

Abstract
Acetycholine-mediated relaxations in phenylephrine-contracted aortas, femoral and mesenteric resistance arteries were studied in vessels from endothelial nitric oxide synthase knock-out (eNOS −/−) and the corresponding wild-type strain (eNOS +/+) C57BL6/SV19 mice.
Aortas from eNOS (+/+) mice relaxed to acetylcholine in an endothelium-dependent NG-nitro-L-arginine (L-NOARG) sensitive manner. Aortas from eNOS (−/−) mice did not relax to acetylcholine but demonstrated enhanced sensitivity to both authentic NO and sodium nitroprusside.
Relaxation to acetylcholine in femoral arteries was partially inhibited by L-NOARG in vessels from eNOS (+/+) mice, but relaxation in eNOS (−/−) mice was insensitive to a combination of L-NOARG and indomethacin and the guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ). The L-NOARG/ODQ/indomethacin-insensitive relaxation to acetylcholine in femoral arteries was inhibited in the presence of elevated (30 mM) extracellular KCl.
In mesenteric resistance vessels from eNOS (+/+) mice, the acetylcholine-mediated relaxation response was completely inhibited by a combination of indomethacin and L-NOARG or by 30 mM KCl alone. In contrast, in mesenteric arteries from eNOS (−/−) mice, the acetylcholine-relaxation response was insensitive to a combination of L-NOARG and indomethacin, but was inhibited in the presence of 30 mM KCl.
These data indicate arteries from eNOS (−/−) mice demonstrate a supersensitivity to exogenous NO, and that acetylcholine-induced vasorelaxation of femoral and mesenteric vessels from eNOS (−/−) mice is mediated by an endothelium-derived factor that has properties of an EDHF but is neither NO nor prostacyclin. Furthermore, in mesenteric vessels, there is an upregulation of the role of EDHF in the absence of NO.
Keywords: eNOS knockout mice, EDHF, NO, smooth muscle, endothelium
Introduction
It is generally believed that endothelium-dependent relaxation to acetylcholine is primarily mediated by the release of nitric oxide (NO). However, other mediators including prostacyclin and a poorly characterized endothelium-dependent hyperpolarizing factor (EDHF) have been proposed. Nonetheless, the existence of EDHF has been questioned and various explanations have been put forward to account for the hyperpolarization and nitric oxide synthase (NOS) inhibitor-resistant relaxations that have been observed in several arteries. These include incomplete pharmacological blockade of NOS (Martin et al., 1992; Cohen et al., 1997), NO or prostacyclin activation of potassium channels (Tare et al., 1990; Parkington et al., 1993; Bolotina et al., 1994), gap junctional communication between endothelial and smooth muscle cells (Davies et al., 1988), and a small increase in extracellular K+ (Edwards et al., 1998). Here we describe acetylcholine-induced relaxation of small peripheral arteries isolated from mice which lack endothelial NOS. This relaxation possesses the characteristics of being mediated by an EDHF.
Methods
Breeding pairs of homozygous endothelial NOS knockout (eNOS−/−) and control (C57BL6/SV19) mice were obtained from Dr Paul Huang (Cardiovascular Research Centre, Massachusetts General Hospital) and bred at the University of Calgary. The generation of eNOS (−/−) mice by gene targeting in embryonic stem cells has been previously described (Huang et al., 1995). In accordance with guidelines established by the University of Calgary Animal Care Committee, mice of either sex (25–37 g) were killed by cervical dislocation and the aorta, femoral and first order branches of the mesenteric artery were dissected out into Krebs' solution of composition (in mM): NaCl, 120; NaHCO3, 25; KCl, 4.8; NaH2PO4, 1.2; MgSO4, 1.2; Dextrose, 11.0; CaCl2, 1.8, bubbled with 95% O2/5% CO2. The aorta was cut into 2 mm rings and placed on wires in an organ bath under a resting tension of 1 g. Isometric force was measured by Grass FT-03 transducers and a Grass model 7D polygraph. Femoral arteries were mounted in a Mulvany-Halpern myograph (Model 400A, J.P. Trading, Denmark) under normalised tension as previously described (Mulvany & Halpern, 1977). Mesenteric arteries were mounted in a pressure myograph (Living Systems, Vermont, U.S.A.) under constant pressure of 60 mmHg, perfusion of 50 μl min−1 and superfusion of 5 ml min−1 and their diameter was measured by videomicroscopy. All experiments were performed at 37°C. For the relaxation/vasodilation experiments, tissues were precontracted with phenylephrine (0.3–10 μM). Care was taken to ensure that a submaximal contraction/constriction was obtained, and that equi-effective concentrations of phenylephrine or KCl were used in all protocols. High [K+] solutions were made by iso-osmolar replacement of Na+ by K+. Data are expressed as pD2 values which are defined as the negative logarithm to base 10 of the EC50 values. Relaxation is expressed as percentage of phenylephrine-induced tone (aorta and femoral arteries) or vasoconstriction (mesenteric artery) ±s.e.mean. In the femoral artery experiments there was a tachyphylaxis between the first and second concentration-response curve to acetylcholine, though subsequent control curves were not different from the second. The significance of differences between mean values was calculated by Student's t-test. All drugs were from Sigma except 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; Tocris, MO, U.S.A.) and NO gas (Union Carbide, Canada).
Results
pD2 values for phenylephrine in wild-type (+/+) and eNOS knockout (−/−) mice aortas were 7.14±0.02 and 6.54±0.06, respectively (n=5, P<0.05). The maximum contractions to phenylephrine were significantly greater in eNOS (−/−) vs eNOS (+/+) (1.51±0.16 Nm−1 vs 1.24±0.15 Nm−1, n=5, P<0.05). Aortas from eNOS (+/+) mice were contracted with phenylephrine and relaxed in response to acetylcholine (10 μM), whereas aortas from endothelial NOS knockout mice did not (67±3.5 vs 6±3.5%, n=5 and 6). However, as illustrated in Figure 1, aortas from both groups of animals were maximally relaxed by exogenous NO, aortas from eNOS (−/−) mice being significantly more sensitive than C57BL6/SV19 mice (control pD2 7.26±0.04 vs 8.07±0.06, n=5 and 6, P<0.001). Aortic tissue from eNOS (−/−) was also more sensitive to sodium nitroprusside than was tissue from eNOS (+/+) (pD2 of 8.46±0.04 vs 10.21±0.02, n=5–6, P<0.001 for eNOS (+/+) and eNOS (−/−) respectively).
Figure 1.
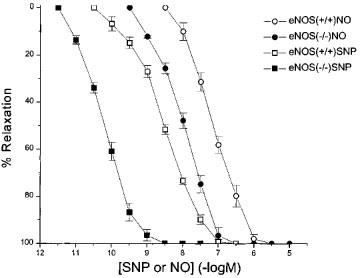
Concentration-relaxation curve to authentic nitric oxide (NO) solution and sodium nitroprusside in phenylephrine-precontracted aorta from eNOS (+/+) and eNOS (−/−) mice.
Femoral arteries isolated from eNOS (−/−) and eNOS (+/+) mice contracted to both KCl and phenylephrine (Figure 2a,b), and there was no significant difference (P>0.05) in the pD2 values obtained for KCl (1.47±0.9 vs 1.56±0.7, n=8, P>0.05 respectively). For phenylephrine in the absence or presence of L-NOARG (100 μM), the pD2 values were 5.89±0.03; 5.92±0.02; 5.60±0.06; 5.87±0.03 for eNOS (−/−) in the absence and presence of L-NOARG and eNOS (+/+) in the absence of L-NOARG, respectively (n=10). In the absence of L-NOARG, femoral arteries from the (−/−) mice were significantly (n=10, P<0.05) more sensitive to phenylephrine, however, the difference in sensitivity was lost after pretreatment with L-NOARG. The maximum contraction to phenylephine in the femoral artery were for eNOS (+/+) 2.05±0.2 Nm−1 (n=3) and for eNOS (−/−) 3.01±0.16 Nm−1 (n=8, P<0.05). In the presence of indomethacin, femoral arteries isolated from eNOS (+/+) mice (D100 357±22 mm, n=16 vessels from eight animals) relaxed in response to acetylcholine (Figure 3a). The pD2 value and maximum response for the concentration-response curve to acetylcholine was 6.82±0.06 and 88.1±3.3% (n=4). Exposure of the vessels to 30 mM K+ containing Krebs' solution elicited a contraction (4.4±0.4 Nm−1) that was not significantly different from that elicited by 3 μM phenylephrine (3.7±0.2 Nm−1, n=4, P>0.05). In the presence of 30 mM K+, acetylcholine relaxed the tissues with a pD2 of 6.42±0.03 (P=0.001 vs phenylephrine-contracted tissue) and a maximum relaxation of 58.8±4.0% (n=4, P>0.05). In the presence of a combination of indomethacin, L-NOARG (100 μM) with nitro-L-arginine methyl ester (L-NAME, 100 μM) the acetylcholine concentration response curve was not shifted to the right (pD2 value of 6.77±0.12, P>0.05) though the maximum response was decreased to 39.4±2.6% (n=3, P<0.001). In the presence of ODQ alone, the maximum response was reduced to 20.0±3.7% (n=7, P<0.001) with the pD2 value of 6.3±0.39. Combination of indomethacin, L-NOARG, L-NAME and 30 mM K+ abolished all relaxations induced by acetylcholine (n=3).
Figure 2.
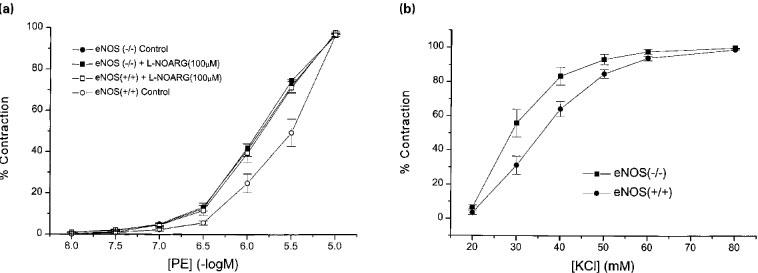
Concentration-response curves to (a) KCl and (b) phenylephine in the absence or presence of L-NOARG (100 μM) in femoral arteries from eNOS (+/+) and eNOS (−/−) mice.
Figure 3.
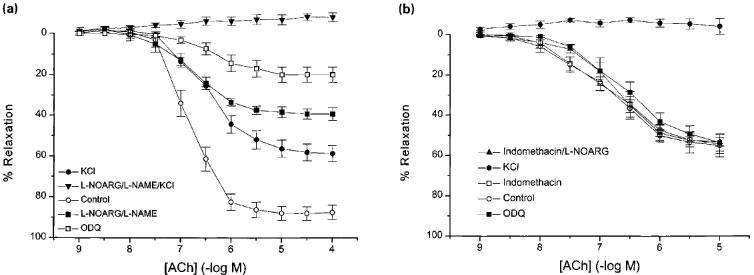
Effect of inhibition of cyclo-oxygenase, NOS, guanylyl cyclase or potassium efflux and combination of these treatments on acetylcholine-induced relaxation of phenylephrine precontracted femoral arteries isolated from (a) eNOS (+/+) and (b) eNOS (−/−) mice.
Femoral arteries isolated from eNOS (−/−) mice (D100 290±15 μm, n=26 vessels from 13 animals) relaxed in response to acetylcholine, though to a lesser maximum than the control mice (pD2 6.73±0.14 and 54.2±4.9%, n=9; P<0.05 vs eNOS (+/+) mice; Figure 3b). These relaxations were insensitive to indomethacin (3 μM), indomethacin plus L-NOARG (100 μM) or indomethacin, L-NOARG and ODQ (3 μM) in combination (pD2 values of 6.78±0.29, 6.53±0.17 and 6.89±0.20 and maximal relaxations of 49.6±4.3, 51.2±3.3 and 45.9±8.3%, n=3 for acetylcholine, P>0.05 for all). Precontraction with 30 mM K+ containing Krebs' solution, in the absence of inhibitors of NOS or guanylyl cyclase, completely abrogated the relaxation to acetylcholine in femoral arteries isolated from mice lacking endothelial NOS.
First order mesenteric arterioles from eNOS (−/−) mice had a resting diameter of 259±18.8 μm (n=8) when pressurized to 60 mmHg. Figure 4a shows that after constriction with phenylephrine (1–10 μM, diameter 112±9.8 μm, n=8), these vessels dilated to acetylcholine with a pD2 value of 6.40±0.15 and a maximum relaxation of 80.7±3.2% (n=11). This relaxation was slightly reduced after incubation with indomethacin (10 μM) with the pD2 value not different at 6.59±0.30 (n=4) and only the relaxation due to 10 μM acetylcholine significantly reduced. The acetylcholine-induced relaxation was virtually abolished by L-NOARG (100 μM) in combination with indomethacin (maximum relaxation of 15.8±6.6%, n=3, P<0.001). Raising external K+ to 30 mM decreased the diameter of these vessels to 188±14.6 μm and subsequent addition of phenylephrine (1 μM) constricted the vessels to 84.1±16.4 μm (n=7). Following this pretreatment, acetylcholine-induced relaxation was also inhibited with a maximum relaxation of 12.6±4.5% (n=3, P<0.001). The potassium channel blocker, tetraethylammonium (TEA, 1 mM), in the absence of other blockers, also inhibited the relaxation, reducing the maximal effect of acetylcholine to 19.6±6.7% (n=4, P<0.001).
Figure 4.
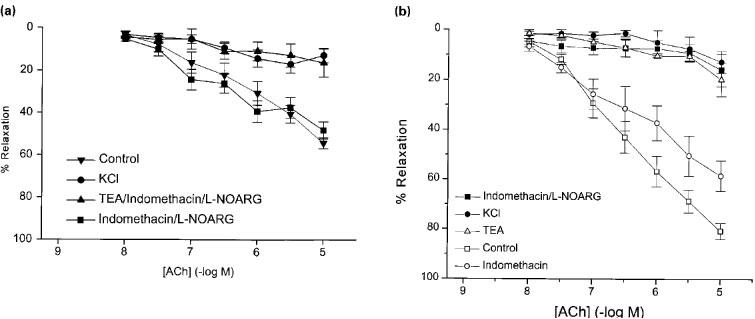
Effect of inhibition of cyclo-oxygenase, NOS or potassium efflux and combination of these treatments of acetylcholine-induced vasodilation of phenylephrine preconstricted mesenteric arteries isolated from (a) eNOS (+/+) and (b) eNOS (−/−) mice.
Mesenteric arteries isolated from mice lacking endothelial NOS possessed a resting diameter of 238±7.4 μm, n=13, P>0.05 compared to wild type). The pD2 values for phenylephrine-induced vasoconstriction in the absence and presence of L-NOARG in mesenteric vessels from eNOS (−/−) mice were 6.36±0.05 and 6.36±0.03 respectively, and thus were not significantly different (P>0.05). In contrast to the aortas from eNOS (−/−) mice, mesenteric arteries, when contracted to 84.9±9.6 μm with phenylephrine, relaxed in response to acetylcholine (pD2 value of 6.40±0.21 and maximum relaxation of 54.2±2.6%; n=9; Figure 4b). However, the combination of L-NOARG and indomethacin did not affect the response (pD2 value of 6.85±0.17 and maximum relaxation of 48.2±4.1%, n=13, P>0.05 for both). Raising extracellular K+ to 30 mM constricted the vessels to 197±19.3 μm and further addition of 1 μM phenylephrine constricted the vessels to 79.3±8.8 μm (n=8). Under these conditions the response to acetylcholine was greatly reduced (16.9±4.2% maximum relaxation, n=4, P<0.01). TEA (1 mM), in the presence of indomethacin and L-NOARG, also blocked the relaxation with a maximum effect of 16.3±6.7%, (n=3, P<0.001).
Discussion
These data indicate that (a) arteries isolated from mice lacking endothelial NOS show a supersensitivity to exogenous NO, and (b) acetylcholine-induced vasorelaxation of arteries can be mediated by a factor that is not NO nor prostacyclin. The relaxation in arteries lacking endothelial NOS is completely sensitive to raised potassium or TEA indicating that a potassium flux is required to elicit this relaxation and hence is presumably mediated by an EDHF.
There is a growing body of evidence that, in addition to NO and prostacyclin, a third class of endothelium-dependent vasodilators, hyperpolarizing factors, can contribute to the regulation of vascular tone (Garland et al., 1995; Mombouli & Vanhoutte, 1997). These factors induce vascular smooth muscle relaxation by activation of a potassium conductance, hyperpolarization of the smooth muscle cells and hence inhibition of calcium influx through voltage-gated calcium channels. Difficulties in bioassaying EDHF and an inconsistent pharmacology between preparations have hampered efforts to identify its molecular nature and led to speculation that a factor separate from NO may not exist. Furthermore, both prostacyclin (Parkington et al., 1993) and NO (Bolotina et al., 1994) can hyperpolarize smooth muscle cells. Recently, the bradykinin evoked EDHF of the porcine coronary circulation has been bioassayed and tentatively identified as an epoxyeicosatrienoic acid (Hecker et al., 1994; Pöpp et al., 1996). In addition, a small increase in extracellular K+ has been tentatively identified as mediating endothelium-dependent hyperpolarization in rat mesenteric and hepatic arteries (Edwards et al., 1998). However, the EDHF of other vascular beds has not been convincingly bioassayed and the pharmacology in other arteries is dissimilar leading to speculation about the nature of the hyperpolarizing factor (Martin et al., 1992; Cohen et al., 1997).
We have utilized the eNOS (−/−) mouse as a way of studying non NO-induced vasodilation without recourse to NOS inhibitors. Some of the cardiovascular consequences of disruption of the endothelial NOS gene have been studied previously (Huang et al., 1995; Meng et al., 1996; Shesely et al, 1996; Faraci et al., 1998). Chataigneau et al. (1999) reported no difference in the maximal contraction of a variety of arteries to 60 mM KCl between eNOS (+/+) and (−/−) animals; however, there was an enhanced vasoconstrictor effect to noradrenaline and U46619 in the mesenteric and coronary arteries of eNOS (−/−) animals. Our results are similar and we also report an enhanced maximum response and sensitivity to phenylephrine in the eNOS (−/−) aortic and femoral artery tissues when compared to tissues from the eNOS (+/+) animals. Faraci (1998) reported a supersensitivity to sodium nitroprusside in the carotid artery of eNOS (−/−) mice and the present results confirm and extend this observation by demonstrating an increase in sensitivity to not only sodium nitroprusside but also to exogenous NO. Supersensitivity to NO (by a factor of 6.5 in the present study) exists in the eNOS (−/−) mice as one might expect if the basal level of endogenous guanylyl cyclase stimulating activity is decreased as was concluded by Moncada et al. (1990) using chronic treatments with NOS inhibitors. However in our study, we report an even greater increase in sensitivity to sodium nitroprusside (48) than to NO (6.5). One explanation we can provide for this result is that tissue sensitivity to NO may be reduced when the source is provided globally to the cell (as in the case of an NO solution) vs the local release from NO that might be expected from an NO donor such as nitroprusside. Another explanation may relate to the different redox states of NO that may predominate in a solution of NO gas vs that locally released by SNP (Goyal & Hee, 1998). Surprisingly, Chataigneau et al. (1999) did not find any significant difference in the hyperpolarization produced by SIN-1 in coronary artery cells from eNOS (−/−) vs (+/+) mice.
Acetylcholine induced relaxation of both femoral and mesenteric arteries isolated from wild type eNOS (+/+) mice. However, the responsible mechanisms varied between these vessels and in the mesenteric artery NO predominantly accounts for the vasodilation whereas in the femoral artery a non-NO, non-prostanoid hyperpolarizing factor also contributes. The mesenteric arteries from other species including rat, guinea-pig and rabbit release EDHF and we have demonstrated acetylcholine-induced vasorelaxation in mesenteric arteries from other strains of mice (C57BL6 and C57BL/KsJ, data not shown) under identical conditions that suggests that this lack of EDHF activity is strain-dependent. Although NO appears to be the predominant endothelium-derived vasorelaxant factor released from mesenteric vessels from eNOS (+/+) mice, it is of interest that vasorelaxation response to acetylcholine is inhibited in the presence of 30 mM KCl or TEA. These data suggest that NO mediates vasorelaxation in the mesenteric resistance vessels via the activation of a K+ channel(s). Further studies are required to determine the nature of the K+ channel activated by NO and whether this process is dependent on the activation of guanylyl cyclase. However, mesenteric vessels from eNOS (−/−) mice did demonstrate a hyperpolarizing factor with different properties from that produced in vessels from eNOS (+/+) mice, as these vessels were insensitive to combined NOS and cyclo-oxygenase inhibition but were sensitive to raising the concentration of K+ to 30 mM.
Unlike mesenteric arteries, femoral arteries demonstrated a non-NO, non-prostanoid relaxing factor in both eNOS (+/+) and eNOS (−/−) mice. In our studies of endothelium-dependent vasorelaxation in femoral vessels, we utilized a combination of NOS inhibitors in eNOS (+/+) mice to ensure complete block of NOS as others have found that a single NOS inhibitor is insufficient (Cohen et al., 1997). In addition, we determined the effect of the guanylyl cyclase inhibitor ODQ on acetylcholine-mediated vasorelaxation in eNOS (+/+) mice. ODQ produced a greater inhibition of vasorelaxation than that observed with the L-NOARG/L-NAME combination, thus indicating that NOS inhibitors alone may not be sufficient to completely inhibit the production of NO. These data are therefore in agreement with those of Cohen et al. (1997) which indicated that NO production could still be detected in rabbit carotid vessels after pre-treatment of the tissues with L-NAME and L-NOARG. Further, in femoral arteries of mice lacking endothelial NOS, a combination of L-NOARG and the inhibitor of soluble guanylyl cyclase, ODQ, had no effect on the acetylcholine-induced relaxation. The lack of effect of NOS inhibition on the relaxation in mesenteric and femoral arteries shows that other isoforms of NOS do not compensate for the lack of endothelial NOS derived NO in the mutant mice as is the situation in the pial arterioles of endothelial NOS (−/−) mice where neuronal (type I) NOS compensates for the lack of endothelial NOS activity (Meng et al., 1996).
Our results are in apparent disagreement with those of Chataigneau et al. (1999) who have recently described the complete loss of the endothelium-dependent vasorelaxation response to acetylcholine in aorta, coronary, carotid as well as mesenteric artery from eNOS (−/−) mice. We note that Chataigneau et al. (1999) did not study femoral vessels and, furthermore, the protocol pursued for the study of mesenteric vessels was different from that which we used. Thus, we investigated endothelium-dependent vasorelaxation in mesenteric arteries that were pressurized to 60 mmHg with drug effects determined by videomicroscopy following superfusion. It is known that the level of stretch applied to the vessel can affect the nature of the response to acetylcholine. For instance, Parkington et al. (1993) have reported that in stretched guinea-pig artery preparations, both a transient and a slow hyperpolarization response to acetylcholine was recorded, whereas in unstretched vessels only the transient hyperpolarization was recorded. Furthermore, Parkington et al. (1993) reported that in stretched, but not in unstretched vessels, a hyperpolarization response was recorded for both NO and prostacyclin. We also report that in the eNOS (+/+) mice, the acetylcholine-mediated vasorelaxation appears to be almost entirely mediated by an L-NOARG/indomethacin sensitive process but is also inhibited in the presence of 30 mM KCl. Thus, in the pressurized mesenteric artery from eNOS (+/+) mice, the acetylcholine-mediated vasorelaxation appears to be entirely mediated by NO/PGI2 and results presumably from K+ channel activation and hyperpolarization of the vascular smooth muscle cell. Of interest is that our data from femoral arteries obtained from eNOS (+/+) mice indicate a less important role for NO-mediated hyperpolarization and the contribution of a non-NO/PGI2 mediator which has the expected properties of a distinct EDHF. Further studies are clearly warranted in order to better understand the cellular processes that determine and regulate the release and the cellular effects of NO vs PGI2 vs EDHF.
The interpretation of gene knockout experiments has been criticized when used to infer that the product of the gene in question is unimportant when the mutant mouse has a similar phenotype as the control as compensatory changes may occur in the mutant animal (Becker et al., 1996). However, just such a compensatory role has been speculated for EDHF. It has been proposed that EDHF acts as a backup to NO in pathophysiological states (e.g. pulmonary hypertension; Kemp et al., 1995). In the present study, it is clear that an EDHF is upregulated in the mesenteric artery of eNOS knockout mice.
In conclusion, these results show that EDHF is not NO, and that EDHF acts in a compensatory manner in some vessels when endothelial NOS is absent.
Acknowledgments
We are very grateful to Dr Paul Huang of the Cardiovascular Research Centre, Massachusetts General Hospital for supplying the breeding pairs of both control and eNOS mice which were used during the study, and also for the discussions regarding our results. The authors would like to thank Dr Michael Hickey, Immunology Research Group, University of Calgary for his help with the mice. This work was supported by the Canadian MRC.
Abbreviations
- EDHF
endothelium-derived hyperpolarizing factor
- eNOS
endothelial nitric oxide synthase
- eNOS (−/−) mice
endothelial nitric oxide synthase knock-out mice
- eNOS (+/+) mice
wild-type (C57BL/SV19) mice
- L-NAME
NG-nitro-L-arginine methyl ester
- L-NOARG
NG-nitro-L-arginine
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- TEA
tetraethylammonium
References
- BECKER K.D., GOTTSHALL K.R., CHIEN K.R. Strategies for studying cardiovascular phenotypes in genetically manipulated mice. Hypertension. 1996;27:495–501. doi: 10.1161/01.hyp.27.3.495. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FELETOU M., HUANG P.L., FISHMAN M.C., DUHAULT J., VANHOUTTE P.M. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br. J. Pharmacol. 1999;126:219–226. doi: 10.1038/sj.bjp.0702300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKY T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES P.F., OLESEN S-P., CLAPHAM D.E., MORREL E.M., SCHOEN F.J. Endothelial communication. State of the art lecture. Hypertension. 1988;11:563–572. doi: 10.1161/01.hyp.11.6.563.a. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTEON A.H. K+ is an endotheluim-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FARACI F.M., SIGMUND C.D., SHESELY E.G., MAEDA N., HEISTAD D.D. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am. J. Physiol. 1998;274:H564–H570. doi: 10.1152/ajpheart.1998.274.2.H564. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-derived hyperpolarizing factor. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GOYAL R.K., HE X.D. Evidence for NO• redox form of nitric oxide as nitrergic inhibitory neurotransmitter in gut. Am. J. Physiol. 1998;275:G1185–G1192. doi: 10.1152/ajpgi.1998.275.5.G1185. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG P.L., HUANG Z., MASHIMO H., BLOCH K.D., MOSKOWITZ M.A., BEVAN J.A., FISHMAN M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- KEMP B.K., SMOLICH J.J., RITCHIE B.C., COCKS T.M. Endothelium-dependent relaxations in sheep pulmonary arteries and veins: resistance to block by NG-nitro-L-arginine in pulmonary hypertension. Br. J. Pharmacol. 1995;116:2457–2467. doi: 10.1111/j.1476-5381.1995.tb15096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN G.R., BOLOFO M.L., GILES H. Inhibition of endothelium-dependent vasorelaxation by arginine analogues: a pharmacological analysis of agonist and tissue dependence. Br. J. Pharmacol. 1992;105:643–652. doi: 10.1111/j.1476-5381.1992.tb09033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENG W., MA J., AYATA C., HARA H., HUANG P.L., FISHMAN M.C., MOSKOWITZ M.A. Acetylcholine dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am. J. Physiol. 1996;271:H1145–H1150. doi: 10.1152/ajpheart.1996.271.3.H1145. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MONCADA S., REES D.D., SCHULZ R., PALMER R.M.J. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 1990;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- PARKINGTON H.C., TARE M., TONTA M.A., COLEMAN H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PÖPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHESELY E.G., MAEDA N., KIM H-S., DESAI K.M., KREGE J.H., LAUBACH V.E., SHERMAN P.A., SESSA W.C., SMITHIES O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarisation and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]