

Abstract
Rat proteinase-activated receptor-2 (PAR2) variants were stably expressed in rat KNRK cells: (a) wild-type (wt)–PAR2; (b) PAR2PRR, with the extracellular loop 2 (EL-2) sequence P231E232E233mutated to PRR and (c) PAR2NET, with the EL-2 sequence, PEEV changed to NETL. Cell lines were evaluated for their sensitivity (calcium signalling) towards trypsin and the receptor-activating peptides, SLIGRL-NH2, SLIGEL-NH2, trans-cinnamoyl(tc)-LIGRLO-NH2, and SFLLR-NH2.
SLIGEL-NH2 exhibited low potency (1 : 200 relative to SLIGRL-NH2) in wild-type PAR2. Its activity was increased 5 fold in PAR2PRR, but it was inactive in PAR2NET.
In PAR2PRR, the potencies of SLIGRL-NH2, tc-LIGRLO-NH2, and SFLLR-NH2 were decreased by 80–100 fold. But, the potency of trypsin was decreased by only 7 fold.
In PAR2NET, highly homologous in EL-2 with proteinase-activated receptor-1 (PAR1), the potency of the PAR1-derived peptide, SFLLR-NH2, was reduced by 100 fold compared with wt-PAR2, whereas the potency of the PAR2-derived AP, SLIGRL-NH2 was reduced 10 fold. In contrast, the potency of trypsin in PAR2NET was almost the same as in wt-PAR2.
We conclude that the acidic EL-2 tripeptide, PEE, in PAR2 plays an important role in governing agonist activity.
The data obtained with the PEEV→NETL mutation suggested: (a) that SLIGRL-NH2 and SFLLR-NH2 interact in a distinct manner with PAR2 and (b) that SFLLR-NH2 may interact differently with PAR2 than it does with PAR1.
The differential reductions in the potencies of SLIGRL-NH2, compared with trypsin in the PAR2PRR and PAR2NET cell lines point to differences between the interactions of the trypsin-revealed tethered ligand and the free receptor-activating peptide with PAR2.
Keywords: Trypsin, proteinase, PAR, proteinase-activated receptor, PAR1, PAR2
Introduction
Proteinases such as thrombin, trypsin and tryptase are now known to regulate target tissues via the activation of proteinase-specific cell surface G-protein-coupled-receptors (Vu et al., 1991; Rasmussen et al., 1991; Nystedt et al., 1994; Ishihara et al., 1997; Xu et al., 1998; Kahn et al., 1998; Dery et al., 1998). The novel mechanism whereby the proteinase-activated receptors (PAR1, PAR3 and PAR4 activated by thrombin; PAR2 activated by trypsin and tryptase) are triggered involves the proteolytic unmasking of a nascent N-terminal receptor-activating sequence that acts as a tethered ligand (Vu et al., 1991; Chen et al., 1994). For three of the four PARs cloned to date, synthetic receptor-activating peptides (PAR-APs) based on the proteolytically revealed tethered ligand are able to act as surrogate receptor agonists, so as to mimic the effects of protease-mediated receptor activation. In our own work, in keeping with the results of others, we have explored the structure-activity relationships (SARs) for short (four to seven amino acids) receptor-activating peptides based on the tethered ligand sequences present in PAR1 and PAR2 (Scarborough et al., 1992; Vassallo et al., 1992; Hollenberg et al., 1992; 1993; 1996; 1997; Blackhart et al., 1996; Chao et al., 1992; Hui et al., 1992; Natarajan et al., 1995; Kawabata et al., 1999). Such SAR studies of the PAR-APs have pointed out the importance of the arginine residue at position 5 of a PAR1-AP such as SFLLR-NH2 (Hollenberg et al., 1996; Natarajan et al., 1995). In contrast, the substitution of an amino acid with an acidic side chain (e.g. glutamic acid) at position 5 of a PAR1-AP (e.g. SFLLEN-NH2) has been found to reduce peptide potency for activating PAR1 by at least two orders of magnitude (Natarajan et al., 1995; Nanevicz et al., 1995). Although derived from the tethered ligand of PAR1, SFLLR-NH2 is also a relatively potent activator of PAR2 (Hollenberg et al., 1997; Blackhart et al., 1996; Kawabata et al., 1999). Nonetheless, the PAR2 derived activating peptide, SLIGRL-NH2, is unable to activate PAR1. As discussed below, extracellular loop 2 is believed to play an important role in governing the selectivity of PAR1 for PAR-APs (Lerner et al., 1996).
As a counterpart to studies of the SARs for the activity of PAR-APs, other work has focused attention on the roles that the extracellular receptor domains of PAR1 and PAR2 play in determining agonist specificity (Nanevicz et al., 1995; Lerner et al., 1996; Gerszten et al., 1994). In particular, the extracellular loop 2 of PAR1 and PAR2 has been singled out as being a primary determinant of peptide agonist specificity (Nanevicz et al., 1995; Lerner et al., 1996). This receptor domain contains a short sequence (CHD) which is conserved amongst all PARs that have been cloned to date; and there is a strikingly high degree of homology between PAR1 and PAR2 for a stretch of about ten amino acid residues (Figure 1).
Figure 1.
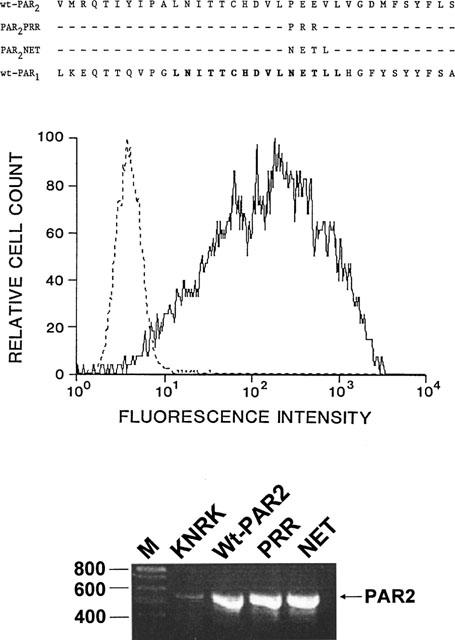
Sequences of PAR2 variants compared with PAR2 and PAR1 (upper panel) and their expression in KNRK cells (middle and lower panel). (Upper panel) The sequences of the extracellular loop 2 of wtPAR2 (PAR2) and the two receptor variants (PAR2PRR, PAR2NET) are compared with the extracellular loop 2 sequence of rat PAR1. The bold print of the PAR1 sequence denotes the region of high sequence homology between PAR1 and PAR2. (Middle panel) Cellular fluorescence intensity detected with the B5 anti-PAR2 antiserum was monitored by fluorescence-activated cell sorting for untransfected KNRK cells (dashed lines; same signal was observed for vector-transfected cells) or for wt-PAR2-expressing cells (solid lines). (Lower panel) The expression of receptor mRNAs for the host wild-type KNRK cells, (Wt-PAR2), PAR2PRR (PRR) and PAR2NET (NET) were determined by RT–PCR. The RT–PCR signal observed for actin was the same in all cell lines (not shown). The positions of the oligonucleotide size markers (in base-pairs) are shown on the left (lane M). The position of the expected PCR product for PAR2 (560 base-pairs) is shown on the right. Identical amounts of RNA obtained from each cell line were subjected to analysis by RT–PCR, as outlined in Methods.
In this extracellular loop 2 sequence, there is a conserved glutamic acid residue (E232 in rat PAR2; E260 in human PAR1) present in PAR1 and PAR2. In human PAR1, E260 (equivalent to E267 in rat PAR1) has been identified as potentially playing either a direct role in agonist binding or a regulatory function in modulating agonist access to a receptor docking site (Nanevicz et al., 1995). In the human PAR1 thrombin receptor, low activity was observed for the PAR1AP analogue, SFLLEN-NH2, wherein an acidic side chain was substituted for the basic arginine side chain at position 5 in the parent agonist, SFLLRN-NH2 (Nanevicz et al., 1995). However, functional complementation was achieved to enhance substantially the activity of SFLLEN-NH2 by a single arginine substitution (E260R) in the extracellular loop 2 of human PAR1. The Activity of the PAR1-derived peptide, SFLLRN-NH2, in the E260R human thrombin PAR1 receptor mutant was only slightly lower than in the wild-type receptor (Nanevicz et al., 1995). Since E260 in the human PAR1 receptor is equivalent to E232 in rat PAR2, we wondered if this amino acid residue might play a comparable role in affecting the activity of PAR2APs. We therefore synthesized and tested the activity of the PAR2AP analogue, SLIGEL-NH2, with a substitution of an acidic residue for a basic arginine side chain at position 5 of the parent PAR2AP (SLIGRL-NH2). Further, we prepared two rat PAR2 mutants, one of which had a switch of acidic to basic residues (PAR2PRR: E232E233→R232R233: Figure 1) in extracellular loop 2 and one of which was changed so that this amino acid portion of the PAR2 extracellular loop 2 would have exactly the same sequence as human PAR1 (PAR2NET: P231E232E233V234→N231E232T233L234). The second substitution provides for an additional potential glycosylation site in the extracellular loop 2 of PAR2. The activity of a number of PAR-APs as well as trypsin were assessed using a calcium signalling assay for the wild-type rat PAR2 (wt-PAR2) and the two receptor mutants (PEE→PRR; PEEV→NETL) expressed in KNRK cells.
In our experiments, we were interested in two main questions: (1) would the PAR2PRR receptor be more sensitive to the PAR2AP, SLIGEL-NH2, compared with wt-PAR2? and (2) would the PAR2NET receptor, having an increased stretch of amino acid sequence homology with the extracellular loop 2 of PAR1 have an increased sensitivity towards the PAR1AP, SFLLR-NH2?
Methods
Preparation of transfected KNRK cell lines
Rat PAR2 (Saifeddine et al., 1996) was cloned into the pcDNA3 mammalian expression vector (In Vitrogen, San Diego, CA, U.S.A.) and the ‘wild-type' and mutated receptors were expressed in Kirsten virus-transformed rat kidney cells (KNRK, American Tissue Type Culture Collection, Bethesda, MD, U.S.A.). The receptor mutants, PAR2PRR and PAR2NET were prepared by the overlapping PCR approach wherein P231E232E233 in rat PAR2 were changed to P231R232R233 and N231E232T233L234 respectively. In the background nontransfected KNRK cell line, PAR2 can be detected by a polymerase chain reaction (PCR) approach, but insufficient receptor is expressed in the nontransfected cells to yield an appreciable intracellular calcium signal in response to high concentrations of either trypsin or PAR2APs (Böhm et al., 1996; Al-Ani et al., 1999). Nor are cell surface PAR2 receptors detectable by immunofluorescence, using a PAR2-targeted receptor antibody (Al-Ani et al., 1999). Cells were transfected using the Lipofectamine® method according to the manufacturer's instructions (Gibco BRL, Gaithersburg, MD, U.S.A.) with 5 μg of each construct used per KNRK cell monolayer (60-mm2 flask, 60% confluent). Transfected cells were subcloned in geneticin (0.6 mg ml−1)-containing medium and receptor-bearing cells were isolated with the use of the anti-receptor B5 antibody (Kong et al., 1997) and with fluorescence-activated cell sorting to yield three permanent cell lines: wild-type KNRK-PAR2 (wt-PAR2), PAR2PRR and PAR2NET, as illustrated in Figure 1. The cell lines were routinely propagated in geneticin (0.6 mg ml−1) containing Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% (v v−1) foetal calf serum, using 80 cm2 plastic T-flasks. Cells were subcultured by resuspension in calcium-free isotonic saline/EDTA solution, without the use of trypsin. Background KNRK cells were similarly grown in geneticin-free medium. A KNRK cell line transfected with an ‘empty' pcDNA3 vector was also subcloned and grown in geneticin-containing medium, as for the other transfected cell lines. The B5 anti-receptor antibody, used in previous work (Kong et al., 1997) was raised in rabbits using a peptide corresponding to rat PAR2 (G30PNSKGRSLIGRLDTP46-YGGC) coupled to keyhole limpet hemocyanin (YCGGC added for conjugation). Cyanine 3-coupled goat anti-rabbit IgG (Cedarlane Laboratory Hornby, ON, Canada) was used to detect cell-surface bound B5 anti-receptor antibody. To assess the presence of increased PAR2 mRNA in the transfected cell lines, total RNA was prepared from confluent T-flasks using the TRI® reagent (Molecular Research Center, Cincinnati, OH, U.S.A.). The RNA was reverse-transcribed (RT) with a first strand cDNA synthesis kit using pd(N)6 primer (Pharmacia LKB Biotechnology, Uppsala, Sweden) according to manufacturer's recommendations at 37°C for 60 min; 3 μl of this solution was used with primer pairs targeted to (1) Rat PAR2 forward primer, PAR2F: 5′- CACCACCTGTCACGATGTGCT-3′ and reverse primer, 5′- CCCGGGCTCAGTAGGAGGTTTTAACAC-3′, and (2) to actin: forward primer, 5′- CGTGGGCCGCCCTAGGCACCA-3′ and reverse primer, 5′- TTGGCCTTAGGGTTCAGGGGG-3′. The detection of an intron-free 243 base pair product using this actin primer pair which spans an intron in genomic DNA, can confirm the absence of DNA-derived intron sequences in the RT product obtained from the cell-derived DNA preparation (Watson et al., 1992). Routinely, amplification was done using 2.5 units of Taq DNA polymerase (Promega, Madison, WI, U.S.A.) in a 10 mM Tris-HCl buffer, pH 9.0 (50 μl, final vol.) Containing 1.5 mM MgCl2, 50 mM KCl, 0.1% v v−1 triton X-100 and 0.2 mM each of deoxynucleotide triphosphates. Amplification was allowed to proceed for 25 cycles beginning with a 1 min denaturing period at 94°C, followd by a 1 min reannealing time at 55°C and a primer extension period of 1 min at 72°C. The PCR products were separated by 1.5% agarose gel electrophoresis and visualized with ethidium bromide. A 560 base-pair product yielded by the PAR2 amplimers has been previously documented by sequence analysis to represent rat PAR2 (Saifeddine et al., 1996). For each transfected cell line, a population isolated by fluorescence-activated cell sorting for subculturing was obtained from a fraction in which >95% of the population was found to exhibit cell surface fluorescence using the B5 anti-receptor antibody. For each cell line so isolated, the approximate number of anti-receptor antibody binding sites per cell was estimated fluorometrically with the use of Quantum Simply Cellular® microbeads (Flow Cytometry Standards Corp, San Juan, PR, U.S.A.) according to the manufacturer's instructions, and in keeping with previous work (Lopez et al., 1992; Zagursky et al., 1995). However, our polyclonal rabbit B5 anti-receptor antibody was used instead of a monoclonal mouse anti-receptor antibody, as described in previously published work (Lopez et al., 1992; Zagursky et al., 1995).
Measurements of calcium signalling using fluorescence emission
Cells to be used of measurements of peptide-stimulated fluorescence emission (reflecting an increase in intracellular calcium) were grown to about 85% confluency in 80 cm2 T-flasks and were disaggregated with calcium-free isotonic phosphate-buffered saline containing 0.2 mM EDTA. Disaggregated cells were pelleted by centrifugation and were resuspended in 1 ml DMEM/10% FCS for loading with the intracellular calcium indicator, Fluo-3 (Molecular Probes Inc., Eugene, OR, U.S.A.) at a final concentration of 22 μM (25 μg ml−1) of fluo-3AM ester. Indicator uptake was established over 20–25 min at room temperature in the presence of 0.25 mM sulphinpyrazone, after which time cells were washed two times by centrifugation and resuspension with the buffer described below, to remove excess dye. Fluo-3-loaded cells were then resuspended to yield a stock solution (about 6×106 cells ml−1) in a buffer of the following composition (mM): NaCl 150, KCl 3, CaCl2 1.5, HEPES 20, glucose 10 and sulphinpyrazone 0.25. Fluorescence measurements, reflecting elevations of intracellular calcium, were conducted at 24°C using a Perkin-Elmer fluorescence spectrometer, with an excitation wavelength of 480 nm and an emission recorded at 530 nm. Cell suspensions (about 2 ml of approx. 3×105 cells ml−1) were maintained in suspension with a stirred (magnetic flea bar) thermostatted cuvette (total volume, 4 ml) and peptide stock solutions were added to monitor peptide-induced changes in fluorescence. To construct concentration-response curves for fluorescence yield, the signals caused by the addition of test peptides were expressed as a percentage (%A23187) of the fluorescence peak height yielded by replicate cell suspensions when treated with 2 μM of the ionophore A23187 (Sigma, St. Louis, MD, U.S.A.). This concentration of A23187 was at the plateau of its concentration-response curve for a fluorescence response. Under the assay conditions, the addition of proteinase inhibitors (e.g. Amastatin) did not potentiate or diminish the fluorescence response caused by the PAR-APs. Thus, routinely, proteinase inhibitors were not added to the assay cuvettes. The relative potencies of the PAR-APs studied, SLIGRL-NH2, trans-cinnamoyl-LIGRLO-NH2, SLIGEL-NH2, SFLLR-NH2, were expressed in two ways: (1) relative to the activity of the parent tethered ligand, SLIGRL-NH2, in each of the three receptor constructs examined (wt-PAR2, PAR2PRR; PAR2NET) and (2) for each peptide agonist acting in the mutant receptors, relative to their activity in the wild-type KNRK-PAR2 cell line. Activity ratios for the PAR-AP peptides REC,SL-NH2 in each of the receptor systems were determined as outlined previously (Hollenberg et al., 1997) by using several points along the linear portions of the concentration-effect curves (e.g. Figures 3, 4 and 5) to estimate for each agonist, an average concentration ratio (REC,SL-NH2=ECpeptide÷ECSL-NH2) related to a concentration of SLIGRL-NH2 that caused the same calcium signal response as did the peptide studied. REC,SLNH2 values greater than unity denote peptides that were lower in potency than SLIGRL-NH2. Similarly an activity ratio (REC,PAR2) for an individual PAR-AP as well as trypsin in each of the three expressed receptor systems was calculated using several points along the calcium signalling concentration-effect curves to estimate for each of the two receptor mutants, an average peptide concentration ratio (REC,PAR2=ECmutant÷ECPAR2) related to a concentration of agonist that in the PAR2 mutant receptor caused a calcium signal equivalent to that observed in the wild-type receptor. Values of REC,PAR2 greater than unity denote a receptor that is less sensitive to the tested agonist.
Figure 3.
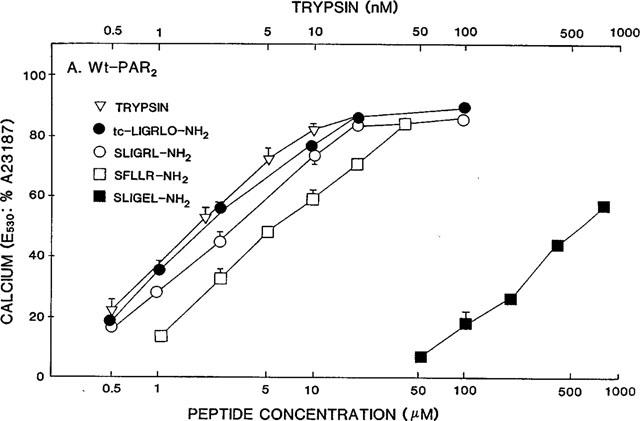
Concentration-effect curves for PAR-APs and trypsin in wt-PAR2 cells. The fluorescence responses relative to the signal caused in identical cell suspensions by 2 μM A23187 (E530: %A23187) were measured in replicate cell suspensions, as outlined in Methods, for increasing concentrations of the indicated PAR-APs and trypsin. Values represent the averages (±s.e.mean, bars) for measurements done with four or more replicate cell suspensions coming from two or more independently grown crops of cells. Error bars smaller than the symbols are not shown.
Figure 4.
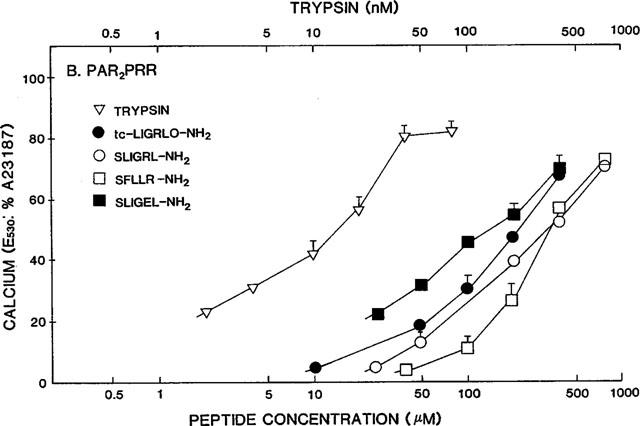
Concentration effect curves for PAR-APs and trypsin in PAR2PRR cells. The fluorescence responses relative to the signal caused by 2 μM A23187 (E530: %A23187) were measured for increasing concentrations of the indicated PAR-APs and trypsin, exactly as described in the legend to Figure 3.
Figure 5.
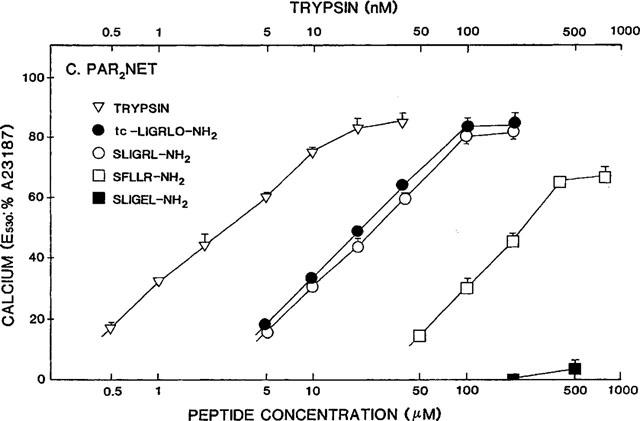
Concentration-effect curves for PAR-APs and trypsin in PAR2-NET cells. Concentration-effect curves for the fluorescence response relative to 2 μM A23187 (E530: %A23187) were obtained for the same PAR-APs and trypsin, as outlined in the legends to Figures 3 and 4.
Peptides and other reagents
All peptides were synthesized by solid phase methods at the peptide synthesis Facility, University of Calgary, Faculty of Medicine (Calgary AB, Canada) (Director, Dr Denis McMaster), or were provided through the courtesy of Dr L. Leblond, via the peptide synthesis Facility at BioChem Therapeutic, Laval PQ, Canada. The composition and purity of all peptides were confirmed by HPLC analysis, mass spectral analysis and quantitative amino acid analysis. Stock solutions, prepared in 25 mM HEPES buffer, pH 7.4 were standardized by quantitative amino acid analysis to verify peptide concentration and purity. Porcine trypsin (14,900 U mg−1, Cat. No. T7418) was from Sigma (St. Louis, MO, U.S.A.). A maximum specific activity of 20,000 U mg−1 was used to calculate the approximate molar concentration of trypsin in the incubation medium.
Results
Receptor expression and comparison of agonist potencies
Using the fluorescence-activated cell sorting approach to obtain receptor-expressing KNRK cell lines and estimate receptor density, we observed, with the B5 anti-receptor antibody, that there was a comparable abundance of cell surface receptors (about 75,000 sites cell−1) in the three cell lines, designated wt-PAR2 (wild-type receptor), PAR2PRR (PEE→PRR mutation), and PAR2NET (PEEV→NETL mutation). In the vector-transfected KNRK cells, and in the background KNRK cells, no fluorescence above that observed with non-immune serum was detected with the use of the B5 anti-receptor antibody (Figure 1, middle panel; and see Al-Ani et al., 1999). In all receptor transfected cell lines, there was a marked shift to the right in the fluorescence intensity curve, using the B5 anti-receptor antibody probe (solid tracing, Figure 1, middle panel). The sequences confirmed for the extracellular loop 2 domains of the three receptor clones are shown in Figure 1 (upper panel), along with the sequence of rat PAR1. A comparable abundance of receptor RNA was found for each receptor cell line using an RT–PCR approach (Figure 1, lower panel). Under the same conditions, the background KNRK cells yielded a very low PCR signal for PAR2 (Figure 1, lower panel). The PCR signal obtained for actin (not shown) was comparable for all cell lines, for which the PAR2 signals are shown in Figure 1 (lower panel). All three receptor-bearing cell lines yielded a calcium signal in response either to trypsin (20–100 nM) or to the PAR2AP, SLIGRL-NH2 (see below). The non-transfected KNRK cells and the vector-transfected KNRK cells did not yield a calcium signal at concentrations of trypsin (20 nM) and SLIGRL-NH2 (50 μM) that were at the plateau for the concentration-effect curves for wt-PAR2 (not shown and see Al-Ani et al., 1999). Thrombin (100 nM) was similarly unable to cause a calcium signal in the non-transfected or vector-transfected cells (not shown).
Since the three receptor-bearing cell lines were observed to have comparable receptor densities according to the fluorescence signal yielded by the B5 anti-receptor antibody, it was possible, as outlined in Methods, to compare the relative potencies of the agonists we tested in two ways: (1) In an individual cell line, the activity of each agonist could be measured relative to the action of the tethered ligand-derived PAR2AP, SLIGRL-NH2 (REC,SL-NH2: Table 1); and (2) for an individual agonist, an activity could be determined in the two mutant receptor cell lines, relative to its activity in the wild-type PAR2KNRK cell line (REC,PAR2: Table 2). The relative activities were determined according to the relative concentrations of agonists that in each cell suspension caused an equivalent calcium signal, normalized to the signal generated by the addition of the ionophore, A23187, as summarized in Tables 1 and 2. Typical comparative responses of the three cell lines (calcium signals) to the PAR2APs, SLIGRL-NH2 and SLIGEL-NH2 are shown in Figure 2. In Figure 2, the concentrations of peptides were selected so as to cause a comparable calcium signal, relative to the ionophore A23187, in each of the cell lines. The PAR2NET cell line responded poorly to SLIGEL-NH2, even at high concentrations (Figures 2f and 5).
Table 1.
Agonist activitiesa relative to SLIGRL-NH2

Table 2.
Relative receptor activitiesa for each agonist

Figure 2.
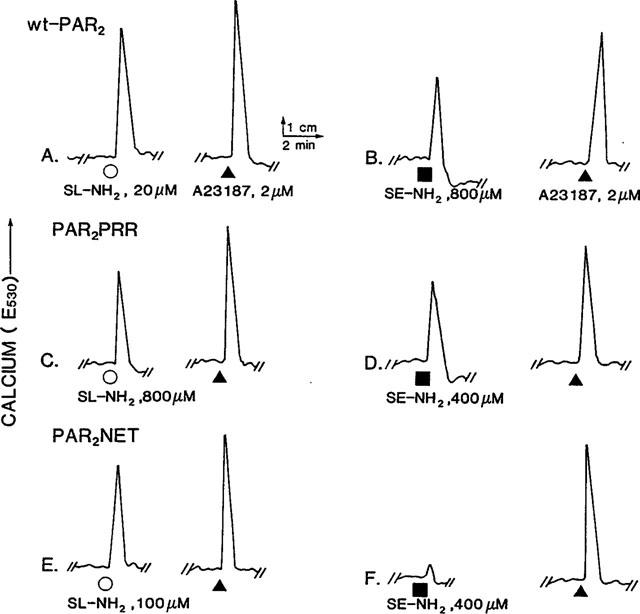
Calcium signalling by wt-PAR2 (a, b), PAR2PRR (c, d) and PAR2NET (e, f). Comparative responses to SLIGRL-NH2 (SL-NH2, ○) and SLIGEL-NH2 (SE-NH2, ▪) [fluorescence (E530), reflecting increases in intracellular calcium] were monitored in fluo-3-loaded cell lines stimulated by either SLIGRL-NH2 (a, c, e) or SLIGEL-NH2 (b, d, f). The concentrations of peptide agonists were adjusted in an attempt to show comparable increases in fluorescence, relative to the signal yielded in each cell sample by 2 μM of the ionophore A23187 (▴). (a, b) wt-PAR2; (c, d) PAR2PRR; (e, f) PAR2NET. The PAR2NET cell line responded poorly to relatively high concentrations of SLIGEL-NH2 (f and Figure 5).
Reduced activity of SLIGEL-NH2, partially compensated by substituting PRR for PEE in PAR2
In wt-PAR2, the PAR2AP analogue, SLIGEL-NH2 was over two orders of magnitude less potent than the parent PAR2AP, SLIGRL-NH2, that has a basic instead of an acidic side chain at the fifth position corresponding to the revealed PAR2 tethered ligand (Figure 3 and Table 1). When the two acidic residues in the P231EE sequence of the extracellular loop of PAR2 were changed to arginines (PAR2PRR), the potency of the PAR2AP analogue, SLIGEL-NH2, in PAR2PRR, was increased about 5 fold, relative to its potency in wild-type-PAR2 (compare Figures 3 and 4 and see Table 2). In contrast, the potency of all other peptide agonists was reduced 80 to 100 fold, relative to their activities in wild-type PAR2 (compare Figures 3 and 4 and see Table 2). Nonetheless, in the mutant PAR2PRR receptor, the potencies relative to SLIGRL-NH2 of the PAR1-derived agonist, SFLLR-NH2, and the PAR2-derived agonist, trans-cinnamoyl-LIGRLO-NH2 were equivalent to their relative potencies in wild-type PAR2 (Table 1). In contrast, relative to the activity of SLIGRL-NH2, SLIGEL-NH2 was over twice as potent in the PAR2PRR receptor, whereas it displayed a 180 fold lower potency than SLIGRL-NH2 in the wild-type receptor (Figures 3 and 4 and see Table 1).
Differential changes in the potencies of trypsin and SLIGRL-NH2 in PAR2PRR
Surprisingly, on a molar basis, the activity of trypsin, which liberates the tethered PAR2 ligand, SLIGRLDTP., was reduced by only about 8 fold in PAR2PRR compared with the wild-type receptor (Table 2). This small reduction in potency contrasted with the 110 fold reduction in the potency of the PAR2AP, SLIGRL-NH2, derived from the tethered ligand sequence itself (Table 2). Thus, expressed relative to the potency of the free peptide, SLIGRL-NH2, trypsin appeared to be almost an order of magnitude more effective in PAR2PRR than it was relative to SLIGRL-NH2 in the wild-type receptor (For trypsin, 103×REC,SL-NH2≈percnt;0.04 in PAR2PRR and ≈percnt;0.6 in wt-PAR2: Table 1). Thus, the PEE→PRR mutation in extracellular loop 2 markedly reduced the potency of all PAR-APs tested that had a basic side chain at position 5, but did not, in the same proportion, appear to affect the activity of the tethered ligand revealed by trypsin. It is to be noted that the tethered ligand also has a basic side chain at position 5.
Relative activities of agonists in PAR2NET: reduced activity of SFLLR-NH2 and lack of effect for trypsin-mediated activation
In the PEEV→NETL mutant receptor (PAR2NET), which has a 15 amino acid sequence stretch identical to PAR1 (Figure 1), both the PAR2AP, SLIGRL-NH2, and the PAR1AP, SFLLR-NH2 displayed lower potencies than in the wild-type receptor (compare Figures 3 and 5, and see Tables 1 and 2). Compared with wt-PAR2, the reduction in the potency of the PAR1AP, SFLLR-NH2 in the PAR2NET receptor was more pronounced (about 40 fold lower, Table 2) than the reduction observed for the PAR2APs, SLIGRL-NH2 and trans-cinnamoyl-LIGRLO-NH2 (about a 10 fold reduction: Table 2). The result of this differential shift in the potencies of SFLLR-NH2 and SLIGRL-NH2 was that the PAR1AP was about 10 fold less potent than the PAR2AP, in PAR2NET, whereas these peptides had comparable potencies in either wild-type PAR2 or the mutant PAR2PRR receptor (Table 1). The PAR2AP analogue with the acidic side chain, SLIGEL-NH2 was essentially inactive in PAR2NET (Figure 5). Significantly, although the potency of the PAR2AP, SLIGRL-NH2 was reduced by about 10 fold in PAR2NET, the potency of trypsin in PAR2NET was close to that in wt-PAR2 itself (Table 2). Thus, as was the case for the PRR receptor mutant, the potency of the free peptide in PAR2NET appeared to be much more affected than the potency of the trypsin-revealed tethered ligand (for trypsin, relative to SLIGRL-NH2 in PAR2NET: 103× REC,SL-NH2=0.10, compared with 0.60 in wt-PAR2, Table 1).
In summary, in PAR2NET, there was a differential shift in the potencies of the PAR-APs, relative to the activity of trypsin. As indicated in Table 1, the relative orders of potencies for all agonists in the three cell lines were: (a) for wt-PAR2: trypsin>> tc-LIGRLO-NH2⩾SLIGRL- NH2>SFLLR-NH2 >>SLIGEL-NH2, (b) for PAR2PRR: trypsin>>>SLIGEL-NH2>tc-LIGRLO-NH2⩾SLIGRL-NH2⩾SFLLR-NH2, and (c) for PAR2NET: trypsin>>>tc-LIGRLO-NH2⩾SLIGRL-NH2>>SFLLR-NH2>>>>SLIGEL-NH2.
Discussion
Complementation of SLIGEL-NH2 activity in the PAR2PRR mutant
One main finding of our study was that the PAR2PRR receptor did exhibit an increased sensitivity towards the PAR2AP, SLIGEL-NH2 compared with wt-PAR2. Previous work (Nanevicz et al., 1995; Lerner et al., 1996) had highlighted the importance in PAR1 and PAR2 of extracellular loop 2 (EL-2) as a key determinant of peptide agonist specificity. Further, the negatively-charged glutamic acid residue at position 260 in the EL-2 of human PAR1 was found to play a governing role for the activity of the PAR1APs, SFLLRN-NH2 and SFLLEN-NH2, in that the E260R mutant of PAR1 displayed a reduced (4 fold) sensitivity towards SFLLRN-NH2, that has a basic side chain, and a substantially increased (about 100 fold) sensitivity towards SFLLEN-NH2, that has an acidic side chain (Nanevicz et al., 1995). Qualitatively, our results with PAR2PRR mirrored the data obtained previously with the human PAR1E260R mutant, in that compared with the wild-type PAR2 receptor, the rat PAR2PRR mutant showed about a 5 fold increased sensitivity for SLIGEL-NH2. Like the PAR1AP, SFLLEN-NH2, the PAR2AP, SLIGEL-NH2 has an acidic side chain (Table 2). Our results were thus consistent with our working hypothesis that the P231EE sequence in the EL-2 of PAR2 plays an agonist docking or access role, akin to that proposed for the N259ET sequence in extracellular loop 2 of human PAR1 (Nanevicz et al., 1995). That said, the complementation of the low activity of SLIGEL-NH2 in the wild-type receptor caused by the substitution of basic for acidic side chains in the PAR2PRR receptor mutant was quite modest (about a 5 fold increase in potency) and was much less impressive than the large increase in the potency (about 100 fold) of SFLLEN-NH2 caused by the E260R mutation in human PAR1 (Nanevicz et al., 1995). Further, the PAR2PRR mutant, relative to wild-type PAR2, exhibited a much more marked reduction (about 100 fold) in sensitivity towards SLIGRL-NH2 than would have been expected from the data obtained with the E260R mutant of PAR1, where there was only a small (3 fold) reduction in sensitivity towards the PAR1AP, SFLLRN-NH2 (Nanevicz et al., 1995). Moreover, the PAR2PRR mutant was slightly less able to discriminate between SFLLR-NH2 and SLIGRL-NH2 than was the wild-type receptor (REC,SL-NH2=1.9 in wt-PAR2, compared with 1.4 in PAR2PRR: Table 1). These quantitative differences between the results obtained with the ‘acidic' PAR-APs (containing glutamic acid) acting on the PAR2 and PAR1 receptor mutants containing E→R substitutions suggest that the mechanism of docking of the ligands with extracellular loop 2 of the PAR1 and PAR2 receptors may differ considerably. Notwithstanding, in general accord with the data obtained with mutant PAR1 receptors, our data support the working hypothesis that in rat PAR2 as in PAR1, the charge properties of extracellular loop 2 (P231E232E233 of PAR2) play an important role in ligand recognition.
Differential effects on peptide and trypsin-mediated activation of PAR2NET and PAR2PRR
Since the PAR1AP, SFLLR-NH2 displays a potency lower than, but close to that of SLIGRL-NH2 for activating PAR2, (Blackhart et al., 1996; Kawabata et al., 1999 and Figure 3) we expected that the PAR2NET mutant, wherein a 15 residue sequence of extracellular loop 2 is identical to PAR1, would be even more sensitive to the PAR1AP, SFLLR-NH2, than was the wild-type PAR2. To our surprise, compared with wild-type PAR2, PAR2NET was about 40 fold less sensitive to SFLLR-NH2 whereas there was only about a 9 fold reduction in sensitivity towards SLIGRL-NH2 and the PAR2-selective peptide analogue, tc-LIGRLO-NH2. It should be noted that SLIGRL-NH2 cannot activate/interact with PAR1, unless the extracellular loop 2 of PAR2 is substituted into PAR1 (Lerner et al., 1996). We were thus able to conclude that although the PAR1 sequence LNITTCHDVLNETLL, is involved in the interaction of PAR1 with either the PAR1AP, SFLLR-NH2 or the PAR2AP, SLIGRL-NH2, it would appear that in general, SFLLR-NH2 seems to dock differently with the extracellular loop 2 of PAR2 than it does with the extracellular loop 2 of PAR1.
It is remarkable that the PEEV→NETL mutation in PAR2NET did not noticeably affect the activity of trypsin (and presumably of the tethered ligand revealed by trypsin), whereas the activity of the synthetic receptor-activating peptide, SLIGRL-NH2, was reduced by about 9 fold (Table 2). This result echoed the much lower reduction in the activity of trypsin (8 fold) compared with SLIGRL-NH2 (110 fold) in PAR2PRR (Table 2). One possibility, although unlikely, was that the accessibility of trypsin to the receptor cleavage/activation site might have been enhanced in PAR2NET and PAR2PRR, so as to compensate for a lowered intrinsic activity of the revealed tethered ligand. Notwithstanding, we believe that, taken together, the data indicate that the rat PAR2 tethered ligand sequence revealed by trypsin cleavage (SLIGRLDTPP), remaining attached to the receptor, interacts differently with the receptor than does the free peptide, SLIGRL-NH2 in solution. These possible differences between the interaction of the free and tethered ligand with the receptor may prove of importance in the context of developing PAR2 receptor antagonists.
In summary, our data obtained from the assay of the activities of PAR-APs and trypsin in PAR2PRR and PAR2NET point to an important role for the acidic extracellular loop 2 tripeptide, PEE, in governing agonist activity in PAR2. The results also suggest that there may be differences in the receptor activation interactions, between those of the tethered ligand and those of the synthetic activating peptides derived from the tethered ligand sequence. Further work with peptide based receptor crosslinking reagents may allow for a more precise analysis of the docking sites at which the activating peptides and the tethered ligand can interact.
Acknowledgments
These studies were supported in large part by an Operating Grant from the Canadian Medical Research Council, with ancillary support from a Canadian National Research Council/Natural Sciences and Engineering Research Council Partnership Grant, in conjunction with BioChem Therapeutic, Laval PQ, Canada. We are grateful to Drs Lorraine Leblond and Denis McMaster for the efficient provision of the peptides used in our studies. We appreciate very much, the help of Laurie Robertson with the FACS analysis.
Abbreviations
- (Amino acids are abbreviated by their one-letter codes) AP
receptor-activating peptide
- EL-2
extracellular loop 2
- KNRK
Kirsten virus-transformed normal rat kidney cells
- PAR
proteinase-activated receptor
- PAR1
proteinase-activated receptor-1 (activated by thrombin)
- PAR2
proteinase-activated receptor-2 (activated by trypsin & tryptase)
- PAR2NET
PEEV→NETL mutant of PAR2 expressed in KNRK cells
- PAR2PRR
PEE→PRR mutant of PAR2 expressed in KNRK cells
- PAR-AP
PAR-activating peptide
- SAR
Structure-activity relationship
- tc
trans-cinnamoyl
- Wt-PAR2
wild-type rat PAR2 expressed in KNRK cells
References
- AL-ANI B., SAIFEDDINE M., HOLLENBERG M.D. Detection of functional receptors for the proteinase-activated receptor-2-activating polypeptide, SLIGRL-NH2 in rat vascular and gastric smooth muscle. Can. J. Physiol. Pharmacol. 1995;73:1203–1207. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- AL-ANI B., SAIFEDDINE M., KAWABATA A., RENAUX B., MOKASHI S., HOLLENBERG M.D.Proteinase activated receptor-2 (PAR2): Development of a ligand binding assay correlating with activation of PAR2 by PAR1- and PAR2-derived peptide ligands J. Pharmacol. Exp. Ther. 1999. in press [PubMed]
- BLACKHART B.D., EMILSSON K., NGUYEN D., TENG W., MARTELLI A.J., NYSTEDT S., SUNDELIN J., SCARBOROUGH R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- BÖHM S.K., KONG W., BRÖMME D., SMEEKENS S.P., ANDERSON D.C., CONNOLLY A., KAHN M., NELKEN N.A., COUGHLIN S.R., PAYAN D.G., BUNNETT N.W. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAO B.H., KALKUNTE S., MARAGANORE J.M., STONE S.R. Essential groups in synthetic agonist peptides for activation of the platelet thrombin receptor. Biochemistry. 1992;31:6175–6178. doi: 10.1021/bi00142a001. [DOI] [PubMed] [Google Scholar]
- CHEN J., ISHII M., WANG L., ISHII K., COUGHLIN S.R. Thrombin receptor activation. Confirmation of the intramolecular tethered liganding hypothesis and discovery of an alternative intermolecular liganding mode. J. Biol. Chem. 1994;269:16041–16045. [PubMed] [Google Scholar]
- DERY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:1429–1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- GERSZTEN R.E., CHEN J., ISHII M., ISHII K., WANG L., NANEVICZ T., TURCK C.W., VU T.K., COUGHLIN S.R. Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature. 1994;368:648–651. doi: 10.1038/368648a0. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D., LANIYONU A.A., SAIFEDDINE M., MOORE G.J. Role of the amino- and carboxyl-terminal domains of thrombin receptor-derived polypeptides in biological activity in vascular endothelium and gastric smooth muscle: evidence for receptor subtypes. Mol. Pharmacol. 1993;43:921–930. [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B. Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol. Pharmacol. 1996;49:229–233. [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B., KAWABATA A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity and receptor selectivity of receptor-activating peptides. Can. J. Phys. Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- HOLLENBERG M.D., YANG S.G., LANIYONU A.A., MOORE G.J., SAIFEDDINE M. Action of thrombin receptor polypeptide in gastric smooth muscle: identification of a core pentapeptide retaining full thrombin-mimetic intrinsic activity. Mol. Pharmacol. 1992;42:186–191. [PubMed] [Google Scholar]
- HUI K.U., JAKUBOWSKI J.A., WYSS V.L., ANGLETON E.L. Minimal sequence requirements of thrombin receptor agonist peptide. Biochem. Biophys. Res. Commun. 1992;184:790–796. doi: 10.1016/0006-291x(92)90659-9. [DOI] [PubMed] [Google Scholar]
- ISHIHARA H., CONNOLLY A.J., ZENG D., KAHN M.L., ZHENG Y.W., TIMMONS C., TRAM T., COUGHLIN S.R. Protease-activated receptor-3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- KAHN M.K., ZHENG Y.-W., HUANG W., BIGORNIA V., ZENG D., MOFF S., FARESE R.V., JR, TAM C., COUGHLIN S.R. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL-ANI B., LEBLOND L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-a (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J. Pharmacol. Exp. Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- KONG W., MCCONALOGUE K., KHITIN L.M., HOLLENBERG M.D., PAYAN D.G., BÖHM S.K., BUNNETT N.W. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:8884–8889. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LERNER D.J., CHEN M., TRAM T., COUGHLIN S.R. Agonist recognition by proteinase-activated receptor 2 and thrombin receptor. J. Biol. Chem. 1996;271:13943–13947. [PubMed] [Google Scholar]
- LOPEZ J.G., CHEW S.J., THOMPSON H.W., MALTER J.S., INSLER M.S., BEUERMAN R.W. EGF cell surface receptor quantitation on ocular cells by an immunocytochemical flow cytometry technique. Invest. Ophthalmol. Vis. Sci. 1992;33:2053–2062. [PubMed] [Google Scholar]
- NANEVICZ T., ISHII M., WANG L., CHEN M., CHEN J., TURCK C.W., COHEN F.E., COUGHLIN S.R. Mechanisms of thrombin receptor agonist specificity. Chimeric receptors and complementary mutations identify an agonist recognition site. J. Biol. Chem. 1995;270:21619–21625. doi: 10.1074/jbc.270.37.21619. [DOI] [PubMed] [Google Scholar]
- NATARAJAN S., RIEXINGER D., PELUSO M., SEILER S.M. ‘Tethered ligand' derived pentapeptide agonists of thrombin receptor: a study of side chain requirements for human platelet activation and GTPase stimulation. Int. J. Pept. Protein. Res. 1995;45:145–151. doi: 10.1111/j.1399-3011.1995.tb01033.x. [DOI] [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTADT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASMUSSEN U.G., VOURET-CRAVIARI V., JALLAT S., SCHLESINGER Y., PAGES G., PAVIRANI A., LECOCQ J.P., POUYSSEGUR J., VAN OBBERGHEN-SCHILLING E. cDNA cloning and expression of a hamster α-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 1991;288:1223–1228. doi: 10.1016/0014-5793(91)81017-3. [DOI] [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (Par2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCARBOROUGH R.M., NAUGHTON M.-A., TENG W., HUNG D.T., ROSE J., VU T.-K.H., WHEATON V.I., TUREK C.W., COUGHLIN S.R. Tethered ligand agonist peptides: structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J. Biol. Chem. 1992;267:13146–13149. [PubMed] [Google Scholar]
- VASSALLO J.R., KIEBER-EMMONS T., CICHOWSKI K., BRASS L.F. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J. Biol. Chem. 1992;267:6081–6085. [PubMed] [Google Scholar]
- VU T.K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WATSON A.J., HOGAN A., HAHNEL A., WIEMER K.E., SCHULTZ G.A. Expression of growth factor ligand and receptor genes in the preimplantation bovine embryo. Mol. Reprod. Dev. 1992;31:87–95. doi: 10.1002/mrd.1080310202. [DOI] [PubMed] [Google Scholar]
- XU W.-F., ANDERSEN H., WHITMORE T.E., PRESNESS S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAGURSY R.J., SHARP D., SOLOMON K.A., SCHWARTZ A. Quantitation of cellular receptors by a new immunocytochemical flow cytometry technique. BioTechniques. 1995;18:504–509. [PubMed] [Google Scholar]