

Abstract
Diabetes mellitus produces marked abnormalities in motor nerve conduction, but the mechanism is not clear. In the present study we hypothesized that in the streptozotocin (STZ)-induced diabetic rat impaired vasodilator function is associated with reduced endoneural blood flow (EBF) which may contribute to nerve dysfunction.
We examined whether diabetes-induced reductions in sciatic nerve conduction velocity and EBF were associated with impaired endothelium-dependent dilation in adjacent arterioles. We measured motor nerve conduction velocity (MNCV) in the sciatic nerve using a non-invasive procedure, and sciatic nerve nutritive blood flow using microelectrode polarography and hydrogen clearance. In vitro videomicroscopy was used to quantify arteriolar diameter responses to dilator agonists in arterioles overlying the sciatic nerve.
MNCV and EBF in 4-week-STZ-induced diabetic rats were decreased by 22% and 49% respectively. Arterioles were constricted with U46619 and dilation to acetylcholine (ACh), aprikalim, or sodium nitroprusside (SNP) examined. All agonists elicited dose-dependent dilation in control and diabetic rats, although ACh-induced dilation was significantly reduced in diabetic rats. Treating vessels from normal or diabetic rats with indomethacin (INDO) alone did not significantly affect ACh-induced relaxation. However, ACh-induced vasodilation was significantly reduced by treatment with KCl or Nω-nitro-L-arginine (LNNA) alone. Combining LNNA and KCl further reduced ACh-induced dilation in these vessels.
Diabetes causes vasodilator dysfunction in a microvascular bed that provides circulation to the sciatic nerve. These studies imply that ACh-induced dilation in these vessels is mediated by multiple mechanisms that may include the endothelial-dependent production of nitric oxide and endothelial-derived hyperpolarizing factor. This impaired vascular response is associated with neural dysfunction.
Keywords: Diabetic neuropathy, endothelium, vascular reactivity, acetylcholine, motor nerve conduction velocity, neural blood flow, nitric oxide, endothelium-derived hyperpolarizing factor
Introduction
The purpose of this study was to examine the relationship between diabetes-induced reduction in sciatic nerve motor nerve conduction velocity, endoneural blood flow, and vascular reactivity of arterioles that provide circulation to the region of the sciatic nerve.
Reduced motor nerve conduction velocity (MNCV) has been described in diabetic neuropathy (Yorek et al., 1993). Several laboratories have reported that reduced MNCV is preceded or associated with a decrease in endoneural blood flow (EBF), suggesting that vascular disease may be a major contributing factor to diabetic neuropathy (Cameron et al., 1991; Wright & Nakada, 1994; Stevens et al., 1994). However, other investigations suggest that metabolic neural abnormalities associated with hyperglycaemia, such as reduced Na+/K+ ATPase activity are responsible for diabetic neuropathy (Gillon et al., 1983; Greene et al., 1984; 1988; Greene & Lattimer, 1984). Adding to this controversy is a study by Williamson and coworkers claiming that EBF is not decreased in diabetic rats (Chang et al., 1997; Ido et al., 1997). To more directly explore the effect of diabetes on vascular function, as it relates to diabetic neuropathy, we examined vascular reactivity in arterioles that provide circulation to the region of the sciatic nerve. The effect of diabetes on endothelium-dependent vasodilation has been examined in the aorta and isolated conduit vessels from the heart and kidney where impaired dilation is observed (Cohen, 1993; Cooper et al., 1997; Stehouwer et al., 1997). To date no studies have examined whether impaired vasodilatory responses in diabetes extend to the arterioles that provide blood flow to peripheral nerves. In this study we found that acetylcholine-induced vasodilation is decreased in arterioles adjacent to the sciatic nerve. However, vasodilation to sodium nitroprusside and to aprikalim was not altered by diabetes. In contrast to larger vessels in the rat where acetylcholine-induced dilation is mediated solely by nitric oxide (NO) (Sobey et al., 1996), dilation of arterioles adjacent to the sciatic nerve is not dependent solely on NO, but involves a combination of endothelial derived factors. Thus, sciatic nerve dysfunction in streptozotocin-induced diabetic rats is associated with intrinsic vasodilator dysfunction in the corresponding microvascular bed. The complex regulation of vascular tone in this vessel bed may help explain the diverse aetiology of diabetic neuropathy and provide opportunities for the treatment of this disorder.
Methods
Animals
Male Sprague-Dawley (Harlan Sprague Dawley, Indianapolis, IN, U.S.A.) rats 8–9 weeks of age were used for these studies. The animals were housed in a certified animal care facility and food and water were provided ad libitum. All institutional and NIH guidelines for use of animals were followed. Diabetes was induced by intravenously injecting streptozotocin (60 mg kg−1 in 0.9% NaCl, adjusted to a pH 4.0 with 0.2 M sodium citrate). Control rats were injected with vehicle alone. The rats were anaesthetized with methoxyflurane before injection. Diabetes was verified 24 h later by evaluating blood glucose levels with the use of glucose oxidase reagent strips (Boehringer-Mannheim, Indianapolis, IN, U.S.A.). All studies were conducted after 4 weeks of diabetes. On the day of the experiment rats were anaesthetized with Nembutal i.p. (50 mg kg−1, Abbott Laboratories, North Chicago, IL, U.S.A.). Following the determination of MNCV and EBF, the abdominal aorta was isolated and occluded 1–2 cm above the branch of the common iliac artery. Distal to the occlusion a solution containing India ink with 2% gelatin (Kuo et al., 1990) was injected to facilitate the identification of the superior gluteal and internal pudendal arteries, which arise from the common iliac artery. The rat was then sacrificed and its body temperature rapidly cooled with ice. Samples of the left sciatic nerve for determination of Na+/K+ ATPase activity and sorbitol and myo-inositol content were then taken. Samples for blood glucose measurements were also taken on the day of the experiment.
Motor nerve conduction velocity
MNCV was determined as previously described using a noninvasive procedure in the sciatic-posterior tibial conducting system in a temperature controlled environment (Yorek et al., 1993). The left sciatic nerve was stimulated first at the sciatic notch and then at the Achilles tendon. Stimulation consisted of single 0.2 ms supra maximal (8 V) pulses through a bipolar electrode (Grass S44 Stimulator, Grass Medical Instruments, Quincy, MA, U.S.A.). The evoked potentials were recorded from the interosseous muscle with a unipolar platinum electrode and displayed on a digital storage oscilloscope (model 54600A Hewlett Packard, Rolling Meadows, IL, U.S.A.). MNCV was calculated by subtracting the distal from the proximal latency measured in milliseconds from the stimulus artifact of the take off of the evoked potential and the difference was divided into the distance between the two stimulating electrodes measured in millimeters using a Vernier caliper. The MNCV was reported in metres per second.
Endoneural blood flow
Immediately after determination of MNCV, sciatic endoneural nutritive blood flow was determined as described by Cameron et al. (1991; 1997). The trachea was intubated for artificial ventilation and a carotid cannula inserted to monitor mean arterial blood pressure. Core temperature was monitored using a rectal probe and temperature regulated between 36 and 37°C using a heating pad and radiant heat. The right sciatic nerve was carefully exposed by a small surgical incision and the surrounding skin sutured to a plastic ring. The isolated area was filled with mineral oil, at 37°C to a depth of 1 cm to minimize diffusion of hydrogen gas from the nerve. The rats were then artificially ventilated. A glass insulated platinum microelectrode (tip=2 μm) was inserted into the sciatic nerve, above the trifurcation, and polarized at 0.25 V with respect to a reference electrode inserted subcutaneously into the flank of the rat. Once the recording had stabilized the inspired air was modified to contain 10% hydrogen gas and this gas flow continued until the hydrogen current recorded by the electrode had maximized, indicating equilibrium with arterial blood. The hydrogen gas supply was then discontinued and the hydrogen clearance curve recorded until a baseline was achieved. The hydrogen clearance data was fitted by computer to a mono- or bi-exponential curve using commercial software (Prism, GraphPad, San Diego, CA, U.S.A.) and nutritive blood flow, (ml−1 min−1 100 g), calculated using the equation described by Young (1980) and vascular conductance, (ml−1 min−1 100 g mmHg) was determined by dividing nutritive blood flow by the average mean arterial blood pressure. Two recordings were made for each rat at different locations along the nerve and the final blood flow value averaged.
Vascular reactivity
Videomicroscopy was used to investigate in vitro vasodilatory responsiveness of arterioles supplying the region of the sciatic nerve (branches of the superior gluteal and internal pudendal arteries). The common iliac was exposed and the branch points of the internal pudendal and superior gluteal arteries identified. The vessels were then clamped, and tissue containing these vessels and its branches dissected en bloc. The block of tissue was immediately submerged in a cooled (4°C), oxygenated (20% O2, 5% CO2 and 75% N2) Krebs Henseleit physiological saline solution (PSS) of the following composition (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 20, Na2EDTA 0.026, and 5.5 glucose for dissection. Branches of the superior gluteal and internal pudendal arteries (50–150 μm internal diameter and 2 mm in length) were carefully dissected and trimmed of fat and connective tissue. Both ends of the isolated vessel segment were cannulated with glass micropipettes filled with PSS (4°C), and secured with 10-0 nylon Ethilon monofilament sutures (Ethicon Inc., Cornelia, GA, U.S.A.). The pipettes were attached to a single pressure reservoir (initially set at 20 mmHg) under condition of no flow. The preparation was transferred to the stage of an inverted microscope (CK2, Olympus, Lake Success, NY, U.S.A.). Attached to the microscope were a CCTV camera (WV-BL200, Panasonic, Secaucus, NJ, U.S.A.), video monitor (Panasonic), and a video caliper (VIA-100K, Boeckeler Instruments Inc., Tucson, AZ, U.S.A.). The organ chamber was connected to a rotary pump (Masterflex, Cole Parmer Instrument Co., Vernon Hills, IL, U.S.A.), which continuously circulated oxygenated PSS at 30 ml min−1 and warmed to 37°C. All pharmacological agents were added to the external bathing solution. The pressure within the vessel was then slowly increased to 40 mmHg. At this pressure we found that KCl gave the maximal constrictor response. Therefore, all the studies were conducted at 40 mmHg. Internal diameter (resolution of 2 μm) was measured by manually adjusting the video micrometer. After 30 min equilibration, KCl was added to the bath to test vessel viability. Vessels which failed to constrict more than 30% were discarded. After washing with PSS, vessels were incubated for 30 min in PSS and then constricted with U46619 (10−8–10−7 M) to 30–50% of passive diameter. There was no significant difference in the amount of U46619 required to induce constriction in control and diabetic vessels. Cumulative concentration-response relationships were evaluated for acetylcholine (10 nM–0.1 mM), sodium nitroprusside (10 nM–0.1 mM) or aprikalim (1 nM–10 μM) with vessels from control and diabetic rats.
Mechanism of acetylcholine-induced dilation in vessels from control and diabetic rats
Vessels were incubated for 30 min in fresh PSS (as a control), PSS with LNNA (0.1 mM), and/or INDO (0.1 mM), and then constricted with U46619 (10–100 nM) or KCl (25–35 mM) to 30–50% of passive diameter. Afterwards, cumulative concentration-response relationships were obtained for acetylcholine (1 μM–0.1 mM). At the end of each concentration-response relationship, sodium nitroprusside (0.1 mM) was given to assess the maximal vasodilatory capacity. This passive diameter was used for calculating the per cent constriction to U46619. The per cent dilation to each dose of administered agonist was calculated using the difference between the constricted diameter and passive diameter as 100% relaxation.
Sciatic nerve myo-inositol and sorbitol content
The left sciatic nerve was removed, desheathed, and weighed for determination of Na+/K+ ATPase activity as described below, and for determination of tissue sorbitol and myo-inositol content (Yorek et al., 1993). Tissue samples were boiled for 10 min in water containing α-D-methylmannopyranoside as an internal standard and deproteinized with 0.5 ml each of 0.19 M Ba(OH)2 and 0.19 M ZnSO4. Following centrifugation the supernatant was collected, frozen, and lyophilized. The samples were derivatized and intracellular contents determined by gas-liquid chromatography as previously described (Yorek et al., 1993).
Na+/K+ ATPase activity
Total and ouabain-inhibited Na+/K+ ATPase activities were measured in crude homogenates of sciatic nerve (Yorek et al., 1993). Sciatic nerves were desheathed and homogenized in a polytron, utilizing three 10 s bursts, at 4°C in 1 ml of 0.2 M sucrose, 0.02 M Tris-HCl buffer, pH 7.5. The samples were then centrifuged at 100×g for 10 min at 4°C. An aliquot of the supernatant (50 μl) was added to two cuvettes containing (mM): NaCl 100, KCl 10, MgCl2 2.5, ethylene glycol-bis(β-aminoethyl ether)-N1-N′-tetraacetic acid (EGTA) 2, Tris-ATP 1, 3-(cyclohexylammonium) phosphoenolpyruvate 1, imidazole-HCl buffer 30 (pH 7.3), NADH 0.15, 50 μg lactate dehydrogenase, 30 μg pyruvate kinase with or without 1 mM ouabain to inhibit the ouabain-sensitive Na+/K+ ATPase fraction. After a 20 min stabilization period, the oxidation of NADH was recorded over a 30 min period. The activity was expressed as μmol ADP/g wet weight h−1. Each assay was conducted in triplicate.
Data analysis
The results are presented as mean±s.e.mean. Comparisons between the groups (control vs diabetic) for MNCV, EBF, Na+/K+ ATPase, and sorbitol and myo-inositol content were conducted using independent unpaired Student's t-tests. Dose response curves for control vs diabetic rats for all agonists, and for non-diabetic rats with and without inhibitors were compared using a two-way repeated measures analysis of variance with autoregressive covariance structure using proc mixed program of SAS. Whenever significant interactions were noted for treatment (control vs diabetic) and dose (highest to lowest), specific treatment-dose-effects were analysed using a Bonferoni adjustment. A P value of less than 0.05 was considered significant. All computations were performed using SAS for Windows version 6.12.
Results
Body weight and plasma glucose levels
Data in Table 1 show that streptozotocin-induced diabetic rats gained less weight than age-matched control rats over the 4-week experimental period of this study. At the time of experimentation plasma glucose levels were increased 4 fold in diabetic compared to control rats.
Table 1.
Body weight and plasma glucose levels

Motor nerve function and biochemical parameters
Data in Table 2 show that motor nerve conduction velocity was significantly decreased by 22% in diabetic compared to control rats. Sciatic nerve ouabain-sensitive Na+/K+ ATPase activity was also significantly reduced by 33% in diabetic rats. The sorbitol content of the sciatic nerve in diabetic rats was significantly increased, whereas the myo-inositol content was significantly decreased.
Table 2.
Motor nerve conduction velocity (MNCV), endoneural blood flow (EBF), and sciatic nerve Na+/K+ ATPase. Activity and sorbitol and myo-Inositol content

Endoneural blood flow and perineural arteriolar vascular reactivity
Endoneural blood flow reported as nutritive flow (ml min −1 100 g−1) or conductance (ml min−1 100 g/mmHg−1) was reduced by about 50% in diabetic compared to control rats (Table 2). In these studies the mean arterial blood pressure was not significantly different between control and diabetic rats (119±4 and 121±4 mmHg, respectively).
Stimulated changes in vascular diameter were measured in vitro by application of acetylcholine. Baseline diameter in these perineural arterioles was 89±7 μm. Vessels were constricted to a similar degree (39±4% and 36±3% of resting diameter for control and diabetic rats, respectively) with U46619 (10–100 μM). Acetylcholine produced concentration-dependent vasodilation (endothelium-dependent) in these arterioles that provide circulation to the region of the sciatic nerve (max. dilation=93±5%, −log ED50=6.7±0.2). In vessels from diabetic rats, acetylcholine-induced dilation was significantly impaired (max. dilation=63±6%, −log ED50=6.7±0.3, Figure 1). In contrast, the concentration-dependent vasodilation induced by sodium nitroprusside (endothelium-independent) and aprikalim (KATP channel opener) was not significantly affected by diabetes in these vessels (Figures 2 and 3, respectively). Mechanical removal of the endothelium resulted in nearly complete loss of acetylcholine-induced vasodilation compared to intact vessels, whereas vasodilation in response to sodium nitroprusside was not impaired by removal of the endothelium (data not shown).
Figure 1.
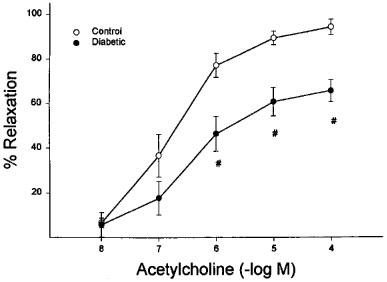
Acetylcholine-induced endothelium dependent relaxation of arterioles adjacent to the sciatic nerve in control and diabetic rats. Pressurized arterioles were constricted with U46619 (30–50%) and incremental doses of acetylcholine were added to the bathing solution while recording steady state vessel diameter. The response to acetylcholine was significantly attenuated in the diabetic rat. Maximal dilation was 94±3 vs 65±5%, −log ED50 was 6.7±0.2 vs 6.7±0.3 (control (n=10) vs diabetic rat (n=8)). #P<0.05 vs control.
Figure 2.
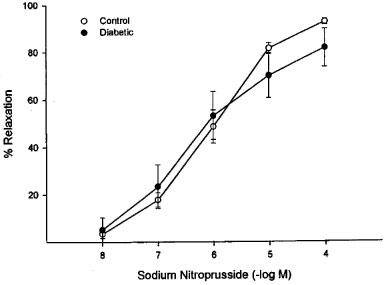
Sodium nitroprusside-induced endothelium independent relaxation of arterioles adjacent to the sciatic nerve in control and diabetic rats. Rat arterioles were mounted on glass pipettes for measurement of internal diameter. After constriction with U46619, graded doses of nitroprusside were added, measuring diameter after each dose. The response to sodium nitroprusside was unchanged between control (n=7) and diabetic rats (n=7). Maximal dilation was 93±1 vs 82±8%, −log ED50 was 6.1±0.1 vs 6.4±0.2, respectively.
Figure 3.
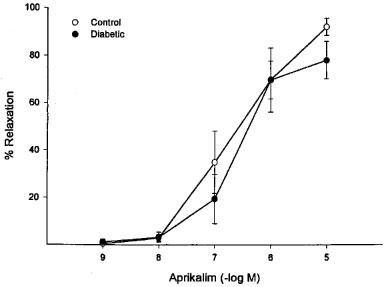
Aprikalim-induced relaxation of arterioles adjacent to the sciatic nerve in control and diabetic rats. The KATP-channel opening agent, aprikalim, was added to the external bathing media of pressurized arterioles constricted with U46619. Aprikalim produced similar concentration dependent relaxations in arterioles from control (n=6) and diabetic rats (n=5). Maximal dilation was 92±4 vs 80±8%, −log ED50 was 6.7±0.3 vs 6.7±0.2, respectively.
Mechanism of acetylcholine-induced vasodilation in arterioles that supply the region of the sciatic nerve
Data in Figure 4 show that pre-exposing vessels from normal rats to LNNA or KCl alone significantly decreased maximal relaxation induced by acetylcholine. The combination of LNNA and KCl caused a significantly greater decrease in acetylcholine-induced vasodilation in these vessels compared to vessels treated with LNNA or KCl alone. In several studies we substituted 100 μM NG-nitro-L-arginine methyl ester (L-NAME) for LNNA and similar results were observed. Pretreatment of vessels from diabetic rats with LNNA or KCl alone also caused a significant decrease in acetylcholine-induced vasodilation (Figure 5). In studies not shown treatment of vessels from normal or diabetic rats with INDO alone did not significantly inhibit acetylcholine-induced vasodilation. In addition, the combination of INDO with LNNA and/or KCl did not further impair acetylcholine-induced vasodilation compared to LNNA and/or KCl alone (data not shown).
Figure 4.
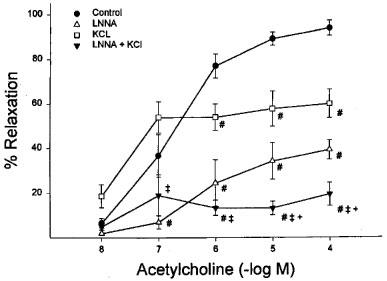
Mechanism of acetylcholine-induced dilation of arterioles adjacent to the sciatic nerve. Arterioles from non-diabetic rats were constricted with U46619 and treated with LNNA or KCl alone or LNNA and KCl prior to application of graded doses of acetylcholine. Acetylcholine induced dilation was significantly decreased by LNNA or KCl alone and LNNA and KCl. #P<0.05 vs control, +P<0.05 vs LNNA alone, ‡+P<0.05 vs KCl alone. Control (n=10), LNNA (n=3), KCl (n=3) and LNNA+KCl (n=4).
Figure 5.
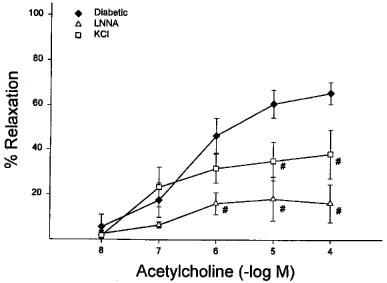
Mechanism of acetylcholine-induced dilation of arterioles adjacent to the sciatic nerve. Arterioles from diabetic rats were constricted with U46619 and treated with LNNA or KCl alone prior to application of graded doses of acetylcholine. Acetylcholine induced dilation was significantly decreased by LNNA or KCl alone. #P<0.05 vs diabetic. Diabetic (n=8), LNNA (n=3) and KCl (n=3).
Discussion
The results from these studies show that endothelium-dependent vasodilation of arterioles that provide circulation to the region of the sciatic nerve is impaired by diabetes. The impaired dilation to acetylcholine is associated with a reduction in motor nerve conduction velocity. Furthermore, impaired vasodilator responsiveness of these vessels is consistent with the observed decrease in endoneural blood flow in the sciatic nerve of diabetic rats.
The effect of diabetes on vascular reactivity has been studied in a variety of vascular tissues. In the aorta from several species, agonist induced endothelium-dependent vasodilation is impaired by diabetes and by acute hyperglycaemia (Tesfamariam et al., 1991; 1993; Tesfamariam & Cohen, 1992; Dorigo et al., 1997). Treating diabetic rats or their aortic rings with insulin, an aldose reductase inhibitor (zopolrestat), hydroxyl radical scavengers (superoxide dismutase, catalase, desferroxamine, or allopurinol) or a nitric oxide donor prevents the diabetes-induced decrease in vascular reactivity (Tesfamariam & Cohen, 1992; Tesfamariam et al., 1993; Dorigo et al., 1997). These studies suggest that hyperglycaemia may induce endothelial cell dysfunction by several mechanisms. Similar alterations in vasodilator responses have been observed in mesenteric arteries from diabetic rats (Taylor & Poston, 1994; Heygate et al., 1995; Tribe et al., 1998; Rodriguez-Manas et al., 1998), where the impaired dilation to acetylcholine is improved by treating the arteries with ponalrestat, an aldose reductase inhibitor, L-arginine, or superoxide dismutase (Taylor & Poston, 1994). Other studies have shown reduced acetylcholine-induced vasodilation of the basilar artery (Fujii et al., 1992; Mayhan et al., 1996), renal interlobar artery (Dai et al., 1993), and coronary artery (Matsunaga et al., 1996; Koltai et al., 1997) in diabetes. Therefore, impaired endothelial-dependent vasodilation of the aorta and arteries from the kidney, heart and as reported in this study, the region of the sciatic nerve, is a common defect in diabetes. However, the mechanisms responsible for the endothelial cell-dependent vascular defect may vary depending on the vasculature being studied.
We have shown that LNNA or KCl alone or the combination of LNNA and KCl reduce acetylcholine-induced vasodilation to different degrees in arterioles that provides circulation to the region of the sciatic nerve. In contrast, INDO alone or in combination with LNNA and/or KCl did not impair acetylcholine-induced vasodilation in these vessels. The latter studies suggest that the production of prostacyclin does not contribute to acetylcholine-induced vasodilation in these vessels. Acetylcholine is thought to cause vasodilation through the release of nitric oxide in many vessels including rat conduit arteries (Sobey et al., 1996). However, our data are more consistent with the idea that acetylcholine-induced dilation in these vessels is mediated by two mechanisms involving the production of nitric oxide and possibly endothelium-derived hyperpolarizing factor (EDHF). One caveat to consider concerning this conclusion is that Kemp & Cocks, 1997, in studies using human coronary arteries, have shown that nitric oxide synthase inhibitors such as NG-nitro-L-arginine do not completely inhibit nitric oxide production by endothelial cells and that nitric oxide itself can cause hyperpolarization. However, we consider that the uninhibited production of nitric oxide is not responsible for the K+-sensitive/hyperpolarization component of vasodilation induced by acetylcholine in arterioles that provide circulation to the sciatic nerve. This conclusion is supported by a study showing that the guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) had no greater effect than LNNA alone in inhibiting acetylcholine mediated vasodilation in these vessels (data not shown). Combined, these results imply that acetylcholine-induced vasodilation in this vessel bed is mediated by a NO-dependent and NO-independent mechanism.
It is not clear which vasodilator mechanism is impaired in diabetes. Others have shown reduced activity of nitric oxide in vessels from diabetic animals (Tribe et al., 1998; Rodriguez-Manas et al., 1998). In addition, a deficiency in nitric oxide release has been suggested as the mechanism for reduced nerve blood flow in diabetic rats (Omawari et al., 1996). In other studies, pathways involving the metabolism of arachidonic acid could also be involved in mediating changes in vascular reactivity. Supplementing diabetic rats with γ-linolenic acid, an arachidonic acid precursor, has been shown to improve motor nerve conduction velocity and neuronal blood flow in the sciatic nerve (Lockett & Tomlinson, 1992; Karasu et al., 1995; Cameron et al., 1996; Cotter & Cameron, 1997). Furthermore, the prostacyclin analogue beraprost sodium has been shown to increase sciatic nerve blood flow in diabetic rats (Hotta et al., 1996). Diabetes causes a defect in the desaturation of essential fatty acids, thereby reducing the availability of arachidonic acid (Cotter & Cameron, 1997). It is possible that γ-linolenic acid supplementation could restore arachidonic acid metabolism in the vasculature and improve vascular relaxation via the cyclooxygenase pathway (by normalizing production of prostacyclin) or through the cytochrome P450-sensitive pathway (via production of endothelium-derived hyperpolarizing factor) (Quilley et al., 1997; Cotter et al., 1993). The activity of the latter pathway may be especially important in vessels that provide circulation to the region of the sciatic nerve. Restoring the bioactivity of these vasodilators could improve neuronal blood flow and motor nerve conduction velocity. Other treatments such as aminoguanidine and antioxidants have also been shown to improve neuronal blood flow and motor nerve conduction velocity in diabetic rats (Kihara et al., 1991; Cameron et al., 1992; 1993). These treatments could improve vascular relaxation by restoring nitric oxide bioactivity (Cameron & Cotter, 1995). Future studies using vessels that provide circulation to the region of the sciatic nerve will be conducted to determine which of these pathways is impaired by diabetes.
Studies of arterioles from diabetic rats demonstrated that pretreating arterioles with LNNA or KCl significantly reduced acetylcholine-induced vasodilation. These results suggest that the diabetes-induced decrease in acetylcholine-induced vasorelaxation is not due to an impairment of any singular pathway but may be due to altering the activity of several pathways that contribute to mediating vasorelaxation in these vessels.
Our studies demonstrated that aprikalim caused vasodilation of arterioles that provide circulation to the region of the sciatic nerve suggesting that ATP-sensitive potassium channels are functional in these vessels. However, unlike the effect of acetylcholine on vasodilation, aprikalim-induced relaxation was not impaired by diabetes. This contrasts with studies of basilar artery dilation in rats where diabetes reduces dilation to aprikalim (Mayhan, 1994). It also contrasts with data from dog coronary arterioles where dilation to aprikalim is enhanced in diabetic dogs (Kersten et al., 1995). The reason for this difference is unknown but suggests that the effect of diabetes on vascular relaxation is heterogeneous among vascular beds and species, and that altered dilation to activation of KATP channels does not contribute to the reduced nerve conduction velocity observed in this model.
In summary, these studies have shown that acetylcholine-induced endothelial cell-dependent vasodilation of arterioles that regulate blood flow to the region of the sciatic nerve is impaired by diabetes. This defect could have a direct impact on neuronal blood flow of the sciatic nerve and motor nerve conduction velocity, which are likewise reduced in diabetic neuropathy.
Acknowledgments
This work was supported by a National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-25295, by a Diabetes Center Grant from the Veterans Affairs and International Juvenile Diabetes Foundation, and by a research grant from the American Diabetes Association. We thank Fausto Loberiza, MD for his expert statistical assistance.
Abbreviations
- EBF
endoneural blood flow
- EDHF
endothelium-derived hyperpolarizing factor
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N1-N′-tetraacetic acid
- INDO
indomethacin
- MNCV
motor nerve conduction velocity
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- Na+/K+-ATPase
sodium/potassium ATPase
- L-NAME
NG-nitro-L-arginine methyl ester
- LNNA
Nω-nitro-L-arginine
- NO
nitric oxide
- PSS
Krebs Henseleit physiological saline solution
References
- CAMERON N.E., COTTER M.A. Effects of chronic treatment with a nitric oxide donor on nerve conduction abnormalities and endoneurial blood flow in streptozotocin-diabetic rats. European J. Clin. Invest. 1995;25:19–24. doi: 10.1111/j.1365-2362.1995.tb01520.x. [DOI] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A., BASSO M., HOHMAN T.C. Comparison of the effects of inhibitors of aldose reductase and sorbitol dehydrogenase on neurovascular function, nerve conduction and tissue polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. 1997;40:271–281. doi: 10.1007/s001250050674. [DOI] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A., DINES K., LOVE A. Effects of aminoguanidine on peripheral nerve function and polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. 1992;35:946–950. doi: 10.1007/BF00401423. [DOI] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A., HOHMAN T.C. Interactions between essential fatty acids, prostanoid, polyol pathway and nitric oxide mechanisms in the neurovascular deficit of diabetic rats. Diabetologia. 1996;39:172–182. doi: 10.1007/BF00403960. [DOI] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A., LOW P.A. Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am. J. Physiol. 1991;261:E1–E8. doi: 10.1152/ajpendo.1991.261.1.E1. [DOI] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A., MAXFIELD E.K. Anti-oxidant treatment prevents the development of peripheral nerve dysfunction in streptozotocin-diabetic rats. Diabetologia. 1993;36:299–304. doi: 10.1007/BF00400231. [DOI] [PubMed] [Google Scholar]
- CHANG K., IDO Y., LEJEUNE W., WILLIAMSON J.R., TILTON R.G. Increased sciatic nerve blood flow in diabetic rats: assessment by molecular vs. particulate microspheres. Am. J. Physiol. 1997;273:E164–E173. doi: 10.1152/ajpendo.1997.273.1.E164. [DOI] [PubMed] [Google Scholar]
- COHEN R.A. Dysfunction of vascular endothelium in diabetes mellitus. Circulation. 1993;87:V67–V76. [Google Scholar]
- COOPER M.E., GILBERT R.E., JERUMS G. Diabetic vascular complications. Clin. Exp. Pharm. Physiol. 1997;24:770–775. doi: 10.1111/j.1440-1681.1997.tb02130.x. [DOI] [PubMed] [Google Scholar]
- COTTER M.A., CAMERON N.E. Effects of dietary supplementation with arachidonic acid rich oils on nerve conduction and blood flow in streptozotocin-diabetic rats. Prostaglandins, Leukotrienes and Ess. Fatty Acids. 1997;56:337–343. doi: 10.1016/s0952-3278(97)90581-0. [DOI] [PubMed] [Google Scholar]
- COTTER M.A., DINES K.C., CAMERON N.E. Prevention and reversal of motor and sensory peripheral nerve conduction abnormalities in streptozotocin-diabetic rats by the prostacyclin analogue iloprost. Arch. Pharm. 1993;347:534–540. doi: 10.1007/BF00166747. [DOI] [PubMed] [Google Scholar]
- DAI F., DIEDERICH A., SKOPEC J., DIEDERICH D. Diabetes-induced endothelial dysfunction in streptozotocin-treated rats: role of prostaglandin endoperoxides and free radicals. J. Am. Soc. Nephrol. 1993;4:1327–1336. doi: 10.1681/ASN.V461327. [DOI] [PubMed] [Google Scholar]
- DORIGO P., FRACCAROLLO D., SANTOSTASI G., MARAGNO I. Impairment of endothelium-dependent but not of endothelium-independent dilatation in guinea-pig aorta rings incubated in the presence of elevated glucose. Br. J. Pharmacol. 1997;121:972–976. doi: 10.1038/sj.bjp.0701203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUJII K., HEISTAD D.D., FARACI F.M. Effect of diabetes mellitus on flow-mediated and endothelium-dependent dilatation of the rat basilar artery. Stroke. 1992;23:1494–1498. doi: 10.1161/01.str.23.10.1494. [DOI] [PubMed] [Google Scholar]
- GILLON K.R.W., HAWTHORNE J.N., TOMLINSON D.R. Myo-inositol and sorbitol metabolism in relation to peripheral nerve function in experimental diabetes in the rat: The effect of aldose reductase inhibition. Diabetologia. 1983;25:365–371. doi: 10.1007/BF00253203. [DOI] [PubMed] [Google Scholar]
- GREENE D.A., LATTIMER S.A. Impaired energy utilization and Na-K-ATPase in diabetic peripheral nerve. Am. J. Physiol. 1984;246:E311–E318. doi: 10.1152/ajpendo.1984.246.4.E311. [DOI] [PubMed] [Google Scholar]
- GREENE D.A., LATTIMER S.A., SIMA A.A.F. Are disturbances of sorbitol, phosphoinositide, and Na+-K+-ATPase regulation involved in pathogenesis of diabetic neuropathy. Diabetes. 1988;37:688–693. doi: 10.2337/diab.37.6.688. [DOI] [PubMed] [Google Scholar]
- GREENE D.A., SOROKU Y., LATTIMER S.A., SIMA A.A.F. Nerve Na+-K+-ATPase, conduction, and myo-inositol in the insulin-deficient BB rat. Am. J. Physiol. 1984;247:E534–E539. doi: 10.1152/ajpendo.1984.247.4.E534. [DOI] [PubMed] [Google Scholar]
- HEYGATE K.M., LAWRENCE I.G., BENNETT M.A., THURSTON H. Impaired endothelium-dependent relaxation in isolated resistance arteries of spontaneously diabetic rats. Br. J. Pharmacol. 1995;116:3251–3259. doi: 10.1111/j.1476-5381.1995.tb15132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOTTA N., KOH N., SAKAKIBARA F., NAKAMURA J., HAMADA Y., HARA T., MORI K., NAKASHIMA E., NARUSE K., FUKASAWA H., KAKUTA H., SAKAMOTO N. Effects of beraprost sodium and insulin on the electroretinogram, nerve conduction, and nerve blood flow in rats with streptozotocin-induced diabetes. Diabetes. 1996;45:361–366. doi: 10.2337/diab.45.3.361. [DOI] [PubMed] [Google Scholar]
- IDO Y., CHANG K., LEJEUNE W., TILTON R.G., MONAFO W.W., WILLIAMSON J.R. Diabetes impairs sciatic nerve hyperemia induced by surgical trauma: implications for diabetic neuropathy. Am. J. Physiol. 1997;273:E174–E184. doi: 10.1152/ajpendo.1997.273.1.E174. [DOI] [PubMed] [Google Scholar]
- KARASU C., DEWHURST M., STEVENS E.J., TOMLINSON D.R. Effects of anti-oxidant treatment on sciatic nerve dysfunction in streptozotocin-diabetic rats; comparison with essential fatty acids. Diabetologia. 1995;38:129–134. doi: 10.1007/BF00400086. [DOI] [PubMed] [Google Scholar]
- KEMP B.K., COCKS T.M. Evidence that mechanisms-dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary arteries. Br. J. Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERSTEN J.R., BROOKS L.A., DELLSPERGER K.C. Impaired microvascular response to graded coronary occlusion in diabetic and hyperglycemic dogs. Am. J. Physiol. 1995;268:H1667–H1674. doi: 10.1152/ajpheart.1995.268.4.H1667. [DOI] [PubMed] [Google Scholar]
- KIHARA M., SCHMELZER J.D., PODUSLO J.F., CURRAN G.L., NICKANDER K.K., LOW P.A. Aminoguanidine effects on nerve blood flow, vascular permeability, electrophysiology, and oxygen free radicals. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6107–6111. doi: 10.1073/pnas.88.14.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLTAI M.Z., HADHAZY P., POSA I., KOCSIS E., WINKLER G., ROSEN P., POGATSA G. Characteristics of coronary endothelial dysfunction in experimental diabetes. Cardiovasc. Res. 1997;34:157–163. doi: 10.1016/s0008-6363(97)00050-3. [DOI] [PubMed] [Google Scholar]
- KUO L., CHILIAN W.M., DAVIS M.J. Coronary arteriolar myogenic response is independent of endothelium. Circ. Res. 1990;66:860–866. doi: 10.1161/01.res.66.3.860. [DOI] [PubMed] [Google Scholar]
- LOCKETT M.J., TOMLINSON D.R. The effects of dietary treatment with essential fatty acids on sciatic nerve conduction and activity of the Na+/K+ pump in streptozotocin-diabetic rats. Br. J. Pharmacol. 1992;105:355–360. doi: 10.1111/j.1476-5381.1992.tb14258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUNAGA T., OKUMURA K., ISHIZAKA H., TSUNODA R., TAYAMA S., TABUCHI T., YASUE H. Impairment of coronary blood flow regulation by endothelium-derived nitric oxide in dogs with alloxan-induced diabetes. J. Cardiovasc. Pharm. 1996;28:60–67. doi: 10.1097/00005344-199607000-00010. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G. Effect of diabetes mellitus on response of the basilar artery to activation of ATP-sensitive potassium channels. Brain Res. 1994;636:35–39. doi: 10.1016/0006-8993(94)90172-4. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., DIDION S.P., PATEL K.P. L-Arginine does not restore dilatation of the basilar artery during diabetes mellitus. J. Cerebral Blood Flow Met. 1996;16:500–506. doi: 10.1097/00004647-199605000-00017. [DOI] [PubMed] [Google Scholar]
- OMAWARI N., DEWHURST M., VO P., MAHMOOD S., STEVENS E., TOMLINSON D.R. Deficient nitric oxide responsible for reduced nerve blood flow in diabetic rats: effects of L-NAME, L-arginine, sodium nitroprusside and evening primrose oil. Br. J. Pharmacol. 1996;118:186–190. doi: 10.1111/j.1476-5381.1996.tb15384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUILLEY J., FULTON D., MCGIFF J.C. Hyperpolarizing factors. Biochem. Pharm. 1997;54:1059–1070. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ-MANAS L., ANGULO J., PEIRO C., LLERGO J.L., SANCHEZ-FERRER A., LOPEZ-DORIGA P., SANCHEZ-FERRER C.F. Endothelial dysfunction and metabolic control in streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1998;123:1495–1502. doi: 10.1038/sj.bjp.0701749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOBEY C.G., HEISTAD D.D., FARACI F.M. Effect of subarachnoid hemorrhage on dilatation of rat basilar artery in vivo. Am. J. Physiol. 1996;40:H126–H132. doi: 10.1152/ajpheart.1996.271.1.H126. [DOI] [PubMed] [Google Scholar]
- STEHOUWER C.D.A., LAMBERT J., DONKER A.J.M., VAN HINSBERGH V.W.M. Endothelial dysfunction and pathogenesis of diabetic angiopathy. Cardiovasc. Res. 1997;34:55–68. doi: 10.1016/s0008-6363(96)00272-6. [DOI] [PubMed] [Google Scholar]
- STEVENS E.J., CARRINGTON A.L., TOMLINSON D.R. Nerve ischaemia in diabetic rats: time-course of development, effect of insulin treatment plus comparison of streptozotocin and BB models. Diabetologia. 1994;37:43–48. doi: 10.1007/BF00428776. [DOI] [PubMed] [Google Scholar]
- TAYLOR P.D., POSTON L. The effect of hyperglycemia on function of rat isolated mesenteric resistance artery. Br. J. Pharmacol. 1994;113:801–808. doi: 10.1111/j.1476-5381.1994.tb17064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., BROWN M.L., COHEN R.A. Elevated glucose impairs endothelium-dependent relaxation by activating protein kinase C. J. Clin. Invest. 1991;87:1643–1648. doi: 10.1172/JCI115179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., COHEN R.A. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am. J. Physiol. 1992;263:H321–H326. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- TESFAMARIAM B., PALACINO J.J., WEISBROD R.M., COHEN R.A. Aldose reductase inhibition restores endothelial cell function in diabetic rabbit aorta. J. Cardiovasc. Pharm. 1993;21:205–211. doi: 10.1097/00005344-199302000-00004. [DOI] [PubMed] [Google Scholar]
- TRIBE R.M., THOMAS C.R., POSTON L. Flow-induced dilatation in isolated resistance arteries from control and streptozotocin-diabetic rats. Diabetologia. 1998;41:34–39. doi: 10.1007/s001250050863. [DOI] [PubMed] [Google Scholar]
- WRIGHT R.A., NUKADA H. Vascular and metabolic factors in the pathogenesis of experimental diabetic neuropathy in mature rats. Brain. 1994;117:1395–1407. doi: 10.1093/brain/117.6.1395. [DOI] [PubMed] [Google Scholar]
- YOREK M.A., WIESE T.J., DAVIDSON E.P., DUNLAP J.A., STEFANI M.R., CONNER C.E., LATTIMER S.A., KAMIJO M., GREENE D.A., SIMA A.A.F. Reduced motor nerve conduction velocity and Na+-K+-ATPase activity in rats maintained on L-fucose diet. Diabetes. 1993;42:1401–1406. doi: 10.2337/diab.42.10.1401. [DOI] [PubMed] [Google Scholar]
- YOUNG W. H2 clearance measurement of blood flow: a review of technique and polarographic principles. Stroke. 1980;11:552–564. doi: 10.1161/01.str.11.5.552. [DOI] [PubMed] [Google Scholar]