as antagonists in the guinea-pig ileum. Life Sci. 1992;51:869–885. doi: 10.1016/0024-3205(92)90615-v. [DOI] [PubMed] [Google Scholar]