

Abstract
Tumour necrosis factor (TNF-α) is involved in the pathogenesis of splanchnic artery occlusion (SAO) shock. On the other hand, inhibition of TNF-α is an important component of the mechanism of action of melanocortins in reversing haemorrhagic shock. We therefore investigated the effects of the melanocortin peptide ACTH-(1–24) (adrenocorticotropin fragment 1–24) on the vascular failure induced by SAO shock.
SAO-shocked rats had a decreased survival rate (0% at 4 h of reperfusion, while sham-shocked rats survived for more than 4 h), enhanced serum TNF-α concentrations (755±81 U ml−1), decreased mean arterial blood pressure, leukopenia, and increased ileal leukocyte accumulation, as revealed by means of myeloperoxidase activity (MPO=9.4±1 U g−1 tissue). Moreover, aortic rings from shocked rats showed a marked hyporeactivity to phenylephrine (PE, 1 nM–10 μM) (Emax and ED50 in shocked rats=7.16 mN mg−1 tissue and 120 nM, respectively; Emax and ED50 in sham-shocked rats=16.31 mN mg−1 tissue and 100 nM, respectively), reduced responsiveness to acetylcholine (ACh, 10 nM-10 μM) (Emax and ED50 in shocked rats=30% relaxation and 520 nM, respectively; Emax and ED50 in sham-shocked rats=82% relaxation and 510 nM, respectively) and increased staining for intercellular adhesion molecule-1 (ICAM-1).
ACTH-(1–24) [160 μg kg−1 intravenously (i.v.), 5 min after SAO] increased survival rate [SAO+ACTH-(1–24)=80% at 4 h of reperfusion], reversed hypotension, reduced serum TNF-α (55±13 U ml−1), ameliorated leukopenia, reduced ileal MPO (1.2±0.2 U g−1 tissue), restored the reactivity to PE, improved the responsiveness to ACh and blunted the enhanced immunostaining for ICAM-1 in the aorta.
Adrenalectomy only in part–but not significantly–reduced the ACTH-induced shock reversal, the survival rate of SAO+ACTH-(1–24) adrenalectomized rats being 60% at 4 h of reperfusion; and methylprednisolone (80 mg−1 i.v., 5 min after SAO) had a non-significant effect (10% survival) at 4 h of reperfusion.
The present data show that melanocortins are effective also in SAO shock, their effect being, at least in part, mediated by reduced production of TNF-α. Furthermore, they demonstrate, for the first time, that this inhibition is responsible for the adrenocorticotropin-induced reversal of vascular failure and leukocyte accumulation.
Keywords: Splanchnic artery occlusion shock, Vascular dysfunction, ACTH(1–24), TNF-α
Introduction
Occlusion of the major splanchnic arteries followed by reperfusion in anaesthetized rats results in an irreversible circulatory failure and shock (splanchnic artery occlusion shock; SAO shock) (Squadrito et al., 1994b). It has been demonstrated that, in SAO shock, a marked and composite vascular dysfunction is present in which tumor necrosis factor-α (TNF-α) plays an important role (Squadrito et al., 1994a). Indeed, aortic rings from shocked rats showed a marked hyporeactivity to phenylephrine (PE), and removal of the endothelium did not restore the PE-induced contractile response to the values of the sham animal, thus suggesting that smooth muscle cells are involved in the hyporesponsiveness to PE. This complex dysfunction is probably the result of an increase in endogenous nitric oxide (NO) produced by the inducible NO synthase (iNOS) activated by TNF-α.
A large number of experimental data indicate that melanocortin peptides have an anti-shock effect in haemorrhagic shock in rats and dogs (Bertolini et al., 1986a,1986b,1986c; Guarini et al. 1990), in another model of hypovolemic shock in rabbits (Ludbrook & Ventura, 1995), as well as in human conditions of haemorrhagic or cardiogenic shock (Bertolini et al., 1987; Pinelli et al., 1989; Noera et al., 1989; 1991). This beneficial activity may be, at least in part, the consequence of a functional antagonism towards endogenous opioids, which are released in shock conditions (Bertolini 1995).
On the other hand, melanocortins are also potent inhibitors of NO overproduction, both in vitro, in cultured macrophages stimulated with bacterial LPS and interferon gamma (Star et al., 1995), and in vivo, in experimental conditions of haemorrhagic and endotoxic shock in rats (Guarini et al., 1997; Abou-Mohamed et al., 1995; Bazzani et al., 1997). The adrenocorticotropin fragment 1–24 [ACTH- (1–24)] seems to inhibit iNOS induction rather than its activity (unpublished data), thus suggesting that antagonism towards NO may be due to other factor(s) involved in the priming of NO production.
Close links exist between TNF-α and iNOS: the pleiotropic cytokine may induce this synthase either in vivo or in vitro and so lead to NO overproduction (Busse & Mulsch, 1990). In the light of these findings, it is thus reasonable to postulate that adrenocorticotropin may firstly inhibit TNF-α and secondly blunt NO production. In accordance with this hypothesis, it has been suggested that inhibition of TNF-α overproduction may be an important component of the anti-shock effect of melanocortins (Altavilla et al., 1998).
The aim of our study was therefore to find out whether ACTH-(1–24) also exerts protective effects on the pathological sequelae associated with SAO shock, both by reducing TNF-α and by modulating vascular dysfunction. We found that ACTH-(1–24) inhibits the inflammatory cytokine, in turn reversing vascular failure and reducing leukocyte accumulation. Our data therefore offer new insights into the mechanism(s) underlying the anti-shock effect of melanocortins.
Methods
Animal preparation
Male Sprague-Dawley rats weighing 200–250 g (Harlan Nossan, Correzzana, Milano, Italy), were kept 4–5 per cage (44×35×20 cm) in temperature-, humidity- and ventilation-controlled colony rooms (temperature: 20±1°C; humidity: 60%), with food in pellets and tap water available ad libitum. Housing conditions and experimental procedures were in strict accordance with the European Community regulations. The animals were accustomed to our housing conditions for at least 1 week before being used. Some of them were bilaterally adrenalectomized by dorsal approach under ethyl ether anaesthesia 4–5 days before use; the adequacy of the adrenalectomy was ascertained at the end of the experiments by carefully autoptical examination. All experiments were performed under general anaesthesia [urethane, 1.3 g kg−1, intraperitoneally (i.p.)]. Urethane (Fluka AG, Buchs, Switzerland) was chosen because it produces long-lasting and quite stable general anaesthesia with minor interference on cardiovascular and respiratory function (Maggi & Meli, 1986a,1986b). After midline laparotomy, the coeliac and superior mesenteric arteries were isolated near their aortic origins. During this procedure, the intestinal tract was maintained at 37°C by placing it between gauze pads soaked with warmed 0.9% NaCl solution. Rats were given heparin [1000 U kg−1, intravenously (i.v.)] and were observed for a 30-min stabilization period prior to either splanchnic ischaemia or sham ischaemia. Splanchnic artery ischaemia-reperfusion injury (SAO) was induced by clamping both the superior mesenteric artery and the coeliac trunk, so as to produce a total occlusion of these arteries for 45 min. The clamps were then removed. Following reperfusion the rats were observed for 4 h. Sham-operated rats were subjected to the same surgical procedures as SAO rats, except that the arteries were not occluded. Allocation to SAO or sham procedure was at random, in parallel (at least one SAO-shocked and one sham-operated rat were used every day).
Survival evaluation and arterial blood pressure monitoring
A first group of animals was used to study survival and arterial blood pressure. ACTH-(1–24) (160 μg kg−1), methylprednisolone (80 mg kg−1) or vehicle (NaCl 0.9%; 1 ml kg−1) were i.v. injected 5 min following the onset of reperfusion. The doses of ACTH-(1–24) and methylprednisolone were chosen on the basis of previous studies in haemorragic shock in rats (Bertolini et al., 1986a,1986b,1986c; Guarini et al., 1990; 1997; Bazzani et al., 1993). Animals were randomly assigned to one of the three treatments, in parallel. Survival was evaluated for 4 h after the onset of reperfusion and expressed both as survival rate and survival time. Animals still surviving at the fourth hour after reperfusion had their surgical wounds sutured and were placed in single cage (20×26×14 cm) with food in pellets and tap water, and kept in a strictly climatized enviroment (temperature: 23±1°C; humidity: 60%). One group of animals was also implanted with cannulae (PE 50) into the left common carotid artery. The arterial catheter was connected to a pressure transducer. The pressure pulse triggered a cardiotachometer, and arterial blood pressure was displayed on a polygraph (Ugo Basile, Varese, Italy). Arterial blood pressure is reported as mean arterial pressure (MAP) in mmHg. Rats were subjected to the same experimental protocol as described above.
Isolated aortic rings
Thoracic aortae were removed 70 min after reperfusion and placed in cold Krebs' solution of the following composition (nM): NaCl 118.4, KCl 4.7, MgSO4 1.2, CaCl2 2.5, KH2PO4 1.2, NaHCO3 25.0 and glucose 11.7; the aortae were then cleaned of adherent connective and fat tissue and cut into rings of approximately 2 mm in length. In some rings, the vascular endothelium was removed mechanically by gently rubbing the luminal surface with a thin wooden stick. Rings were then placed under 9.81 mN of tension in an organ bath containing 10 ml of Krebs' solution at 37°C and bubbled with 95% O2 and 5% CO2 (pH 7.4). All experiments were carried out in the presence of indomethacin (10 μM) in order to exclude the involvement of prostaglandins and their metabolites. The tension developed was measured with an isometric force transducer and recorded on a polygraph (Ugo Basile, Varese, Italy). After an equilibration period of 60 min during which time the rings were washed with fresh Krebs' solution at 15–20 min intervals and basal tension was readjusted to 9.81 mN, the tissue was exposed to phenylephrine (PE, 100 nM). When the contraction was stable, the functional integrity of the endothelium was assessed by a relevant response to acetylcholine (ACh, 100 nM). The tissue was then washed occasionally for 30 min. Concentration-response curves were obtained by cumulative concentrations of PE (1 nM–10 μM) applied to intact or endothelium denuded aortic rings. The results are expressed as mN of tension mg−1 tissue.
Biological assay for tumour necrosis factor-α activity
A third group of animals was used to measure TNF-α, myeloperoxidase (MPO) activity, leukocyte count, vascular reactivity and intercellular adhesion molecule-1 (ICAM-1) expression on vascular endothelium.
The killing of L929 mouse tumour cells was used to measure TNF-α levels in serum and in peritoneal macrophage supernatants on the basis of a standard microelisa assay (Ruff & Gifford, 1980). L929 cells in RPMI 1640 medium containing 5% foetal calf serum were seeded at 3×104 cells per well in 96-well microdilution plates and incubated overnight at 37°C in an atmosphere of 5% CO2 in air. Serial 1 : 2 dilution of serum (drawn 70 min following the onset of reperfusion) and supernatants of peritoneal macrophages, harvested at the same time as the serum using a previously described method (Altavilla et al., 1989), were made in the above-described medium containing 1.0 μg of actinomycin D per ml and 100 μl volumes of each dilution were added to the wells. One TNF-α unit was defined as the amount affording 50% cell cytotoxicity. The TNF-α content in the sample was calculated by comparison with a calibration curve obtained with recombinant murine TNF-α (Nuclear Laser Medicine, Milan, Italy). To test whether the cytotoxicity was due to the presence of TNF-α or to other factor(s), we preincubated our samples for 2 h at 37°C with an excess of rabbit anti-recombinant murine TNF-α polyclonal antibodies (Nuclear Laser Medicine, Milan, Italy), or with control rabbit serum. Our results showed that cytotoxicity against L929 cells was completely neutralized by rabbit anti-recombinant TNF-α polyclonal antibodies, but not by control rabbit serum.
Myeloperoxidase activity and leukocyte count
Leukocyte accumulation was investigated using MPO activity. MPO activity was determined in intestinal mucosa, as previously reported (Mullane et al., 1985). The samples of intestinal mucosa were obtained at 0 min before occluding the splanchnic arteries and at 70 min following the onset of reperfusion. The samples were first homogenized in a solution containing 20 mM of potassium phosphate buffer (pH 7.4), 0.01 M EDTA, 50 U ml−1 of a protease inhibitor (aprotinin) in proportions of 1 : 10 (w v−1) and then centrifuged for 30 min at 20,000×g at 4°C. The supernatant of each sample was then discarded and the pellet was immediately frozen on dry ice. The samples were kept at a temperature of 0°C for 14 h before sonication. After thawing, the resulting pellet was added to a buffer solution consisting of 0.5% hexacyltrimethylammonium bromide (Sigma Chemical Co., St. Louis, MO, U.S.A.) dissolved in 50 mM potassium phosphate buffer (pH 6) containing 30 U ml−1 of protease inhibitor. Each sample was then sonicated (intensity 2) for 1 min at a temperature of 4°C. After sonication the samples were allowed to chill on ice for approximately 30 min and then centrifuged for 30 min at 40,000×g at 4°C. An aliquot of the supernatant was allowed to react with 0.167 mg ml−1 o-dianisidine dihydrochloride (Sigma Chemical Co., St. Louis, MO, U.S.A.) and 0.001% H2O2 and the rate of change in absorbance was measured at 405 nm on a microtitre plate reader. MPO activity was defined as the quantity of enzyme degrading 1 μmol of peroxide min−1 at 25°C and was expressed in units g weight (U g−1 of tissue). Tail vein blood samples for the leukocyte count were taken at 0 min before initiating reperfusion and at 70 min after the onset of reperfusion.
Immunohistochemistry
ICAM-1 expression was studied in thoracic aortae and in superior mesenteric arteries collected 70 min following declamping. Immunohistochemical evaluation was accomplished by staining 5 μm-thick cryostat sections, following the avidin-biotin-peroxidase complex procedure (Hsu et al., 1981). An average of seven sections per immunohistochemical stain was cut from each sample, air-dried for 30 min and then fixed in cold acetone for 10 min. Endogenous peroxidases were blocked with horse serum for 15 min at room temperature prior to incubation with primary antibodies. Monoclonal antibodies consisted of mouse monoclonal antibodies raised against rat ICAM-1 (clone: IA 29, subclass IgG1) and were obtained from British Bio-technology Products Ltd (Abingdon, U.K.). A monoclonal mouse IgG1 antibody was used for controls. Biotinylated, species-specific second-layer reagents were then applied, followed by avidin-biotin-horseradish peroxidase complex as a chromogenic substrate, as previously reported (Ioculano et al., 1994). The experiments were carried out by two observers (PC, GF) unaware of the experimental protocol. The microscope image, sent to a computer-assisted image analyser software (Bio-Profil, Celbio, Milano, Italy), was subjected to a densitometric analysis to evaluate the changes in staining.
Drugs
Acetylcholine chloride, phenylephrine hydrochloride, methylprednisolone hemisuccinate and indomethacin were obtained from Sigma Chemical Co., St. Louis, MO (U.S.A). ACTH-(1–24) was obtained from Novartis Pharma AG, Basel (Switzerland). All drugs were dissolved in NaCl 0.9% immediately before use.
Statistical analysis
Data are expressed as means±s.d.mean and were analysed by analysis of variance for multiple comparison of results; Duncan's multiple range test was used to compare group means. Where appropriate, Student's t-test was also used. For survival rate, statistical analysis was done with Fisher's exact probability test. In all cases, a probability error of less than 0.05 was selected as criterion for statistical significance.
Results
Survival
Table 1 summarizes survival rate, percentage survival and survival time for the groups of rats subjected to SAO shock or sham shock. All sham rats survived the entire 4-h observation period. In contrast, in rats treated with the vehicle (NaCl 0.9%), occlusion and reperfusion of the splanchnic region produced a profound shock state characterized by a high mortality rate: no rat was still surviving at 2 h of reperfusion (survival time=78±10 min; n=10). Administration of ACTH-(1–24) increased survival rate and time in SAO rats: 100% animals were still surviving at 2 h of reperfusion, and 80% at 4 h. On the other hand, methylprednisolone had a non-significant effect: 20% of animals were surviving at 2 h of reperfusion, and only 10% (=1 rat) at 4 h.
Table 1.
Effects of ACTH-(1–24) or methylprednisolone (MP) on survival rate, percentage survival and survival time in splanchnic artery occlusion (SAO) shock

Bilateral adrenalectomy only in part–but not significantly–reduced the effect of ACTH-(1–24) (Table 2): 80% of adrenalectomized and ACTH-treated animals were still surviving at 2 h of reperfusion, and 60% at 4 h (P>0.05 versus non-adrenalectomized group; Fisher's test). Animals surviving at the fourth hour of reperfusion were still alive 24 h following the induction of SAO shock.
Table 2.
Effects of ACTH-(1–24) on survival rate and time in adrenalectomized splanchnic artery occlusion (SAO) shocked rats

Serum and macrophage TNF-α
Serum and macrophage levels of TNF-α were undetectable in sham-operated rats treated either with vehicle or ACTH-(1–24). TNF-α was significantly increased in both serum and macrophages collected from SAO rats at the end of the reperfusion period (Table 3). The administration of ACTH-(1–24) significantly blunted the macrophage and serum levels of this cytokine.
Table 3.
Effects of ACTH-(1–24) on serum and macrophage tumour necrosis factor-α (TNF-α) in splanchnic artery occlusion (SAO) shock

Leukocyte infiltration
Leukocyte infiltration was determined by measuring of MPO activity in rats at different times: 0 min before occlusion (basal; at the beginning of the experiment) and 70 min following the onset of reperfusion. MPO levels were significantly increased in the ileum (9.4±1 U g−1 tissue) at 70 min after reperfusion (Table 4) in SAO rats treated with the vehicle.
Table 4.
Effects of ACTH-(1–24) on myeloperoxidase (MPO) activity of ileum and on white blood cell count (WBC) of rats subjected to splanchnic artery occlusion (SAO) shock
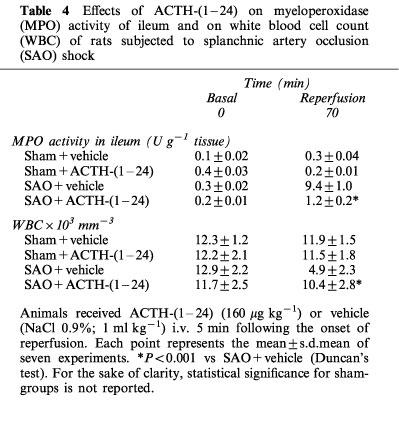
Administration of ACTH-(1–24) significantly lowered the increase in ileal (1.2±0.2 U g−1 tissue) MPO activity (Table 4).
Leukocyte count
The administration of vehicle or ACTH-(1–24) did not modify the white blood-cell count in sham-operated rats (Table 4). In contrast, SAO shock produced a marked leukopenia. Leukocyte count was markedly decreased at the end of reperfusion (70 min). The administration of ACTH-(1–24) significantly ameliorated this leukopenia (Table 4).
ICAM-1 staining on vascular endothelium
The presence of ICAM-1 was verified in thoracic aortae and in superior mesenteric arteries collected 70 min following the release of occlusion. Immunohistochemical evaluation indicated that a very low constitutive staining of ICAM-1 was present in sham-operated animals (Figure 1). By contrast, samples obtained from SAO-shocked rats showed an increase in ICAM-1 staining. Aortic and mesenteric endothelium obtained from SAO shocked rats treated with ACTH-(1–24) showed a marked reduction in ICAM-1 immunostaining (Figure 1).
Figure 1.
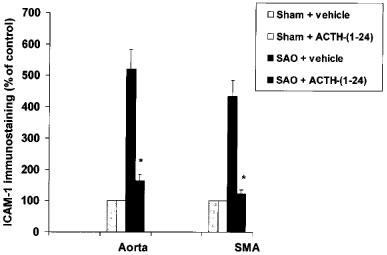
Effects of ACTH-(1–24) (160 μg kg−1) or vehicle (NaCl 0.9%; 1 ml kg−1), both i.v. injected 5 min following the onset of reperfusion, on immunohistochemical staining for ICAM-1 in aortic (Aorta) and superior mesenteric artery (SMA) endothelium from rats subjected to splanchnic artery occlusion (SAO) shock. Each point represents the mean±s.d.mean of seven experiments. *P<0.01 vs SAO+vehicle (Duncan's test). For the sake of clarity, statistical significance for sham-groups is not reported.
Vascular reactivity of aortic rings
Addition of PE (100 nM) to the organ bath caused intact aortic rings to contract (80–90% of the maximum response). These rings were relaxed in a concentration-dependent manner by ACh (10 nM–10 μM). The relaxant effect of ACh was significantly weaker in aortic rings obtained from SAO rats [potency (ED50)=520 nM; Emax=30% relaxation] than in those from sham-operated rats [potency (ED50)=510 nM; Emax=82% relaxation] (Figure 2). Administration of ACTH-(1–24) significantly improved the responsiveness of aortic rings obtained from SAO-shocked rats to ACh [potency (ED50)=500 nM; Emax=62% relaxation] (mean values; s.d.<15% in all cases) (Figure 2).
Figure 2.
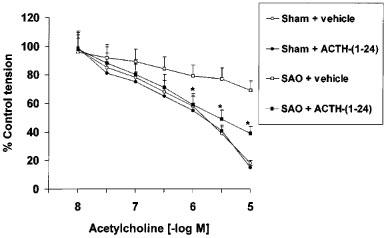
Relaxant effects of acetylcholine (ACh) in aortic rings (contracted with phenylephrine, 100 nM) of sham-operated rats and rats subjected to splanchnic artery occlusion (SAO) shock treated with ACTH-(1–24) (160 μg kg−1) or vehicle (NaCl 0.9%; 1 ml kg−1), both i.v. injected 5 min following the onset of reperfusion. Each point represents the mean±s.d.mean of six experiments. *P<0.01 vs SAO+vehicle (Duncan's test). For the sake of clarity, statistical significance for sham-groups is not reported.
In intact aortic rings prepared from SAO-shocked rats, the contractile response to PE (1 nM–10 μM) was significantly reduced. The maximum force of contraction induced by 10 μM PE in aortic rings from sham rats was 16.68±1.96 mN mg−1 tissue, whereas it was 6.87±1 mN mg−1 tissue in rings from SAO-shocked rats. Removal of the endothelium did not increase the constrictor response elicited by PE in rat aortic rings obtained from both SAO-shocked rats and sham-operated animals (Figure 3). However, the contractile response to PE in endothelium denuded aortic rings was also significantly smaller in SAO-shocked rats [potency (ED50)=120 nM; Emax=7.16 mN] than in sham operated animals [potency (ED50)=100 nM; Emax=16.31 mN]. Administration of ACTH-(1–24) improved the impaired contractile response to PE in SAO rats [potency (ED50)=100 nM; Emax=13.73 mN] (mean values; s.d.<15% in all cases) (Figure 3).
Figure 3.
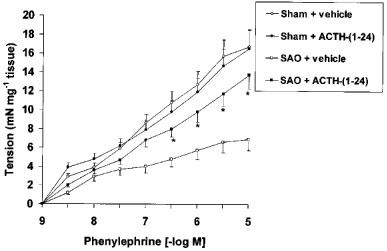
Contractile response to cumulative doses of phenylephrine (PE) in endothelium-denuded aortic rings from sham-operated rats and rats subjected to splanchnic artery occlusion (SAO) shock treated with ACTH-(1–24) (160 μg kg−1) or vehicle (NaCl 0.9%; 1 ml kg−1), both i.v. injected 5 min following the onset of reperfusion. Each point represents the mean±s.d.mean of seven experiments. P<0.02 vs SAO+vehicle (Duncan's test). For the sake of clarity, statistical significance for sham-groups is not reported.
Mean arterial blood pressure
Occlusion of the splanchnic arteries produced a marked increase in MAP. Subsequently, MAP decreased upon declamping (Figure 4). The administration of ACTH-(1–24) significantly blunted the reduction in MAP (Figure 4).
Figure 4.
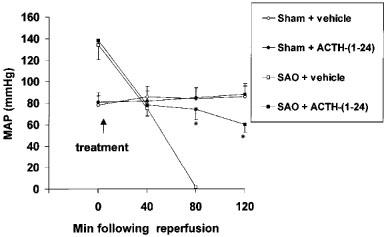
Effects of ACTH-(1–24) (160 μg kg−1) or vehicle (NaCl 0.9%; 1 ml kg−1), both i.v. injected 5 min following the onset of reperfusion, on mean arterial blood pressure (MAP) of sham-operated rats and rats subjected to splanchnic artery occlusion (SAO) shock. Each point represents the mean±s.d.mean of six experiments. *P<0.01 vs SAO+vehicle (Duncan's test). For the sake of clarity, statistical significance for sham-groups is not reported.
Discussion
It has been suggested that the pleiotropic cytokine TNF-α plays an important role in the pathogenesis of SAO shock (Squadrito et al., 1992). In fact, TNF-α may induce vascular dysfunction and cause leukocytes to adhere to the vascular endothelium where they discharge deleterious mediators (i.e. oxygen free radicals, leukotrienes, cytokines, etc.) capable of compounding the vascular damage. This latter phenomenon involves interaction between several adhesive receptors (adhesion molecules) present on both the endothelial and the leukocyte surfaces.
As far as vascular dysfunction is concerned, it has been suggested that TNF-α may impair the release of NO from endothelial cells (Aoki et al., 1989), thus leading to a reduced production of endothelial-derived relaxing factor. However, administration of recombinant human TNF-α in conscious rats has been reported to induce a decrease in MAP and to produce vascular hyporesponsiveness to contractile agents that is reversed by inhibitors of NO synthesis (Takahashi et al., 1992). This phenomenon is probably due to TNF-α-induced stimulation of the Ca2+-independent NO synthase in vascular smooth muscle (Busse & Mulsch, 1990).
Recent evidence has suggested that adhesion mechanisms supporting leukocyte adhesion and accumulation to the endothelium are present in ischaemic states (Ioculano et al., 1994). SAO shock is an experimental model characterized by the presence of adhesion mechanisms for leukocyte accumulation and by a marked vascular dysfunction (Squadrito et al., 1996). We have shown that these two important pathological aspects of this type of experimental model of circulatory shock are due to increased production of TNF-α.
ACTH-(1–24) has been shown to inhibit the increase in serum levels of the pleiotropic cytokine in experimental haemorrhagic shock (Altavilla et al., 1998). This finding prompted us to investigate the effects of this melanocortin on the pathological sequelae of SAO shock. Our results indicate that ACTH-(1–24) reduced the SAO shock-induced increase in macrophage and serum levels of TNF-α.
The administration of ACTH-(1–24) also reduced ICAM-1 staining, ameliorated leukopenia and decreased MPO activity, an index of leukocyte accumulation. Since leukocyte-endothelial interaction (more specifically, the ICAM-1-dependent leukocyte adhesion) is primed by TNF-α, it can be proposed that this melanocortin, by inhibiting this inflammatory cytokine, limits leukocyte accumulation at the ischaemic sites and finally protects against SAO shock.
Aortic rings from rats subjected to SAO shock displayed markedly reduced responsiveness to the vasorelaxant effects of ACh: this finding would indicate the presence of a reduced NO production in this type of experimental shock. However, our results also showed the presence of reduced vascular sensitivity to vasoconstrictor stimuli. This impaired vascular reactivity, as suggested for other models of experimental shock (Moncada et al., 1991; Thiemermann et al., 1993; Szabò & Thiemermann, 1994), may be a consequence of overproduction of NO by the iNOS.
These data might thus combine to suggest that, in SAO shock, (i) NO generated by the endothelial NO synthase (eNOS) is blunted, while (ii) NO produced by the iNOS is increased. These opposite effects in splanchnic ischaemia-reperfusion injury could be induced by TNF-α, which both inhibits eNOS and stimulates iNOS. This hypothesis is supported by the previously obtained evidence that an inhibitor of TNF-α synthesis is able to reverse this complex SAO shock-induced vascular dysfunction (Squadrito et al., 1994a). In the present experiments, aortic rings collected from rats subjected to SAO shock and treated with ACTH-(1–24) exhibited a greater contractile response to PE and improved responsiveness to ACh when compared with vehicle-treated rats. Thus, it can be hypothesized that ACTH-(1–24) improves vascular dysfunction by inhibiting the detrimental vascular effects of TNF-α.
The fact that bilateral adrenalectomy only in part (20%)–but not significantly–reduced the effect of ACTH-(1–24) on the survival rate of SAO shocked rats, together with the fact that methylprednisolone had a non-significant effect (10% survival at 4 h of reperfusion, compared with 80% survival after the same time-lapse in ACTH-treated rats) suggests that the action of ACTH is not adrenal-mediated, as first reported in the case of haemorrhagic shock (Bertolini et al., 1986a,1986c; Bazzani et al., 1993). On the other hand, we previously showed (Altavilla et al., 1998) that ACTH-(1–24) dose-dependently (25–100 nM) reduces the in vitro production of TNF-α by LPS-stimulated rat macrophages. Moreover, it is known that another melanocortin, α-melanocyte stimulating hormone [α-MSH=ACTH-(1–13)], inhibits production of TNF-α by macrophages (Eberle, 1988), and it has been recently shown that α-MSH also inhibits–via MC-1 receptors–the TNF-α- or interferon γ-induced expression of the inducible macrophage isoform of NO synthase (Star et al., 1995). Macrophages contain mRNA for the MC-1 melanocortin receptor (Star et al., 1995), and direct effects of melanocortin peptides on other functions of macrophages have been observed in in vitro experiments (Genedani et al., 1990, 1992).
In conclusion, we have shown that ACTH-(1–24) inhibits TNF-α overproduction in SAO shock, in rats: the melanocortin-induced inhibition of this inflammatory cytokine increases survival, reduces ICAM-1 expression and leukocyte infiltration in the ileum and improves vascular dysfunction. These findings would suggest that TNF-α inhibition may contribute, at least in part, to the acute vasoprotective effects of ACTH. Finally, our data indicate that melanocortins may reverse vascular derangement and reduce leukocyte accumulation during shock states, a hitherto unreported finding.
Acknowledgments
This work was supported in part by grants from Ministero dell'Università e della Ricerca Scientifica e Tecnologica and Consiglio Nazionale delle Ricerche, Roma.
Abbreviations
- ACh
acethylcoline
- ACTH-(1–24)
adrenocorticotropin fragment 1–24
- eNOS
endothelial nitric oxide synthase
- ICAM-1
intercellular adhesion molecule-1
- iNOS
inducible nitric oxide synthase
- i.p.
intraperitoneal
- i.v.
intravenous
- MAP
mean arterial pressure
- MPO
myeloperoxidase
- α-MSH
α-melanocyte stimulating hormone
- NO
nitric oxide
- PE
phenylephrine
- SAO
splanchnic artery occlusion
- TNF-α
tumour necrosis factor-α
References
- ABOU-MOHAMED G., PAPAPETROPOULOS A., ULRICH D., CATRAVAS J.D., TUTTLE R.R., CALDWELL R.W. HP-228, a novel synthetic peptide, inhibits the induction of nitric oxide synthase in vivo but not in vitro. J. Pharmacol. Exp. Ther. 1995;275:584–591. [PubMed] [Google Scholar]
- ALTAVILLA D., BERLINGHIERI M.C., SEMINARA S., IANNELLO D., FOCA' A., MASTROENI P. Different effects of bacterial lipopolysaccharide on superoxide anion production by macrophages from normal and tumor bearing rats. Immunopharmacology. 1989;17:99–105. doi: 10.1016/0162-3109(89)90055-6. [DOI] [PubMed] [Google Scholar]
- ALTAVILLA D., CAINAZZO M.M., SQUADRITO F., GUARINI S., BERTOLINI A., BAZZANI C. Tumour necrosis factor-α as a target of melanocortins in haemorrhagic shock, in the anaesthetized rats. Br. J. Pharmacol. 1998;124:1587–1590. doi: 10.1038/sj.bjp.0702038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AOKI N., SIEGFRIED M., LEFER A.M. Anti-EDRF effect of tumor necrosis factor in isolated perfused cat carotid arteries. Am. J. Physiol. 1989;256:H1509–H1512. doi: 10.1152/ajpheart.1989.256.5.H1509. [DOI] [PubMed] [Google Scholar]
- BAZZANI C., BALUGANI A., BERTOLINI A., GUARINI S. Comparison of the effects of ACTH-(1–24), methylprednisolone, aprotinin, and norepinephrine in a model of hemorragic shock in rats. Resuscitation. 1993;25:219–226. doi: 10.1016/0300-9572(93)90118-a. [DOI] [PubMed] [Google Scholar]
- BAZZANI C., BERTOLINI A., GUARINI S. Inhibition of nitric oxide synthases enhances the effect of ACTH in hemorrhagic shock. Life Sci. 1997;61:1889–1897. doi: 10.1016/s0024-3205(97)00828-x. [DOI] [PubMed] [Google Scholar]
- BERTOLINI A. The opioid/anti-opioid balance in shock: a new target for therapy in resuscitation. Resuscitation. 1995;30:29–42. doi: 10.1016/0300-9572(94)00863-b. [DOI] [PubMed] [Google Scholar]
- BERTOLINI A., GUARINI S., FERRARI W. Adrenal-independent, anti-shock effect of ACTH-(1–24) in rats. Eur. J. Pharmacol. 1986a;122:387–388. doi: 10.1016/0014-2999(86)90424-3. [DOI] [PubMed] [Google Scholar]
- BERTOLINI A., GUARINI S., FERRARI W., NOERA G., MASSINI C., DI TIZIO S. ACTH-(1–24) restores blood pressure in acute hypovolaemia and haemorrhagic shock in humans. Eur. J. Clin. Pharmacol. 1987;32:537–538. doi: 10.1007/BF00637684. [DOI] [PubMed] [Google Scholar]
- BERTOLINI A., GUARINI S., FERRARI W., ROMPIANESI E. Adrenocorticotropin reversal of experimental hemorrhagic shock is antagonized by morphine. Life Sci. 1986b;39:1271–1280. doi: 10.1016/0024-3205(86)90188-8. [DOI] [PubMed] [Google Scholar]
- BERTOLINI A., GUARINI S., ROMPIANESI E., FERRARI W. α-MSH and other ACTH fragments improve cardiovascular function and survival in experimental hemorrhagic shock. Eur. J. Pharmacol. 1986c;130:19–26. doi: 10.1016/0014-2999(86)90179-2. [DOI] [PubMed] [Google Scholar]
- BUSSE R., MULSCH A. Induction of nitric oxide synthase by cytokines in vascular smooth muscle cells. FEBS Lett. 1990;275:87–90. doi: 10.1016/0014-5793(90)81445-t. [DOI] [PubMed] [Google Scholar]
- EBERLE A.N. The melanotropins, Chemistry, Physiology and Mechanisms of Action. Basel: Karger; 1988. [Google Scholar]
- GENEDANI S., BERNARDI M., BALDINI M.G., BERTOLINI A. ACTH (1–24) stimulates the migration of human monocytes in vitro. Peptides. 1990;11:1305–1307. doi: 10.1016/0196-9781(90)90164-z. [DOI] [PubMed] [Google Scholar]
- GENEDANI S., BERNARDI M., BERTOLINI A. Neuropeptides of the stress response and monocyte motility. Ann. N.Y. Acad. Sci. 1992;663:494–496. doi: 10.1111/j.1749-6632.1992.tb38713.x. [DOI] [PubMed] [Google Scholar]
- GUARINI S., BINI A., BAZZANI C., MATTERA RICIGLIANO G., CAINAZZO M.M., TOMASI A., BERTOLINI A. Adrenocorticotropin normalizes the blood levels of nitric oxide in hemorrhage-shocked rats. Eur. J. Pharmacol. 1997;336:15–21. doi: 10.1016/s0014-2999(97)01210-7. [DOI] [PubMed] [Google Scholar]
- GUARINI S., TAGLIAVINI S., BAZZANI C., FERRARI W., BERTOLINI A. Early treatment with ACTH-(1–24) in a rat model of hemorrhagic shock prolongs survival and extends the time-limit for blood reinfusion to be effective. Crit. Care Med. 1990;18:862–865. doi: 10.1097/00003246-199008000-00014. [DOI] [PubMed] [Google Scholar]
- HSU S.M., RAINE L., FANGER H. A comparative study of the peroxidase-antiperoxidase method and an avidin-biotin complex method for studying polypeptide hormones with radioimmunoassay. Am J. Clin Path. 1981;75:734–737. doi: 10.1093/ajcp/75.5.734. [DOI] [PubMed] [Google Scholar]
- IOCULANO M., SQUADRITO F., ALTAVILLA D., CANALE P., CAMPO G.M., SAITTA A., CAPUTI A.P. Antibodies against intercellular adhesion molecule-1 protect against myocardial ischaemia-reperfusion injury in the rat. Eur. J. Pharmacol. 1994;264:143–149. doi: 10.1016/0014-2999(94)00452-8. [DOI] [PubMed] [Google Scholar]
- LUDBROOK J., VENTURA S. ACTH- (1–24) blocks the decompensatory phase of the haemodynamic response to acute hypovolaemia in conscious rabbits. Eur. J. Pharmacol. 1995;275:267–275. doi: 10.1016/0014-2999(95)00003-4. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., MELI A. Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part 1: general considerations. Experientia. 1986a;42:109–114. doi: 10.1007/BF01952426. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., MELI A. Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part 2: cardiovascular system. Experientia. 1986b;42:292–297. doi: 10.1007/BF01942510. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MULLANE K.M., KRAEMER M.R., SMITH B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J. Pharmacol. Methods. 1985;14:156–157. doi: 10.1016/0160-5402(85)90029-4. [DOI] [PubMed] [Google Scholar]
- NOERA G., ANGIELLO L., BIAGI B., PENSA P. Haemorrhagic shock in cardiac surgery. Pharmacological treatment with ACTH-(1–24) Resuscitation. 1991;22:123–127. doi: 10.1016/0300-9572(91)90002-g. [DOI] [PubMed] [Google Scholar]
- NOERA G., PENSA P., GUELFI P., BIAGI B., CURCIO C., BENTINI B. ACTH-(1–24) and hemorrhagic shock: Preliminary clinical results. Resuscitation. 1989;18:145–147. doi: 10.1016/0300-9572(89)90010-5. [DOI] [PubMed] [Google Scholar]
- PINELLI G., CHESI G., DI DONATO C., REVERZANI A., MARANI L. Preliminary data on the use of ACTH-(1–24) in human shock conditions. Resuscitation. 1989;18:149–150. doi: 10.1016/0300-9572(89)90011-7. [DOI] [PubMed] [Google Scholar]
- RUFF M.R., GIFFORD G.E. Purification and physicochemical characterization of rabbit tumor necrosis factor. J. Immunol. 1980;125:1671–1675. [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., CANALE P., IOCULANO M., CAMPO G.M., AMMENDOLIA L., FERLITO M., ZINGARELLI B., SQUADRITO G., SAITTA A., CAPUTI A.P. Participation of tumour necrosis factor and nitric oxide in the mediation of vascular dysfunction in splanchnic artery occlusion shock. Br. J. Pharmacol. 1994a;113:1153–1158. doi: 10.1111/j.1476-5381.1994.tb17118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., CANALE P., IOCULANO M., CAMPO G.M., AMMENDOLIA L., SQUADRITO G., SAITTA A., CALAPAI G., CAPUTI A.P. Contribution of intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of splanchnic artery occlusion shock in the rat. Br. J. Pharmacol. 1994b;113:912–916. doi: 10.1111/j.1476-5381.1994.tb17079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., IOCULANO M., CALAPAI G., ZINGARELLI B., SAITTA A., CAMPO G.M., RIZZO A., CAPUTI A.P. Passive immunization with antibodies against tumor necrosis factor TNF-α protects from the lethality of splanchnic artery occlusion shock. Circ. Shock. 1992;37:236–244. [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., SQUADRITO G., CAMPO G.M., IOCULANO M., CANALE P., ROSSI F., SAITTA A., CAPUTI A.P. Effects of S-ethylisothiourea, a potent inhibitor of nitric oxide synthase, alone or in combination with a nitric oxide donor in splanchnic artery occlusion shock. Br. J. Pharmacol. 1996;119:23–28. doi: 10.1111/j.1476-5381.1996.tb15672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAR R.A., RAJORA N., HUANG J., STOCK R.C., CATANIA A., LIPTON J.M. Evidence of autocrine modulation of macrophage nitric oxide synthase by α-melanocyte-stimulating hormone. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8016–8020. doi: 10.1073/pnas.92.17.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ' C., THIEMERMANN C. Invited opinion: role of nitric oxide in hemorrhagic, traumatic and anaphylactic shock and thermal injury. Shock. 1994;2:145–155. [PubMed] [Google Scholar]
- TAKAHASHI K., ANDO K., ANO A., SHIMOSAWA T., OGATA E., FUJITA T. Tumor necrosis factor-α induces vascular hyporesponsiveness in Sprague-Dawley rats. Life. Sci. 1992;50:1437–1444. doi: 10.1016/0024-3205(92)90262-n. [DOI] [PubMed] [Google Scholar]
- THIEMERMANN C., SZABO' C., MITCHELL J.A., VANE J.R. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 1993;90:267–271. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]