

Abstract
We have investigated the effects of methylenedioxymethamphetamine (MDMA, ‘ecstasy') on peripheral noradrenergic neurotransmission in the rat.
In rat atrial slices pre-incubated with [3H]-noradrenaline and in the presence of desipramine (1 μM) to prevent effects of MDMA on basal outflow of tritium, MDMA (10 μM) significantly inhibited the release of tritium evoked by short trains of six pulses at 100 Hz every 10 s for 3 min. This effect did not occur in the presence of the α2-adrenoceptor antagonist yohimbine (1 μM).
In epididymal portions of rat vas deferens in the presence of nifedipine (10 μM), MDMA produced a concentration-dependent inhibition of single pulse nerve stimulation-evoked contractions with a pD2 of 5.88±0.16 (n=4). Inhibitory effects of MDMA were antagonized by the α2-adrenoceptor antagonist yohimbine (0.3 μM), but not by the 5-hydroxytryptamine receptor antagonist cyanopindolol in a concentration (1 μM) which markedly antagonized the inhibitory actions of the 5-HT-1 receptor agonist 5-carboxamidotryptamine.
In prostatic portions of rat vas deferens in the presence of cocaine (3 μM), MDMA produced a concentration-dependent inhibition of single pulse nerve stimulation-evoked contractions with a pD2 of 5.12±0.21 (n=4). In the absence of cocaine, only the highest concentration of MDMA (30 μM) produced an inhibition, but the α2-adrenoceptor antagonist yohimbine (0.3 μM) converted the response to MDMA from inhibition to potentiation of the stimulation-evoked contraction.
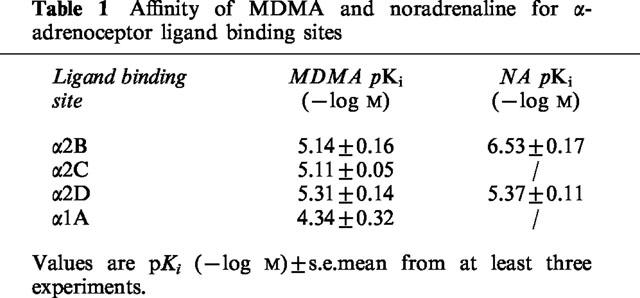
In radioligand binding studies, MDMA showed similar affinities for α2B, α2C and α2D-adrenoceptor sites, with pKi values of 5.14±0.16, 5.11±0.05 and 5.31±0.14, respectively.
It is concluded that MDMA has significant α2-adrenoceptor agonist actions.
Keywords: Rat atrium, rat vas deferens, α2-adrenoceptors, α2D-adrenoceptors, MDMA, desipramine, cocaine
Introduction
Cocaine and other substance abuse is widespread, and as a result, cocaine related cardiovascular and autonomic complications have mushroomed (Rubin & Neugarten, 1992; Das, 1993). Ecstasy (3,4-methylenedioxymethamphetamine; MDMA) has been much less studied than cocaine and classical amphetamines, but is reported to have cardiac stimulant actions in rats resulting in tachycardia (Gordon et al., 1991) and is also reported to facilitate vasoconstriction in the rat (Fitzgerald & Reid, 1994). Tachycardia and hypertension (Hayner & McKinney, 1986), and cardiovascular mortality (Dowling et al., 1987) have been reported in man. In addition, MDMA has been linked to intracerebral haemorrhage (Harries & De Silva, 1992), and cerebral hyperperfusion can be demonstrated in rats (Kelly et al., 1994). Certainly, chronic use of methamphetamine may also result in serious cardiovascular changes including tachycardia and palpitations (Chan et al., 1994), and another amphetamine derivative, fenfluramine has been linked to valvular heart disease (Connolly et al., 1997).
The object of this study was to examine the effects of MDMA on peripheral noradrenergic neurotransmission and how these might relate to cardiovascular actions. However, results obtained led us to turn our attention to prejunctional actions at α2-adrenoceptors. Some of these results have been published in abstract form (Lavelle et al., 1999).
Methods
Male Wistar rats (200–300 g) were obtained from Trinity College Dublin, and atrium, vas deferens, kidney and submandibular gland were employed as outlined below
Radioactive overflow experiments
Rat right and left atrial segments were used, six segments from each animal, at least one serving as a vehicle control. The Krebs-Henseleit solution employed had the following composition (mM): NaCl 119; NaHCO3 25; D-glucose 11.1; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.0; EDTA 0.03, ascorbic acid 0.28. Isolated tissues were pre-incubated for 1 h in 1 ml of Krebs-Henseleit medium containing [3H]-noradrenaline (0.5 μM, Specific activity 39 Ci mmol−1), before being placed in a Brandel Superfusor and superfused at 37°C with [3H]-noradrenaline-free Krebs-Henseleit solution at a rate of 0.33 ml min−1. The Krebs-Henseleit solution had the composition stated above, with additionally corticosterone (30 μM) and propranolol (1 μM). Cocaine (3–30 μM) or desipramine (1 μM) was present after pre-incubation with [3H]-noradrenaline, except where otherwise stated.
In all experiments, tissues were stimulated for 3 min (supramaximal voltage, 0.5 ms pulses) at 5 Hz or for 3 min with short bursts of six pulses at 100 Hz every 10 s, three times (S0-S2) at intervals of 27 min. After 99 min of superfusion the tissues were stimulated to remove excess counts (S0). Effluent samples were collected in 1 ml aliquots, beginning after 120 min of superfusion, so that sampling began 6 min before the control stimulation period at 126 min (S1). Test drugs or an equivalent volume (1%) of distilled water (vehicle) were superfused in the Krebs-Henseleit solution beginning 12 min before the test stimulation period at 153 min (S2). At the end of the experiment, tissues were dissolved in 0.5 ml of tissue solubilizer (Soluene, Packard). A volume of 1 ml superfusate or dissolved tissue was added to 3 ml of liquid scintillant (Ecoscint A) and the radioactivity was measured by liquid scintillation spectroscopy in a LKB 1214 RackBeta counter with on average 48% counting efficiency for tritium and automatic quench correction.
The basal efflux of tritium was expressed as a percentage rate, i.e. the efflux of total tritiated compounds per min was expressed as a percentage of the tritium content of the tissue at the time of collection. To quantify the effects of a drug on the basal efflux, the rate of efflux in the 3 min before stimulation in the presence of the drug (S2) was divided by the rate of efflux in the 3 min before the control stimulation period (S1), and compared with the equivalent ratio obtained in vehicle experiments without the test drug (B2/B1 ratios).
The stimulation-evoked overflow of total tritium was calculated by subtraction of the basal overflow and was expressed as a percentage of the tritium content of the tissue at the onset of the respective stimulation periods (see Smith & Docherty, 1992). To quantify the effects of a drug on stimulation-evoked overflow of tritium, the evoked overflow in the presence of the drug (S2) was divided by the overflow evoked in the control stimulation period (S1), and compared with the equivalent ratio obtained in vehicle experiments without the test drug (S2/S1 ratios).
Rat vas deferens
Whole vas deferens, prostatic or epididymal portions were obtained. Tissues were attached to myograph transducers under 1 g tension in organ baths at 37°C in Krebs-Henseleit solution of the following composition: (mM): NaCl 119; NaHCO3 25; D-glucose 11.1; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.0; EDTA 0.03, ascorbic acid 0.28.
In experiments investigating the ability of MDMA and other agonists to inhibit or potentiate the isometric twitch of vas deferens (in the presence or absence of cocaine 3 μM), tissues were placed between platinum electrodes and stimulated every 5 min with a single stimulus (0.5 ms pulses, supramaximal pulses) to produce isometric contractions, and nifedipine (10 μM) was present to block the non-noradrenergic component of the twitch when using epididymal portions. Test drugs or vehicle were added cumulatively in 0.5 or 1 log unit increments at 5 min intervals. An isometric twitch was obtained following 5 min exposure to each agonist concentration, or following exposure to the vehicle. Distilled water vehicle did not significantly affect stimulation-evoked contractions. In some experiments, interaction with antagonists was investigated. In these experiments, the antagonist or vehicle was presence for 15 min before beginning cumulative additions of the agonist.
Radioligand binding studies
Preparation of rat kidney membranes was carried out as described in Connaughton & Docherty (1990), and preparation of rat submandibular gland and rat vas deferens membranes were as described for rat kidney. Membranes of Sf9 cells expressing human recombinant α2C-adrenoceptors were purchased from Research Biochemicals. The resultant pellets were used immediately or stored at −20°C for later use. Pellets were reconstituted in 5 volumes (submandibular), 10 volumes (kidney and vas deferens) or 25 volumes (Sf9 cells) of incubation buffer.
In saturation experiments, aliquots of membrane suspension were incubated with various concentrations of [3H]-yohimbine (specific activity: 81 Ci mmole−1, Amersham) at 25°C (rat kidney: 0.5–30 nM; Sf9 cells: 0.2–20 nM; rat submandibular gland: 1.0–40 nM; incubation buffer: Tris-HCl 50 mM, EDTA 5 mM, pH 7.4 at 25°C) or with various concentrations of [3H]-prazosin (specific activity: 77.9 Ci mmole−1, NEN) at 25°C (rat vas deferens: 0.25-20 nM; incubation buffer as above). In competition studies, [3H]-yohimbine (5 or 10 nM) or [3H]-prazosin (2 nM) was incubated with competing ligands in concentrations from 0.1 to 1 mM in 0.5 log unit increments for 30 min. Non-specific binding was determined in the presence of phentolamine (10 μM). Specific binding of [3H]-yohimbine or [3H]-prazosin was 70–90% of total binding at the concentration used in displacement experiments. Assays were terminated by washing with ice-cold incubation buffer, followed by rapid vacuum filtration through Whatman GF/C filters, using a Brandel Cell Harvester. Radioactivity retained on filters was determined by liquid scintillation spectroscopy.
The inhibition constant (Ki) for inhibition of radiolabelled ligand binding was determined from the formula:
where IC50 is the concentration of competing ligand that inhibits radioligand specific binding by 50%, KD is the dissociation constant for the radioligand yohimbine (rat kidney: 8.80±0.62 nM, n=7; human recombinant α2C-adrenoceptors: 8.7±3.5 nM, n=3; rat submandibular gland: 23.7±2.0 nM, n=4) or prazosin (rat vas deferens: 0.91±0.34, n=11), and 3H is the concentration of tritiated yohimbine (5 nM: rat kidney; 10 nM: Sf9 cells and rat submandibular gland) or tritiated prazosin (2 nM: rat vas deferens).
Drugs
5-Carboxamidotryptamine (Research Biochemicals, U.S.A.); cocaine hydrochloride (Sigma, U.K.); corticosterone (Sigma); cyanopindolol (gift: Sandoz, Switzerland); desipramine hydrochloride (Research Biochemicals); methylenedioxy-methamphetamine hydrochloride (MDMA: Research Biochemicals); noradrenaline bitartrate (Research Biochemicals); propranolol hydrocholoride (Sigma); yohimbine hydrochloride (Sigma).
Drugs were dissolved in distilled water, except for corticosterone (100% ethanol) and cyanopindolol (few drops of HCl).
Statistics
Values are mean±s.e.mean. Agonist effects were compared with the effects of vehicle using a Student's t-test for unpaired or paired data, where appropriate, and by ANOVAR analysis. Agonist pEC50, pIC50, or pKi values were compared between tissues, and were compared following vehicle or antagonist using a Student's t-test for unpaired or paired data, where appropriate.
Results
Stimulation-evoked overflow in rat atrium; 5 Hz stimulation
In rat atrial slices, basal outflow of tritium was 0.198±0.022% of tissue tritium/min (n=15) and 0.152±0.007% of tissue tritium/min (n=27), and stimulation at a frequency of 5 Hz for 3 min produced a stimulation-evoked release of tritium of 2.84±0.18 and 4.40±0.19% of tissue tritium content, in the absence and presence of the re-uptake blocker cocaine (3 μM), respectively. MDMA (3 and 10 μM) tended to increase the 5 Hz stimulation-evoked overflow in the absence of re-uptake blockers (although the increase did not reach statistical significance) (Figure 1), but this may have been due to a significant increase in the basal release of tritium (B2/B1 ratios, vehicle: 1.00±0.04, n=7; MDMA 10 μM 1.24±0.06, n=11; P<0.05). In the presence of cocaine (3 μM), MDMA (30 μM) still significantly increased basal release of tritium (1.23±0.06, n=13, P<0.01), and MDMA (3–30 μM) did not significantly affect the stimulation-evoked release of tritium, although the tendency was to reduce stimulation-evoked release (Figure 1). The effects of MDMA (10 μM) were significantly different when compared with and without cocaine (P<0.01, see Figure 1). Even cocaine (30 μM) did not prevent MDMA (30 μM) from increasing the basal release of tritium (n=2).
Figure 1.
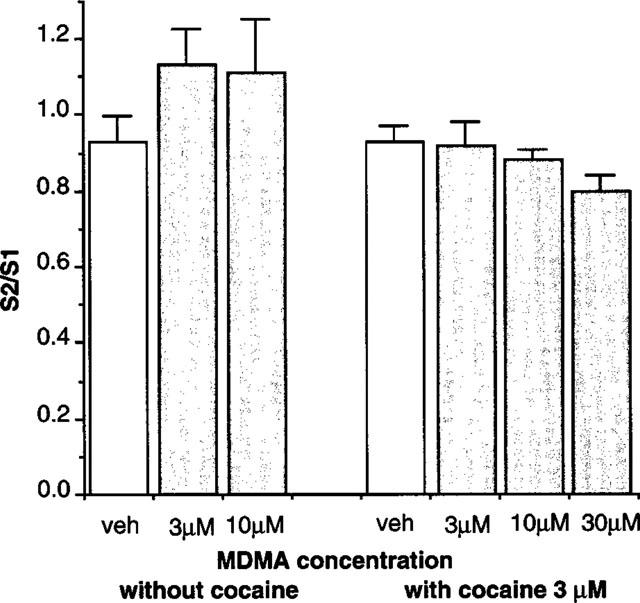
Effects of vehicle or MDMA (3–30 μM) on release of tritium evoked by 3 min stimulation at 5 Hz in rat atrium in the absence or presence of cocaine (3 μM). Vertical bars indicate s.e.mean from at least six (without cocaine) or 12 (with cocaine) experiments.
Stimulation-evoked overflow in rat atrium; stimulation with short trains at 100 Hz
Further experiments were carried out using short trains of pulses at 100 Hz and the re-uptake blocker desipramine. In the presence of desipramine (1 μM), basal outflow was 0.120±0.004% of tissue tritium/min (n=37), and stimulation with six pulses at 100 Hz every 10 s for 3 min produced a stimulation-evoked release of tritium of 0.62±0.02% of tissue tritium content. MDMA no longer increased the basal release of tritium (e.g. vehicle 1.01±0.02, n=22; MDMA 30 μM 0.98±0.06, n=7), and under these conditions, MDMA (10 μM) significantly reduced the stimulation-evoked release of tritium (Figure 2). However, the effects of MDMA (30 μM) did not reach significance. In experiments examining the interaction of the α2-adrenoceptor antagonist yohimbine with MDMA, yohimbine (1 μM) and vehicle increased the stimulation-evoked release of tritium to 203±16% of control (n=8). The combination of MDMA (10 μM) and yohimbine (1 μM) had a similar effect to yohimbine and vehicle (192±18% of control, n=8).
Figure 2.
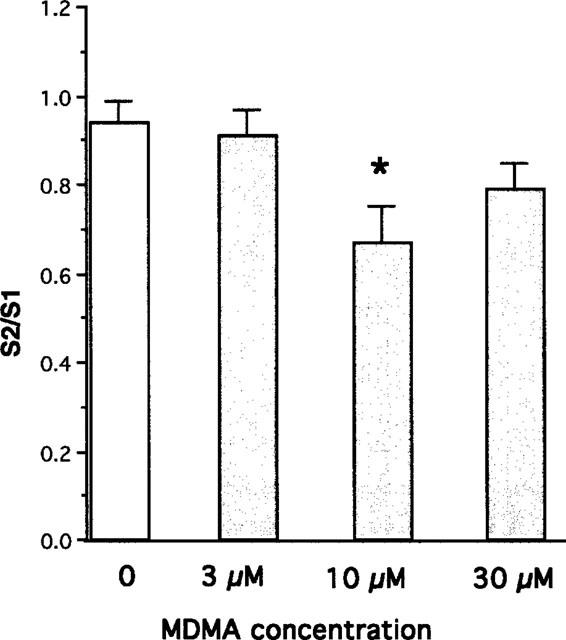
Effects of vehicle or MDMA (3–30 μM) on release of tritium evoked by stimulation for 3 min with six pulses at 100 Hz every 10 s in rat atrium in the presence of desipramine (1 μM). Vertical bars indicate s.e.mean from at least five experiments. Asterisks denote significance of effects of MDMA as compared with the effects of vehicle (Analysis of Variance and Dunnett test: *P<0.01).
Rat whole vas deferens
In rat whole vas deferens, the initial biphasic twitch to a single electrical stimulus was 0.74±0.11 g (n=6). MDMA (0.1–10 μM) produced a concentration-dependent increase in the stimulation-evoked contraction, with a maximum potentiation to 176.8±18.9% of control (n=4) (Figure 3).
Figure 3.
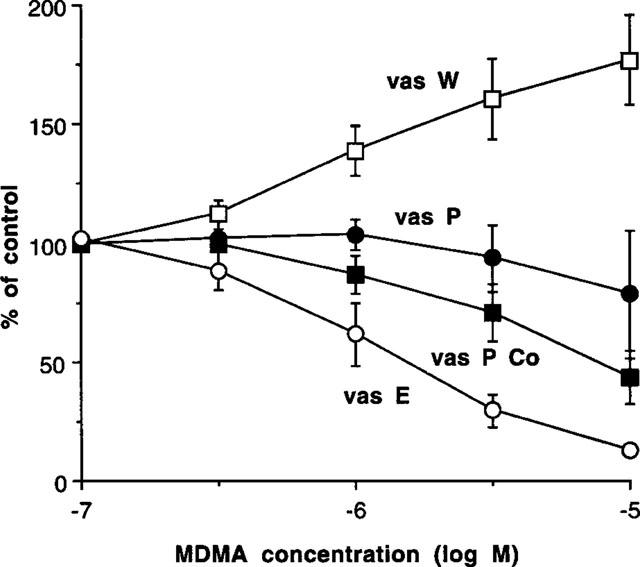
Effects of MDMA to potentiate or inhibit contractions induced by single pulse electrical stimulation in whole vas deferens (vas W), in prostatic portions (vas P), in prostatic portions (in the presence of cocaine 3 μM) (vas P Co), or in epididymal portions (in presence of nifedipine 10 μM) (vas E). Vertical bars indicate s.e.mean from at least four experiments.
Prostatic portions of rat vas deferens
In prostatic portions of vas deferens, the initial monophasic twitch to a single electrical stimulus was 1.12±0.10 g (n=30). MDMA (0.1–10 μM) produced inconsistent effects against nerve-stimulation evoked contractions: in some of experiments MDMA produced a concentration-dependent inhibition, but in other experiments, no inhibition, or even a potentiation occurred, especially at low concentrations. The overall effect was that there was no significant change in the stimulation-evoked contraction (Figure 3).
Cocaine (0.3–3 μM) produced a potentiation of stimulation-evoked contractions, but cocaine (30 μM) began to inhibit contractions (Figure 4). However, in the presence of cocaine (3 μM), MDMA produced a concentration-dependent inhibition of the stimulation-evoked contraction with a pD2 (−log IC50) of 5.12±0.21 (n=4) (Figure 3).
Figure 4.
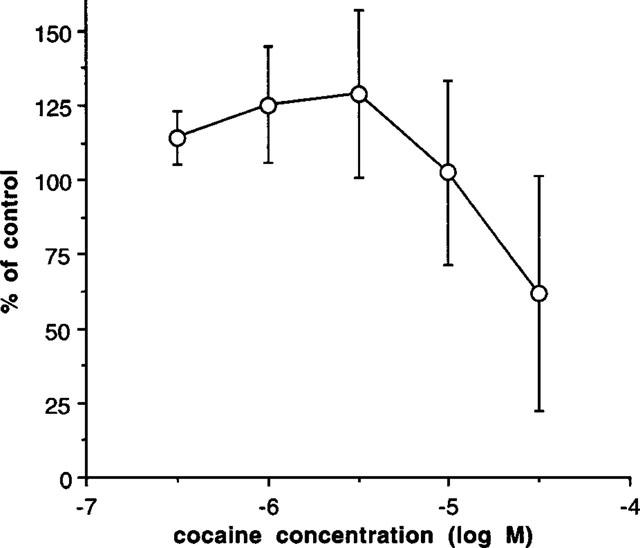
Effects of cocaine on the contraction induced by single pulse electrical stimulation in prostatic portions of rat vas deferens. Vertical bars indicate s.e.mean from at least six experiments.
In further experiments in the absence of cocaine, tissues were treated with yohimbine (0.3 μM) or vehicle 15 min before beginning a concentration-response curve to MDMA. As before, MDMA (0.1–10 μM) produced variable effects, but the higher concentration of 30 μM significantly reduced contractions to nerve stimulation (Figure 5). In contrast, yohimbine prevented the inhibitory effects of MDMA and converted the inhibitory effect to a significant potentiation (Figure 5).
Figure 5.
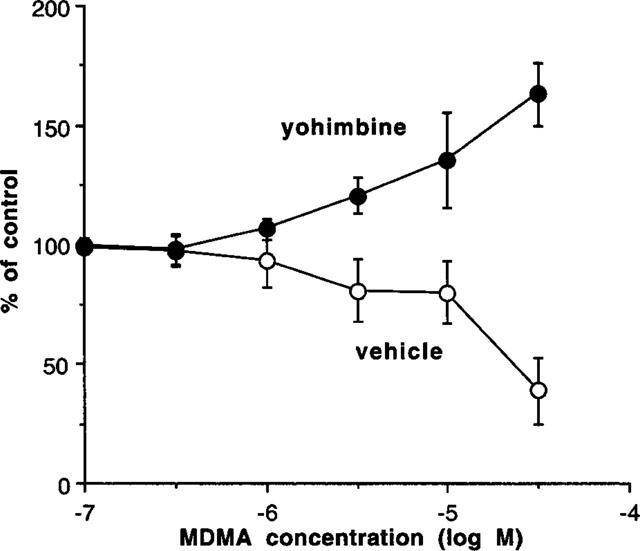
Effects of pre-exposure to yohimbine (0.3 μM) or vehicle on the ability of MDMA to inhibit the contraction induced by single pulse electrical stimulation in prostatic portions of rat vas deferens. Vertical bars indicate s.e.mean from at least five experiments.
Epididymal portions of rat vas deferens
In epididymal portions of rat vas deferens, the initial biphasic twitch to a single electrical stimulus was reduced to a monophasic response by nifedipine (10 μM), which eliminates the first (non-noradrenergic) phase, leaving the second noradrenergic phase. Nifedipine also prevents the postjunctional contractile actions of α-adrenoceptor agonists. Under these conditions, the twitch to a single stimulus was 0.70±0.08 g (n=27), and MDMA (0.1–10 μM) reduced the size of the twitch in a concentration-dependent manner (Figure 3). MDMA virtually abolished nerve-stimulation evoked contractions with a pD2 (−log IC50) of 5.88±0.16.
In further experiments, epididymal portions in the presence of nifedipine were treated with vehicle or yohimbine (0.3 μM) 15 min prior to beginning a concentration-response curve to MDMA. Yohimbine significantly reduced the potency of MDMA (MDMA pD2; vehicle: 5.76±0.11, n=7; yohimbine: 5.27±0.17, n=7, P<0.05).
Epididymal portions in the presence of nifedipine, were treated with vehicle or cyanopindolol (1.0 μM) 15 min prior to beginning a concentration-response curve to MDMA. Cyanopindol did not significantly affect the potency of MDMA (Figure 6). By comparison, this concentration of cyanopindolol virtually abolished the inhibitory effects of the 5-HT-1 receptor agonist 5-carboxamidotryptamine (Figure 6).
Figure 6.
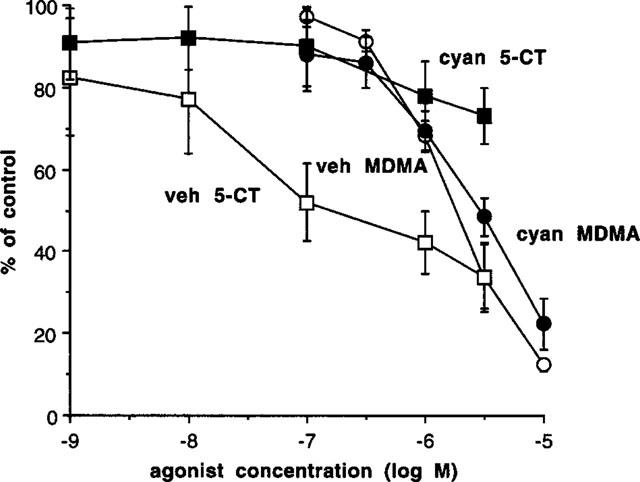
Effects of pre-exposure to cyanopindolol (cyan 1.0 μM) (filled symbols) or vehicle (veh) (open symbols) on the ability of MDMA (squares) or 5-carboxamidotryptamine (5-CT) (circles) to inhibit the contraction induced by single pulse electrical stimulation in epididymal portions of rat vas deferens in the presence of nifedipine (10 μM). Vertical bars indicate s.e.mean from at least six experiments.
Ligand binding studies
The affinities of MDMA and noradrenaline for α2-adrenoceptor ligand binding sites were examined in competition experiments employing [3H]-yohimbine labelled membrane preparations of rat kidney α2B, recombinant human α2C, and rat submandibular α2D-adrenoceptors. The pKi values (−log M) obtained are shown in Table 1. MDMA showed similar affinity for all three subtypes of α2-adrenoceptor and had similar affinity to noradrenaline at α2D-adrenoceptors, but lower affinity at α2B-adrenoceptors (Table 1). MDMA had approximately ten times lower affinity for α1A-adrenoceptors in competition studies employing [3H]-prazosin labelled membrane preparations of rat vas deferens (Table 1).
Table 1.
Affinity of MDMA and noradrenaline for α-adrenoceptor ligand binding sites
Discussion
In this study, we have investigated the effects of MDMA on peripheral noradrenergic neurotransmission. The major finding was a previously unreported α2-adrenoceptor agonist action of MDMA. This and other findings are discussed below.
In radioactive overflow studies carried out in rat atrium, MDMA significantly increased basal release of tritium in the absence of uptake blockers. This effect of MDMA was reduced by cocaine (3–30 μM) and abolished by desipramine (1 μM), and is due to entry of MDMA into nerve terminals via the noradrenaline transporter to displace noradrenaline (see Fitzgerald & Reid, 1993). Desipramine is reported to have greater potency as a noradrenaline uptake blocker than cocaine (see Docherty & McGrath, 1978).
In the presence of cocaine to reduce effects of MDMA on basal release of tritium, MDMA did not significantly affect stimulation-evoked release of tritium using the 5 Hz stimulation parameters. We employ these stimulation parameters in studies to investigate the ability of α2-adrenoceptor antagonists to increase stimulation-evoked overflow (see Smith & Docherty, 1992): hence, MDMA is not an α2-adrenoceptor antagonist. To study possible agonist actions of MDMA, we examined short trains of six pulses at 100 Hz every 10 s for 3 min, where negative feedback by noradrenaline should be greatly reduced (see Bohmann et al., 1993). Under these conditions, MDMA did significantly inhibit stimulation-evoked release of tritium, and this action was prevented by the α2-adrenoceptor antagonist yohimbine. To examine prejunctional inhibitory actions of MDMA further, we turned to the rat vas deferens.
In rat whole vas deferens, the electrical stimulation-evoked contraction to a single stimulus is biphasic, the first phase is non-adrenoceptor mediated and predominates in prostatic portions, and the second phase is α1-adrenoceptor mediated, and predominates in the epididymal portion (see Brown et al., 1983). The second α1-noradrenergic phase can be examined in isolation in the epididymal portion in the presence of the calcium entry blocker nifedipine which eliminates the first non-noradrenergic response and prevents the postjunctional contractile actions of exogenous α1-adrenoceptor agonists (see Brown et al., 1983). MDMA significantly potentiated contractions in whole vas deferens, had variable effects in prostatic portions, and inhibited contractions in epididymal portions (in presence of nifedipine). α1-Adrenoceptor agonists potentiate stimulation-evoked contractions in rat vas deferens (see Docherty & O'Malley, 1983), so that the potentiating action of MDMA probably involves postjunctional stimulation. In prostatic portions in the presence of cocaine to block neuronal uptake (cocaine was preferred in the rat vas deferens due to the α-adrenoceptor antagonist properties of desipramine: McCulloch & Story, 1972), MDMA produced a concentration-dependent inhibition of contractions. These results are consistent with two actions of MDMA. The first action is to potentiate nerve-stimulation-evoked contractions, and this effect predominates in the epididymal portion (as demonstrated in the whole vas deferens) and is due to entry of MDMA into the nerve terminal via the neuronal uptake transporter for noradrenaline to displace noradrenaline. The second action is to inhibit nerve stimulation-evoked contractions by a prejunctional action and is most easily seen when the former action is prevented by cocaine or nifedipine.
We next examined the interaction of MDMA with antagonists. In prostatic portions, the α2-adrenoceptor antagonist yohimbine (0.3 μM) converted the inhibitory effect of MDMA (30 μM) to a potentiation. In epididymal portions, yohimbine significantly reduced the potency of MDMA. By contrast, the 5-HT-1 receptor antagonist cyanopindolol did not affect the inhibitory actions of MDMA in a concentration which virtually abolished the inhibitory actions of 5-CT (see also Borton & Docherty, 1990). This suggests that the major prejunctional actions of MDMA are by agonism at α2-adrenoceptors and not 5-HT-1 receptors.
Ligand binding studies confirm that MDMA has significant affinity for α2-adrenoceptor subtypes, and has equal affinity with noradrenaline for the α2D-adrenoceptor subtype, which is the predominant prejunctional α2-adrenoceptor (see Docherty, 1998). It should be noted that a pKi value of around 5.3 may suggest low affinity for an antagonist, but for a high efficacy agonist which may need to occupy only a small percentage of receptors to produce the maximum effect, this pKi probably means than the agonist binds to the receptors in the range 7.00–5.00 (−log M). Such affinity fits with potency in the functional studies discussed above: threshold effect of around 0.3 μM.
Our results can be compared with effects of MDMA reported in the literature. Central actions of MDMA have been studied in much greater detail and include acute effects to release 5-hydroxytryptamine which can result in hyperthermia in experimental animals and man (Green et al., 1995). A second action is to cause neuronal damage in rodent brain, particularly to serotonergic nerves (Battaglia et al., 1987), and this action may involve free radical generation (Colado et al., 1997). Possible effects of MDMA involving the noradrenergic system have been largely overlooked. Depletion of noradrenaline can be shown in mouse heart in response to MDMA (Steele et al., 1989). Fitzgerald & Reid (1993), in radioactive overflow studies, found that MDMA increased basal release of noradrenaline (and indeed dopamine and 5-hydroxytryptamine) and increased stimulation-evoked release. In the presence of desipramine (1 μM) to block noradrenaline uptake, MDMA no longer increased 2 Hz stimulation-evoked release in rat atrium, but there was no inhibition (Fitzgerald & Reid, 1994). In ligand binding studies, MDMA has been previously reported to bind to α1- and α2-adrenoceptors with Ki values of 18.4 and 3.6 μM, respectively (Battaglia et al., 1988), although these authors discussed MDMA as a possible α2-adrenoceptor antagonist.
Finally, it is worth considering how our findings that MDMA has significant effects as an agonist at α2-adrenoceptors impact on the recreational abuse and toxic effects of MDMA. Certainly, such actions have not been considered in reviewing the effects of MDMA in man. However, it is feasible that α2-adrenoceptor agonism contributes to the central abusive and peripheral cardiovascular and autonomic side effects of MDMA. For instance, it has been reported that MDMA decreases firing rates of serotonergic and noradrenergic but not dopaminergic neurones in the rat dorsal and median raphe (Piercey et al., 1990), and although these authors dismissed an action at α2-autoreceptors, such an action is likely. Actions of MDMA at postjunctional α2-adrenoceptors on vascular and non-vascular smooth muscle may also have to be considered. Further research is needed to elucidate these effects.
In conclusion, MDMA has significant α2-adrenoceptor agonist actions which may contribute to its abusive potential and cardiovascular and autonomic side effects.
Acknowledgments
Supported by the Forbairt Science and Technology Against Drugs Initiative and Royal College of Surgeons in Ireland.
Abbreviations
- MDMA
methylenedioxymethamphetamine
References
- ABOUD R.W., SHAFII M., DOCHERTY J.R. Investigation of the subtypes of α1-adrenoceptor mediating contraction of rat aorta, vas deferens and spleen. Br. J. Pharmacol. 1993;109:80–87. doi: 10.1111/j.1476-5381.1993.tb13534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATTAGLIA G., BROOKS B.P., KULSAKDINUM C., DE SOUZA E.B. Pharmacological profile of MDMA (3,4-methylenedioxymethamphetamine) at various brain recognition sites. Eur. J. Pharmacol. 1988;149:159–163. doi: 10.1016/0014-2999(88)90056-8. [DOI] [PubMed] [Google Scholar]
- BATTAGLIA G., YEH S.Y., O'HEARN E., MOLLIVER M.E., KUHAR K.J., DE SOUZA E.B. 3,4-Methylenedioxymethamphetamine destroys serotonergic terminals in rat brain: quantification of neurodegeneration by measurement of [3H]-paroxetine-labelled serotonin uptake sites. J. Pharmacol. exp. Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- BOHMANN C., SCHOLLMEYER P., RUMP L.C. α2-Autorecpetor subclassification in rat isolated kidney by use of short trains of electrical stimulation. Br. J. Pharmacol. 1993;108:262–268. doi: 10.1111/j.1476-5381.1993.tb13472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORTON M., DOCHERTY J.R. The effects of ageing on prejunctional 5-hydroxytryptamine receptors in the rat vas deferens. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:130–135. doi: 10.1007/BF00166954. [DOI] [PubMed] [Google Scholar]
- BROWN D.A., DOCHERTY J.R., FRENCH A.M., MACDONALD A., MCGRATH J.C., SCOTT N.C. Separation of noradrenergic and non-noradrenergic contractions to field stimulation in the rat vas deferens. Br. J. Pharmacol. 1983;79:379–393. doi: 10.1111/j.1476-5381.1983.tb11010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN P., CHEN J.H., LEE M.H., DENG J.F. Fatal and nonfatal methamphetamine intoxication in the intensive care unit. J. Toxicol.-Clin. Toxicol. 1994;32:147–155. doi: 10.3109/15563659409000444. [DOI] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., MURRAY T.K., GREEN A.R. In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA (‘ecstasy') and p-chloroamphetamine but not the degeneration following fenfluramine. Br. J. Pharmacol. 1997;121:889–900. doi: 10.1038/sj.bjp.0701213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNAUGHTON S., DOCHERTY J.R. Functional evidence for heterogeneity of peripheral prejunctional alpha-2 adrenoceptors. Br. J. Pharmacol. 1990;101:285–290. doi: 10.1111/j.1476-5381.1990.tb12702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOLLY H.M., CRARY J.L., MCGOON M.D., HENSRUD D.D., EDWARDS B.S., EDWARDS W.D., SCHAFF H.V. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- DAS G. Cardiovascular effects of cocaine abuse. Int. J. Clin. Pharmac. 1993;31:521–528. [PubMed] [Google Scholar]
- DOCHERTY J.R. Subtypes of functional α1-and α2-adrenoceptor adrenoceptors. Eur. J. Pharmacol. 1998;361:1–15. doi: 10.1016/s0014-2999(98)00682-7. [DOI] [PubMed] [Google Scholar]
- DOCHERTY J.R., MCGRATH J.C. Sympathomimetic effects of pancuronium bromide on the cardiovascular system of the pithed rat. Br. J. Pharmacol. 1978;64:589–600. doi: 10.1111/j.1476-5381.1978.tb17321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOCHERTY J.R., O'MALLEY K. An examination of age-related changes in pre- and postsynaptic alpha-adrenoceptors in the rat isolated vas deferens. Eur. J. Pharmacol. 1983;95:171–175. doi: 10.1016/0014-2999(83)90631-3. [DOI] [PubMed] [Google Scholar]
- DOWLING G.P., MCDONOUGH E.T., BOST R.O. ‘Eve' and ‘ecstasy'. A report of five deaths associated with the use of MDEA and MDMA. J. Am. Med. Assoc. 1987;257:1615–1617. doi: 10.1001/jama.257.12.1615. [DOI] [PubMed] [Google Scholar]
- FITZGERALD J.L., REID J.J. Interactions of methylenedioxymethamphetamine with monoamine transmitter release mechanisms in rat brain slices. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;347:313–323. doi: 10.1007/BF00167451. [DOI] [PubMed] [Google Scholar]
- FITZGERALD J.L., REID J.J. Sympathomimetic actions of methylenedioxymethamphetamine in rat and rabbit isolated cardiovascular tissues. J. Pharm. Pharmacol. 1994;46:826–832. doi: 10.1111/j.2042-7158.1994.tb03738.x. [DOI] [PubMed] [Google Scholar]
- GORDON C.J., WATKINSON W.P., O'CALLAGHAN J.P., MILLER D.B. Effects of 3,4-methylenedioxymethamphetamine on autonomic thermoregulatory responses of the rat. Pharmacol. Biochem. Behav. 1991;38:339–344. doi: 10.1016/0091-3057(91)90288-d. [DOI] [PubMed] [Google Scholar]
- GREEN A.R., CROSS A.J., GOODWIN G.M. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or ‘ecstasy') Psychopharmacology. 1995;119:247–260. doi: 10.1007/BF02246288. [DOI] [PubMed] [Google Scholar]
- HARRIES D.P., DE SILVA R. ‘Ecstasy' and intracerebral haemorrhage. Scot. Med. J. 1992;37:150–152. doi: 10.1177/003693309203700508. [DOI] [PubMed] [Google Scholar]
- HAYNER G.N., MCKINNEY H. The dark side of ecstasy. J. Psychoactive Drugs. 1986;18:341–346. doi: 10.1080/02791072.1986.10472367. [DOI] [PubMed] [Google Scholar]
- KELLY P.A.T., RITCHIE I.M., SANGRA M., CURSHAM M.J.A., DICKSON E.M., KELLY B., NEILSON R.F.P., REIDY M.J., STEVENS M.C. Hyperaemia in rat neocortex produced by acute exposure to methylenedioxymethamphetamine. Brain Research. 1994;665:315–318. doi: 10.1016/0006-8993(94)91354-4. [DOI] [PubMed] [Google Scholar]
- LAVELLE A., HONNER V., DOCHERTY J.R. Investigation of the effects of MDMA at α-adrenoceptors in the rat. Br. J. Pharmacol. 1999;126:33P. doi: 10.1038/sj.bjp.0702875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCULLOCH M.W., STORY D.F. Antagonism of noradrenaline and histamine by desipramine in the isolated artery of the rabbit ear. Br. J. Pharmacol. 1972;46:140–150. doi: 10.1111/j.1476-5381.1972.tb06856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIERCEY M.F., LUM J.T., PALMER J.R. Effects of MDMA (‘ecstasy') on firing rates of serotonergic, dopaminergic and noradrenergic neurones in the rat. Brain Research. 1990;526:203–206. doi: 10.1016/0006-8993(90)91222-3. [DOI] [PubMed] [Google Scholar]
- RUBIN R.B., NEUGARTEN J. Medical complications of cocaine: changes in pattern of use and spectrum of complications. J. Toxicol.-Clin. Toxicol. 1992;30:1–12. doi: 10.3109/15563659208994441. [DOI] [PubMed] [Google Scholar]
- SMITH K., DOCHERTY J.R. Are the prejunctional alpha-2 adrenoceptors of the rat vas deferens and submandibular gland of the alpha-2A or alpha-2D-subtype. Eur. J. Pharmacol. 1992;219:203–210. doi: 10.1016/0014-2999(92)90297-h. [DOI] [PubMed] [Google Scholar]
- STEELE T.D., NICHOLS D.E., YIM G.K.W. MDMA transiently alters biogenic amines and metabolites in mouse brain and heart. Pharmacol. Biochem. Behav. 1989;34:223–227. doi: 10.1016/0091-3057(89)90303-1. [DOI] [PubMed] [Google Scholar]