

Abstract
In monkey lingual artery strips partially contracted with prostaglandin F2α, acetylcholine-induced, concentration-related relaxations were abolished by removal of the endothelium. The response was not significantly influenced by indomethacin but attenuated by NG-nitro-L-arginine (L-NOARG); the effect of the nitric oxide (NO) synthase inhibitor was reversed by L-arginine.
The response to acetylcholine resistant to L-NOARG was suppressed in the strips exposed to high K+ media. Charybdotoxin partially inhibited the relaxation, and the remaining relaxation was abolished by additional treatment with apamin, whereas glibenclamide, iberiotoxin or apamin alone was without effect. Relaxations induced by sodium nitroprusside were not influenced by charybdotoxin.
The L-NOARG-resistant acetylcholine-induced relaxation was inhibited by metyrapone, proadifen and 17-octadecynoic acid, non-selective cytochrome P450 mono-oxygenase (CYP) inhibitors, and progesterone and ketoconazole, inhibitors selective to CYP3A. The inhibitors did not affect the nitroprusside-induced relaxation. Selective inhibitors of other CYP isoforms, such as debrisoquine and lauric acid, did not reduce the response to acetylcholine.
Reaction mixture containing human liver microsome rich in CYPs, arachidonic acid and NADPH incubated at 37°C and filtrated relaxed endothelium-denuded monkey lingual artery strips, used as bioassay tissues. This response was abolished in the strips exposed to high K+ media. The response was also suppressed by combined treatment of the assay tissue with charybdotoxin plus apamin, but was not affected by treatment with iberiotoxin. The reaction mixture co-incubated with ketoconazole failed to relax the strips.
It is concluded that the monkey lingual arterial relaxation dependent on the endothelium is mediated by NO and also by a charybdotoxin plus apamin-sensitive but iberiotoxin-insensitive Ca2+-activated K+ channel opening substance(s) that may be a CYP3A-derived arachidonic acid metabolite(s).
Keywords: Cytochrome P450 mono-oxygenase, K+ channel, endothelium-dependent relaxation, lingual artery, acetylcholine
Introduction
It is widely recognized that endogenous relaxant substances derived from the endothelium include nitric oxide (NO), (Moncada et al., 1991), endothelium-derived hyperpolarizing factor (EDHF) and vasodilator prostanoids (Vanhoutte & Mombouli, 1996). EDHF is defined as a substance(s) from the endothelium that hyperpolarizes cell membrane and induces smooth muscle relaxation. The K+ channel subtypes involved are quite different in blood vessels, animal species and EDRF-releasing agonists used. It has been reported that ATP-sensitive K+ channels are involved in the response of cerebral arteries and abdominal aortae from rabbits in response to acetylcholine (Brayden, 1990; Cowan et al., 1993), apamin-sensitive Ca2+-activated K+ channels of small conductance are responsible for the relaxation by bradykinin and acetylcholine of the porcine coronary and rat mesenteric arteries (Hecker et al., 1994; Chen & Cheung, 1997; White & Hiley, 1997), and charybdotoxin-sensitive Ca2+-activated K+ channels of intermediate and large conductance participate in the response of rabbit carotid arteries and aortae when stimulated by acetylcholine (Cowan et al., 1993; Dong et al., 1997). Endothelium-dependent relaxations caused by acetylcholine in rat hepatic, and guinea-pig cerebral and carotid arteries are abolished by combined treatment with apamin plus charybdotoxin, but not affected by iberiotoxin, a selective large conductance Ca2+-activated K+ channel blocker (Zygmunt et al., 1997a; Petersson et al., 1997; Chataigneau et al., 1998). Such a diverse mechanism of action may indicate that EDHF is not a single molecule but includes various substances responsible for different modes of action.
Despite the numerous studies on the analysis of ion channels involved, EDHF has not been chemically identified. Although in some blood vessels, NO reportedly acts as a hyperpolarizing factor (Bolotina et al., 1994; Zhao et al., 1997), the majority of papers have demonstrated that EDHF is a substance(s) other than NO and prostanoids. From studies using pharmacological inhibitors, cytochrome P450 mono-oxygenase (CYP)-derived arachidonic acid metabolites are thought to be candidates of EDHF (Hecker et al., 1994; Campbell et al., 1996; Dong et al., 1997). However, Chataigneau et al. (1998) and Eckman et al. (1998) have postulated that EDHF(s) is not thought to be epoxyeicosatrienoic acids. Endogenous cannabinoids are suggested to be EDHF; however, papers against this hypothesis have also been reported (Mombouli & Vanhoutte, 1997; White & Hiley, 1997). Thus, the identity of EDHF appears to differ between vascular beds and species.
During screening experiments in various monkey isolated arteries, we found that lingual arteries responded to acetylcholine with endothelium-dependent relaxations that were mediated partly by substance(s) other than NO and prostanoids. In the present study, we therefore determined the mechanisms of action of acetylcholine with reference to K+ channels, to analyse pharmacologically the nature of substance(s) liberated from the endothelium using inhibitors of CYP isoforms and to elucidate whether human liver microsomes rich in CYPs were capable of synthesizing a vasodilator substance(s) that was responsible for K+ channel opening.
Methods
The Animal Care and Use Committee at Shiga University of Medical Science approved the use of monkey materials along with the experimental protocol in this study.
Preparation
Japanese monkeys (macaca fuscata) of either sex, weighing 7–12 kg, were killed by exanguination from the common carotid arteries under anaesthesia with an intramuscular injection of ketamine (40 mg kg−1) and intravenous injection of thiopental sodium (20 mg kg−1). The tongue was rapidly removed, and the deep lingual artery (0.3–0.4 mm outside diameter) in the middle portion between the tip and the root of the tongue was isolated, cleaned of surrounding tissue, and cut into helical strips approximately 20 mm long, taking special care to preserve the endothelium. The strips were fixed vertically between hooks in a muscle bath (20 ml capacity) containing a modified Ringer-Locke solution which was maintained at 37±0.3°C and aerated with a mixture of 95% O2-5% CO2.The hook anchoring the upper end of the strips was connected to the lever of a force-displacement transducer (Nihon-Kohden Kogyo Co., Tokyo, Japan). The resting tension was adjusted to 1 g, which was optimal for inducing the maximal contraction. The integrity of endothelial function was determined by a relaxant response to 0.1 μM A23187, a Ca2+ ionophore. In preparations used for bioassay of cytochrome P450 metabolites, the endothelium was removed by gently rubbing the intimal surface with a cotton ball. Constituents of the Ringer-Locke solution were (in mM) NaCl 120, KCl 5.4, CaCl2 2.2, MgCl2 1.0, NaHCO3 25.0 and 5.6 dextrose. The pH of the solution was 7.36–7.43. Before the start of the experiments, all of the strips were allowed to equilibrate for 60–90 min in the bathing media, during which time the fluid was replaced every 10 to 15 min.
Recordings of mechanical responses
Isometric contractions and relaxations were displayed on an ink-writing oscillograph. The contractile response to 30 mM K+ was obtained first, and the arterial strips were repeatedly washed with fresh media and equilibrated. The strips were partially contracted with prostaglandin F2α (0.2–1 μM), the contraction being in a range between 32–45% of the contraction induced by 30 mM K+. Concentration-response curves for acetylcholine, sodium nitroprusside (SNP), and arachidonylethanolamide (anandamide) were obtained by adding the drug directly to the bathing media in cumulative concentrations. After the reproducibility of the response to acetylcholine and SNP was determined, the strips were treated with blocking agents. At the end of each series of experiments, papaverine (0.1 mM) was applied to attain the maximum relaxation. Relaxations induced by acetylcholine, SNP, and anandamide are expressed as a percentage of those by papaverine. Contraction induced by the addition of 5 mM K+ is expressed as a percentage of that by 30 mM K+.
Bioassay of cytochrome P450 metabolites
A cytochrome P450-rich microsome fraction from the human liver was prepared as described by Imaoka et al. (1990). A 0.25 ml reaction mixture containing 200 μg human liver microsome, 0.8 mM reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH) and 20 μM arachidonic acid in 100 mM sodium phosphate buffer (pH 7.5) was incubated at 37°C for 30 min. After incubation with or without ketoconazole (10 μM), the reaction mixture was filtered with microcon 30 (Amicon, Inc., Beverly, MA, U.S.A.), and 100 μl of the filtrate was used for bioassay. The solution was directly applied to the bathing media (20 ml) and the mechanical response was obtained in deendothelialized lingual arterial strips treated with indomethacin (1 μM) and NG-nitro-L-arginine (L-NOARG, 0.1 mM) and precontracted with PGF2α.
Statistics and drugs used
The results shown in the text, tables, and figures are expressed as mean values±s.e.mean. Statistical analyses were made using Student's unpaired t-test for two groups and Tukey's test after one-way analysis of variance for three or more groups. Drugs used were NG-nitro-L-arginine (L-NOARG), charybdotoxin, iberiotoxin (Peptide Institute, Minoh, Japan), L-arginine (Nacalai Tesque, Kyoto, Japan), prostaglandin F2α (PGF2α, Pharmacia-Upjohn, Tokyo), acetylcholine chloride (Daiichi Pharmaceutical, Tokyo), sodium nitroprusside (SNP, Merck, Darmstadt, Germany), indomethacin, arachidonic acid, progesterone, glibenclamide, ketoconazole, sodium laurate, L-phenylephrine hydrochloride, 17-octadecynoic acid (Sigma Chemical, St Louis, MO, U.S.A.), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide, sodium salt (carboxy PTIO) (Dojindo Laboratories, Kumamoto, Japan), debrisoquine (Daiichi Pure Chemicals, Tokyo), proadifen, apamin and arachidonylethanolamide (anandamide) (Research Biochemicals Int., Natick, MA, U.S.A.), 2-methyl-1, 2-di-3-pyridyl-1-propanone (metyrapone) (Aldrich Chemical Co. Inc., Milwaukee, WI, U.S.A.), reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH, Oriental Yeast Co., Ltd., Tokyo), and papaverine hydrochloride (Dainippon, Osaka, Japan).
Results
Relaxation sensitive to L-NOARG
In monkey lingual arterial strips contracted with PGF2α, the addition of acetylcholine (10 nM–10 μM) induced a concentration-related relaxation. Removal of the endothelium almost abolished the response but did not inhibit the relaxation caused by sodium nitroprusside (SNP; 1 nM–1 μM) (Figure 1). Treatment with indomethacin (1 μM) did not or only slightly reduced the acetylcholine-induced relaxation; mean values of the response at 10 and 100 nM acetylcholine were 26.3±6.5 and 78.4±2.3%, respectively, in control media and 19.3±4.5 (P>0.05, unpaired t-test, n=7) and 58.1±6.4% (P<0.05), respectively, in the media containing indomethacin. In the strips treated with indomethacin, acetylcholine-induced relaxations were moderately attenuated by treatment with L-NOARG (10 μM), and L-arginine (1 mM) reversed the inhibition (Figure 1). Carboxy PTIO (0.3 mM), a NO scavenger (Akaike et al., 1993), did not inhibit further the response to acetylcholine in 0.1 mM L-NOARG- and 1 μM indomethacin-treated strips (Figure 1). In the remainder of this study to analyse mechanisms of the response to acetylcholine mediated by a substance(s) other than NO and vasodilator prostanoids, the strips were treated with 1 μM indomethacin and 0.1 mM L-NOARG which was considered to be a supramaximal concentration to depress the NO synthesis.
Figure 1.
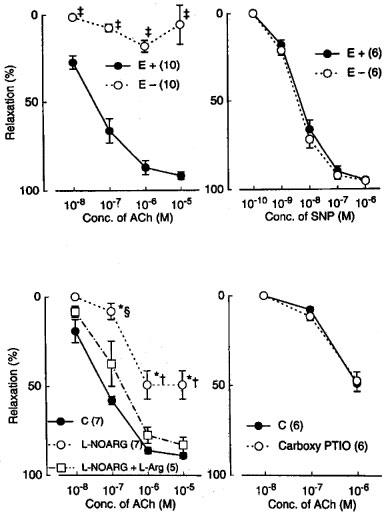
Modification of relaxations induced by acetylcholine (ACh) and sodium nitroprusside (SNP) by endothelium denudation (E−), L-NOARG, L-arginine, (L-Arg), and carboxy PTIO in monkey lingual arteries. The ordinate denotes relaxations relative to those induced by papaverine (0.1 mM). Numbers in parentheses indicate the number of strips from individual monkeys. Upper left and right, effect of endothelium denudation on responses to acetylcholine and nitroprusside, respectively. Lower left, inhibition by L-NOARG (10 μM) of acetylcholine action and reversal by L-arginine (1 mM); lower right, effect of carboxy PTIO (0.3 mM) on acetylcholine action in L-NOARG (100 μM)-treated strips. Significantly different from control (E+), ‡P<0.001 (unpaired t-test). Significantly different from control, *P<0.01; significantly different from the value with L-NOARG+L-arginine, †P<0.01; ≈rcub;P<0.05 (Tukey's test). Vertical bars represent s.e.mean.
Relaxation resistant to L-NOARG and indomethacin
In the L-NOARG- and indomethacin-treated lingual arterial strips contracted with PGF2α, acetylcholine-induced relaxations were abolished by exposure to high K+ solutions (15+5.4 mM), but were not affected by treatment with glibenclamide (1 μM) (Figure 2). Treatment with iberiotoxin (30 and 100 nM) or apamin (1 μM) alone failed to inhibit the response, which was however attenuated significantly by charybdotoxin (30 nM). Raising the concentration of charybdotoxin to 0.1 μM did not produce additional inhibition (n=6). The charybdotoxin-resistant relaxation was abolished by additional treatment with apamin (1 μM) (Figure 3). Relaxations induced by SNP were not reduced by charybdotoxin plus apamin (Table 1). The relaxations caused by acetylcholine did not significantly differ in the L-NOARG- and indomethacin-treated strips contracted with PGF2α or phenylephrine; mean values of the response at 0.1 and 1 μM acetylcholine were 14.3±6.2 and 50.3±9.5%, respectively, in the strips contracted with PGF2α and 14.2±5.2 (P>0.05, unpaired t-test, n=5) and 49.4±9.2% (P>0.05), respectively, in the strips contracted with phenylephrine. Apamin (1 μM), iberiotoxin (0.1 μM), and charybdotoxin (30 nM) did not change either the basal tone or prostaglandin F2α-induced contraction (n=6).
Figure 2.
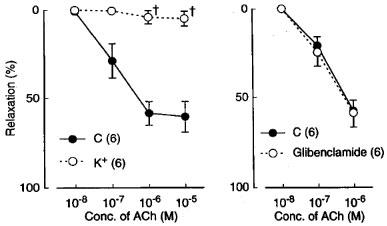
Modifications by high K+ (addition of 15 mM K+) and glibenclamide (1 mM) of relaxant responses to acetylcholine in monkey lingual arteries with intact endothelium in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). The ordinate denotes relaxations relative to those induced by papaverine (0.1 mM). Numbers in parentheses indicate the number of strips from individual monkeys. Significantly different from control (C), †P<0.05 (unpaired t-test). Vertical bars represent s.e.mean.
Figure 3.
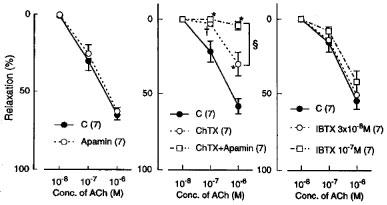
Modifications by apamin (1 μM) alone, charybdotoxin (ChTX; 30 nM) plus apamin, and iberiotoxin (IBTX; 30 and 100 nM) of relaxations induced by acetylcholine in monkey arteries with intact endothelium in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). The ordinate denotes relaxations relative to those induced by papaverine (0.1 mM). Numbers in parentheses indicate the number of strips from individual monkeys. Significantly different from control, *P<0.01; †P<0.05; significantly different from the value with ChTX, ≈rcub;P<0.05 (Tukey's test). Vertical bars represent s.e.mean.
Table 1.
Modifications by inhibitors of the relaxation induced by sodium nitroprusside (SNP, 10 nM) in monkey lingual arterial strips contracted with PGF2α
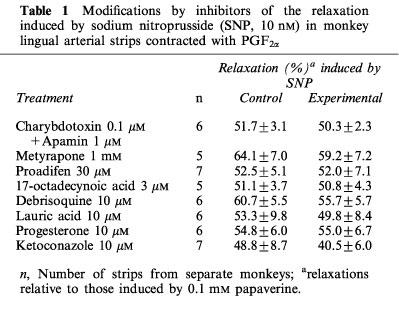
Metyrapone (1 mM) and proadifen (10 μM) significantly inhibited the relaxation to acetylcholine. A higher concentration of proadifen (30 μM) abolished the relaxation. Quantitative data are summarized in Figure 4. Seventeen-octadecynoic acid (3 μM) significantly inhibited the response to acetylcholine; mean values of the response at 0.1 and 1 μM acetylcholine were 27.5±10.0 and 63.0±7.2%, respectively, in the control strips and 1.1±1.1 (P<0.05, unpaired t-test, n=5) and 12.4±3.7% (P<0.001), respectively, in the strips treated with 17-octadecynoic acid. SNP-induced relaxation was unaffected by metyrapone, proadifen or 17-octadecynoic acid (Table 1). Neither lauric acid (1 and 10 μM) nor debrisoquine (1 and 10 μM) affected the response to acetylcholine (Figure 4). Treatment with progesterone (1 and 10 μM) or ketoconazole (1 and 10 μM) significantly impaired the acetylcholine actions in a concentration-dependent manner (Figure 5). Typical tracings of the response to acetylcholine and the effects of these inhibitors are illustrated in Figure 6. Neither progesterone nor ketoconazole reduced the response to SNP (Table 1). Metyrapone, proadifen, lauric acid (10 μM), debrisoquine (10 μM), progesterone (10 μM), ketoconazole (10 μM), and 17-octadecynoic acid (3 μM) had no effect on basal tension (n=6).
Figure 4.
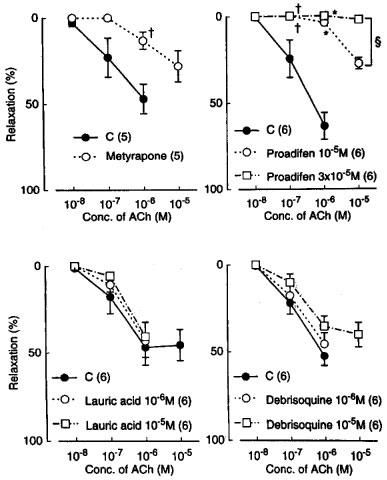
Modifications by metyrapone (1 mM), proadifen (10 and 30 μM), lauric acid (10 μM), and debrisoquine (10 μM) of relaxations induced by acetylcholine in monkey lingual arteries with intact endothelium in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). The ordinate denotes relaxations relative to those induced by papaverine (0.1 mM). Numbers in parentheses indicate the number of strips from individual monkeys. Significantly different from control, †P<0.05 (unpaired t-test for upper left). Significantly different from control, *P<0.01; †P<0.05 (Tukey's test for upper right); significantly different between the values with proadifen 10 and 30 μM, ≈rcub;P<0.001 (unpaired t-test). Vertical bars represent s.e.mean.
Figure 5.
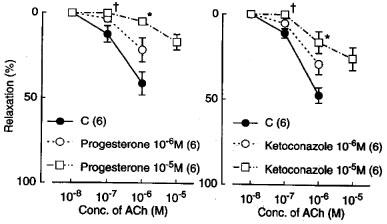
Modifications of progesterone (1 and 10 μM) and ketoconazole (1 and 10 μM) of relaxations induced by acetylcholine in monkey lingual arteries with intact endothelium in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). Numbers in parentheses indicate the number of strips from individual monkeys. Significantly different from control, *P<0.01; †P<0.05 (Tukey's test). Vertical bars represent s.e.mean.
Figure 6.
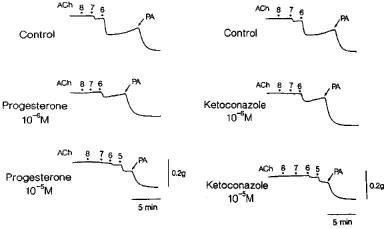
Tracings of the response to acetylcholine as affected by progesterone and ketoconazole in monkey lingual artery strips with intact endothelium in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). Two strips were obtained from the same monkey and partially contracted with PGF2α. Concentrations of acetylcholine from 8 to 5, 10 nM, 100 nM, 1 μM, and 10 μM, respectively. PA represents papaverine (0.1 mM) that produced the maximal relaxation.
In endothelium-denuded strips, anandamide did not produce relaxation at 1 μM (n=5), reported to be an effective concentration in rat isolated mesenteric arteries (pEC50=6.31, White & Hiley, 1997), and elicited slight (32.5±4.4%, n=5), slowly-developing relaxation at 10 μM. The stabilized response was obtained 20–30 min after the drug application, whereas the response to acetylcholine (10 nM inducing about 30% relaxation) in endothelium-intact strips levelled off 1–3 min later.
Bioassay of P450 product
The filtrated reaction mixture (volume of 100 μl) applied to the L-NOARG- and indomethacin-treated endothelium-denuded lingual arterial strips, the bioassay tissue, elicited relaxation. Removal of NADPH or the enzyme preparation abolished the ability of the filtrate to relax the bioassay tissue (control vs NADPH-free; 13.1±2.2 vs 0±0%, n=6, P<0.001, unpaired t-test) and (control vs enzyme-free; 14.8±2.5 vs 4.2±1.7%, n=4, P<0.05, unpaired t-test). The relaxations were abolished in the strips exposed to high K+ solution (Figure 7). Treatment of the assay tissue with charybdotoxin (30 nM) alone moderately inhibited the relaxation (control vs charybdotoxin-treated; 19.7±1.4 vs 9.6±1.7%, n=4, P<0.01, unpaired t-test). Raising the concentration of charybdotoxin to 0.1 μM did not produce additional inhibition (10.0±1.9%, n=4). Combined treatment of the assay tissue with charybdotoxin (30 nM) plus apamin (1 μM) suppressed the relaxation, while treatment with iberiotoxin (0.1 μM) did not influence the response (Figure 7). The reaction mixture co-incubated with ketoconazole (10 μM; the final concentration in the organ bath, 50 nM) failed to relax the strips (Figure 7). Typical tracings of the response to the filtrated reaction mixture and the effects of inhibitors are illustrated in Figure 8.
Figure 7.
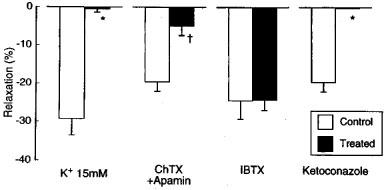
Responses to CYP products of endothelium-denuded monkey lingual artery strips in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM) as affected by exposure to high K+ media (K+ 15 mM) and treatment of the strips with charbdotoxin (ChTX; 30 nM plus apamin (1 μM), iberiotoxin (IBTX; 100 nM), and by treatment of the reaction mixture with ketoconazole (10 μM). The ordinate denotes relaxations relative to those induced by papaverine (0.1 mM). The ‘control' denotes the response to the reaction mixture with full components (liver microsome as enzyme preparation, aracidonic acid and NADPH). n, the number of strips from individual monkeys. Significantly different from control, *P<0.001; †P<0.01 (unpaired t-test). Vertical bars represent s.e.mean.
Figure 8.
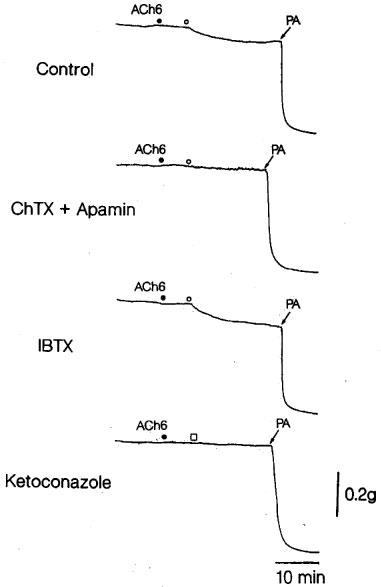
Tracings of the response to acetylcholine and CYP products as affected by treatment of the strips with charybdotoxin (ChTX; 30 nM) plus apamin (1 μM) or iberiotoxin alone (IBTX; 100 nM), and by treatment of the reaction mixture with ketoconazole (1 μM) in endothelium-denuded monkey artery strips in the presence of indomethacin (1 μM) and L-NOARG (0.1 mM). Four strips were obtained from the same monkey and partially contracted with PGF2α. ACh6 •, the addition of 1 μM acetylcholine; ○, the addition of CYP products; □, the addition of CYP products co-incubated with 10 μM ketoconazole. PA represents papaverine (0.1 mM) that produced the maximal relaxation.
The addition of K+ (5–15 mM) produced dose-dependent contraction (15.9±3.0% at 5 mM K+, n=6) in the L-NOARG- and indomethacin-treated endothelium-denuded lingual arterial strips contracted with PGF2α.
Discussion
Monkey lingual arterial strips responded to acetylcholine with concentration-dependent relaxations solely when the endothelium was intact. The acetylcholine-mediated response was only partially due to prostacyclin release, whereas NO release played a greater role. However, there was a large residual relaxation to acetylcholine that may be due to the release of endothelium-derived hyperpolarizing factor (EDHF). This is in contrast to other monkey arteries such as the coronary and mesenteric arteries where the acetylcholine-mediated relaxation can be fully abolished with methylene blue alone or in combination with indomethacin (Okamura et al., 1989). This is the first report of an endothelium-dependent and NO/prostacyclin-independent response in monkey arteries.
The release of the vasodilator substance by acetylcholine was blocked by the combination of charybdotoxin plus apamin, but not iberiotoxin, a profile consistent with activation of small and intermediate conductance, but not large conductance, calcium-activated K+ channels. We confirmed that the concentration of iberiotoxin used (100 nM) was sufficient to significantly inhibit an endothelium-dependent and NO/prostacyclin-independent acetylcholine-induced relaxation in isolated rabbit carotid artery (Ayajiki et al., unpublished observation), as reported by Dong et al. (1997). In studies with patch-clamp technique, coexistence of apamin- and charybdotoxin-sensitive K+ channel has been reported in rat renal arteriolar smooth muscle cells (Gebremedhin et al., 1996).
Metabolites of arachidonic acid by a catalysis of CYP are considered to be candidates of EDHF. Hecker et al. (1994) have reported that relaxations induced by bradykinin of coronary arteries treated with L-NOARG are attenuated by TBA, apamin and proadifen, and suggested that an epoxide, a CYP-derived arachidonate metabolite, is involved in the response. Epoxyeicosatrienoic acids, CYP products, are also speculated to be EDHFs in bovine coronary arteries stimulated by methacholine (Campbell et al., 1996). Relaxations by acetylcholine of isolated rat cremaster muscle arterioles, possibly mediated by opening of K+ channels, are reduced by miconazole and proadifen, CYP inhibitors (Bakker & Sipkema, 1997). Many inhibitors to CYP isoforms have been determined from biochemical studies with enzyme preparations. Ketoconazole and progesterone, inhibitors selective to CYP3A (Senciall et al., 1988; Chiba et al., 1996), were clearly effective in attenuating the response to acetylcholine but were without effect on the SNP-induced relaxation. Although Ca2+-activated K+ channel blocking actions of ketoconazole and progesterone have been reported in the cells other than vascular smooth muscle cells, the concentration (10 μM) of both agents used in the present study is reportedly ineffective (Ishii et al., 1997; Ehring et al., 1998). Seventeen-octadecynoic acid, another type of CYP inhibitor (Dong et al., 1997) without K+ channel inhibitory action, also attenuated the response to acetylcholine. These results indicate that impairment by these drugs of acetylcholine-induced relaxations is due to inhibitory effects on CYP activity but not on Ca2+-activated K+ channel. Metyrapone and proadifen, non-selective inhibitors of CYPs (Iribarne et al., 1996), were also effective. On the other hand, debrisoquine, an inhibitor of CYP2D6 (Marre et al., 1992), and lauric acid, an inhibitor of CYP4A1 (Lawson, 1991), did not significantly reduce the acetylcholine action. The concentrations used are evidenced to be sufficient to inhibit activities of the corresponding CYP isoforms (Chiba et al., 1996; Senciall et al., 1988; Iribarne et al., 1996). These findings indicate a possible involvement of CYP3A product(s) with K+ channel opening property in the acetylcholine-induced relaxation. Filtrated media incubated in the CYP preparation, NADPH and arachidonic acid produced relaxation of endothelium-denuded monkey lingual arterial strips only when all components were present in the incubation media. Treatment of the assay tissue with charybdotoxin alone partially inhibited the response, and that with high K+ or charybdotoxin plus apamin abolished the response. On the other hand, treatment with iberiotoxin did not affect the response. The filtrated media co-incubated with ketoconazole failed to relax the strips. Therefore, the relaxation is likely to be induced by CYP3A product(s) responsible for the opening of small and intermediate conductance, but not large conductance, Ca2+-activated K+ channel, as seen in the acetylcholine-induced relaxation described above.
It is unknown which CYP product is responsible for endothelium-dependent and NO/prostacyclin-independent responses, since several epoxyeicosatrienoic acids do not induce relaxations in guinea-pig carotid arteries (Chataigneau et al., 1998), whereas 11,12-epoxyeicosatrienoic acid induces relaxation in the guinea-pig coronary artery (Eckman et al., 1998). This epoxide does not seem to be involved in the endothelium-dependent, L-NOARG plus indomethacin-resistant relaxation caused by acetylcholine in the monkey lingual artery, because the relaxation observed in this monkey artery was not affected by iberiotoxin, while 11,12-epoxyeicosatrienoic acid-induced relaxation in guinea-pig coronary arteries was inhibited by iberiotoxin.
Edwards et al. (1998) have reported that EDHF is K+ released from endothelial cells through charybdotoxin- and apamin-sensitive K+ channels, then the resulting increase in myoendothelial K+ concentration hyperpolarizes and relaxes adjacent smooth muscle cells by activating Ba2+-sensitive K+ channels and Na+/K+ ATPase in the rat hepatic artery. However, this is not the case in the monkey lingual artery because the addition of 5 mM K+ contracted the endothelium-denuded artery treated with L-NOARG and indomethacin.
Anandamide (1 μM) in a concentration sufficient to produce relaxation (White & Hiley, 1997; Plane et al., 1997; Zygmunt et al., 1997b) did not change arterial tone, and relaxations induced at a ten times higher concentration were clearly slower than those exerted by an equipotent concentration (10 nM) of acetylcholine. Therefore, this substance does not seem to be involved in the endothelium-dependent, L-NOARG plus indomethacin-resistant relaxation in monkey lingual arteries.
Our conclusion is that endothelium-derived vasodilator substance(s) other than NO and prostanoids appears to be CYP3A metabolite(s) from arachidonic acid other than epoxyeicosatrienoic acids and to open the charybdotoxin- and apamin-sensitive Ca2+ activated K+ channel with small or intermediate conductance on the smooth muscle, resulting in relaxation. Monkey lingual arteries are expected to relax in response to acetylcholine by mediations of NO and the CYP-derived arachidonic acid metabolite(s) liberated from the endothelium.
Acknowledgments
This work was supported in part by the Grant-in Aid for Scientific Research (C) from the Ministry of Education, Science, Culture and Sports, Japan.
Abbreviations
- Anandamide
arachidonylethanolamine
- Carboxy PTIO
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide sodium salt
- CYP
cytochrome P450 mono-oxygenase
- EDHF
endothelium-derived hyperpolarizing factor
- L-NOARG
NG-nitro-L-arginine
- Metyrapone
2-methyl-1,2-di-3-pyridyl-1-propanone
- NADPH
nicotinamide-adenine dinucleotide phosphate
- NO
nitric oxide
- SNP
sodium nitroprusside
References
- AKAIKE T., YOSHIDA M., MIYAMOTO Y., SATO K., KOHNO M., SASAMOTO K., MIYAZAKI K., UEDA S., MAEDA H. Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor/.NO through a radical reaction. Biochemistry. 1993;32:827–832. doi: 10.1021/bi00054a013. [DOI] [PubMed] [Google Scholar]
- BAKKER E.N.T.P., SIPKEMA P. Components of acetylcholine-induced dilation in isolated rat arterioles. Am. J. Physiol. 1997;273:H1848–H1853. doi: 10.1152/ajpheart.1997.273.4.H1848. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- BRAYDEN J.E. Membrane hyperpolarization is a mechanism of endothelium-dependent cerebral vasodilation. Am. J. Physiol. 1990;259:H668–H673. doi: 10.1152/ajpheart.1990.259.3.H668. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FÉLÉTOU M., DUHAULT J., VANHOUTTE P.M. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., CHEUNG D.W. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. Am. J. Physiol. 1997;272:H2306–H2312. doi: 10.1152/ajpheart.1997.272.5.H2306. [DOI] [PubMed] [Google Scholar]
- CHIBA M., HENSLEIGH M., NISHIME J.A., BALANI S.K., LIN J.H. Role of cytochrome P450 3A4 in human metabolism of MK-639, a potent human immunodeficiency virus protease inhibitor. Drug Metab. Dispos. 1996;24:307–314. [PubMed] [Google Scholar]
- COWAN C.L., PALACINO J.J., NAJIBI S., COHEN R.A. Potassium channel-mediated relaxation to acetylcholine in rabbit arteries. J. Pharmacol. Exp. Ther. 1993;266:1482–1489. [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., GALIPEAU D., COLE W.C., TRIGGLE C.R. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br. J. Pharmacol. 1997;120:695–701. doi: 10.1038/sj.bjp.0700945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECKMAN D.M., HOPKINS N., MCBRIDE C., KEEF K.D. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EHRING G.R., KERSCHBAUM H.H., EDER C., NEBEN A.L., FANGER C.M., KHOURY R.M., NEGULESCU P.A., CAHALAN M.D. A nongenomic mechanism for progesterone-mediated immunosupression: inhibition of K+ channels, Ca2+ signaling, and gene expression in T lymphocytes. J. Exp. Med. 1998;188:1593–1602. doi: 10.1084/jem.188.9.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEBREMEDHIN D., KALDUNSKI M., JACOBS E.R., HARDER D.R., ROMAN R.J. Coexistence of two types of Ca2+-activated K+ channels in rat renal arterioles. Am. J. Physiol. 1996;270:F69–F81. doi: 10.1152/ajprenal.1996.270.1.F69. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMAOKA S., ENOMOTO K., ODA Y., ASADA A., FUJIMORI M., SHIMADA T., FUJITA S., GUENGERICH F.P., FUNAE Y. Lidocaine metabolism by human cytochrome P-450s purified from hepatic microsomes: comparison of those with rat hepatic cytochrome P-450s. J. Pharmacol. Exp. Ther. 1990;255:1385–1391. [PubMed] [Google Scholar]
- IRIBARNE C., BERTHOU F., BAIRD S., DREANO Y., PICARD D., BAIL J.P., BEAUNE P., MENEZ J.F. Involvement of cytochrome P450 3A4 enzyme in the N-demethylation of methadone in human liver microsomes. Chem. Res. Toxicol. 1996;9:365–373. doi: 10.1021/tx950116m. [DOI] [PubMed] [Google Scholar]
- ISHII T.M., SILVIA C., HIRSCHBERG B., BOND C.T., ADELMAN J.P., MAYLIE J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWSON T.A. Involvement of lauric acid hydroxylase in the activation of beta-substituted nitrosamines. Cancer Lett. 1991;59:177–182. doi: 10.1016/0304-3835(91)90184-j. [DOI] [PubMed] [Google Scholar]
- MARRE F., FABRE G., LACARELLE B., BOURRIE M., CATALIN J., BERGER Y., RAHMANI R., CANO J.P. Involvement of the cytochrome P-450IID subfamily in minaprine 4-hydroxylation by human hepatic microsomes. Drug Metab. Dispos. 1992;20:316–321. [PubMed] [Google Scholar]
- MOMBOULI J.-V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- OKAMURA T., MINAMI Y., TODA N. Endothelium-dependent and -independent mechanisms of action of acetylcholine in monkey and dog isolated arteries. Pharmacology. 1989;38:279–288. doi: 10.1159/000138548. [DOI] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HÖGESTÄTT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., HOLLAND M., WALDRON G.J., GARLAND C.J., BOYLE J.P. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br. J. Pharmacol. 1997;121:1509–1511. doi: 10.1038/sj.bjp.0701361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENCIALL I.R., RAHAL S.R., ROBERTS R. Ketoconazole inhibition of progesterone oxidation by the rabbit. Biochem. Pharmacol. 1988;37:3647–3651. doi: 10.1016/0006-2952(88)90397-8. [DOI] [PubMed] [Google Scholar]
- VANHOUTTE P.M., MOMBOULI J.-V. Vascular endothelium: vasoactive mediators. Prog. Cardiovasc. Dis. 1996;39:229–238. doi: 10.1016/s0033-0620(96)80003-x. [DOI] [PubMed] [Google Scholar]
- WHITE R., HILEY C.R. A comparison of EDHF-mediated and anandamide-induced relaxations in the rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1573–1584. doi: 10.1038/sj.bjp.0701546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Y.-J., WANG J., RUBIN L.J., YUAN X.-J. Inhibition of KV and KCa channels antagonizes NO-induced relaxation in pulmonary artery. Am. J. Physiol. 1997;272:H904–H912. doi: 10.1152/ajpheart.1997.272.2.H904. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., LARSSON B., HÖGESTÄTT E.D. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997a;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D., WALDECK K., EDWARDS G., KIRKUP A.J., WESTON A.H. Studies on the effects of anandamide in rat hepatic artery. Br. J. Pharmacol. 1997b;122:1679–1686. doi: 10.1038/sj.bjp.0701601. [DOI] [PMC free article] [PubMed] [Google Scholar]