

Abstract
The antagonist activity of a series of diinosine polyphosphates (IpnI, where n=3, 4, 5) was assessed against ATP-activated inward currents at rat P2X1–4 receptors expressed in Xenopus oocytes and studied under voltage-clamp conditions.
Diinosine polyphosphates were prepared by the enzymatic degradation of their corresponding diadenosine polyphosphates (e.g., Ap5A into Ip5I) using 5′-adenylic deaminase, and purified using reverse-phase chromatography.
Against ATP-responses at rP2X1 receptors, the potency order for antagonism was (pIC50): Ip5I (8.5)>Ip4I (6.3)>Ip3I (>4.5). Ip5I (10–100 nM) caused a concentration-dependent rightwards displacement of the ATP concentration-response curve without reducing the maximum ATP effect. However, the Schild plot was non-linear which indicated Ip5I is not a competitive antagonist. Blockade by micromolar concentrations of Ip5I was not surmountable. Ip4I also behaved as a non-surmountable antagonist.
Against ATP-responses at rP2X3 receptors, the potency order for antagonism was (pIC50): Ip4I (6.0)>Ip5I (5.6)>Ip3I (>4.5). Blockade by Ip4I (pA2, 6.75) and Ip5I (pA2, 6.27) was surmountable at micromolar concentrations.
Diinosine polyphosphates failed to inhibit ATP-responses at rP2X2 receptors, whereas agonist responses at rP2X4 were reversibly potentiated by Ip4I and Ip5I. None of the parent diadenosine polyphosphates behave as antagonists at rP2X1–4 receptors.
Thus, Ip5I acted as a potent and relatively-selective antagonist at the rP2X1 receptor. This dinucleotide pentaphosphate represents a high-affinity antagonist for the P2X1 receptor, at which it acts in a competitive manner at low (⩽100 nM) concentrations but has more complex actions at higher (>100 nM) concentrations.
Keywords: P2X receptor, ion channel, ATP, dinucleotide, Ip5I, diinosine polyphosphate, antagonist, Xenopus oocyte
Introduction
P2X receptors are ATP-gated cation channels composed of oligomeric assemblies of three, or possibly four, receptor protein subunits (Kim et al., 1997; Nicke et al., 1998; Torres et al., 1999). Seven P2X subunits (P2X1–7) have been cloned thus far, the operational profiles and pharmacological characteristics of homomeric assemblies of P2X1–7 receptors having been well documented (for reviews, see: Evans et al., 1998; Humphrey et al., 1998; King, 1998). Transcripts for P2X1–4 receptor proteins are abundant in excitable tissues (neurons, smooth muscle, cardiac muscle) and in secretory epithelia, while the distribution of P2X5 and P2X7 mRNA is highly restricted. P2X6 transcripts are especially abundant in the neuraxis (Collo et al., 1996), but it remains controversial that the P2X6 subunit protein is capable of forming homomeric assemblies (Torres et al., 1999).
The correspondence between homomeric P2X receptors and native P2X receptors has been hampered by a paucity of selective ligands for P2X subunits. Operational profiles of homomeric P2X receptors have been matched loosely to some examples of native P2X receptors (Evans & Surprenant, 1996), but the possibility of subpopulations of homomeric and heteromeric P2X receptors in any pool of native P2X receptors cannot be discounted. Some progress has been made with agonists showing reasonable P2X subunit selectivity, e.g. L-β,γ-meATP at P2X1 receptors (Evans et al., 1995), D-β,γ-meATP at P2X3 receptors (Rae et al., 1998), and activity series of the diadenosine polyphosphates (ApnA, where n=2–6) at P2X1–4 receptors (Wildman et al., 1999a). However, differentiating homomeric P2X receptors by agonist activity alone is a lengthy and laborious process (Humphrey et al., 1998). Further progress has been made with P2X subunit-selective antagonists: the anion transport inhibitor, DIDS, is relatively selective for P2X1 receptors at micromolar concentrations (Evans et al., 1995); the PPADS derivative, MRS 2220, is wholly selective for P2X1 receptors at micromolar concentrations (Jacobson et al., 1998); the suramin analogue, NF023, is a potent antagonist at P2X1 receptors at submicromolar concentrations, whilst 35–138 fold less potent at species orthologues of P2X3 receptors (Soto et al., 1999); TNP-ATP is a potent antagonist at both P2X1 and P2X3 receptors at nanomolar concentrations (Virginio et al., 1998); KN-62 is a potent antagonist of P2X7 receptors, although it shows differential activity at species orthologues of the P2X7 receptor (Humphreys et al., 1998). Neither suramin, PPADS nor Reactive blue 2 (RB-2) readily discriminate between P2X subunits (Evans et al., 1998; King, 1998; Ralevic & Burnstock, 1998).
Diinosine polyphosphates (abbreviated IpnI, where n is the number of phosphates) comprise two ribosylated inosine molecules bridged by a phosphate chain. These dinucleotides are synthesized by deaminating diadenosine polyphosphates with the non-specific AMP-deaminase of Aspergillus sp. (Guranowski et al., 1995; Pintor et al., 1997). One member of this family, P1,P5-bis(5′-inosyl) pentaphosphate (Ip5I), has already shown interesting pharmacological properties, being a potent antagonist at: (i) a specific dinucleotide receptor for diadenosine polyphosphates in rat brain synaptosomes (IC50 value, 4 nM); (ii) a P2X receptor in the same preparation (IC50 value, 30 μM); (iii) the P2X1-like receptor in guinea-pig isolated vas deferens (pA2 value, 6.5) (Hoyle et al., 1997; Pintor et al., 1997; 1999). In this paper, we describe the antagonist properties of Ip5I and two related dinucleotides, Ip4I and Ip3I, on homomeric P2X1–4 receptors. The activity profile of dinucleotide pentaphosphate reveals selectivity for the P2X1 receptor subtype at nanomolar concentrations.
Methods
Diinosine polyphosphate synthesis
Diinosine pentaphosphate (Ip5I) was prepared by enzymatic degradation of diadenosine pentaphosphate (Ap5A). Ip4I and Ip3I were also prepared in the same manner from Ap4A and Ap3A, respectively. 5′-adenylic acid deaminase (0.12 U) from Aspergillus sp. was incubated with 10 mM Ap5A in a final volume of 1 ml of 50 mM HEPES (pH 6.5) for 90 min (at 37°C). Aliquots (10 μl) were taken at different times, placed in 100°C water bath for 5 min to stop the enzymatic reaction, and diluted 1 : 100 with distilled water to monitor the production of Ip5I by HPLC techniques. After 90 min, the reaction was stopped by boiling the incubation medium at 100°C for 5 min, after which protein debris was removed by filtration through a Millex-G5 filter (0.22 μm; from Millipore). The reaction product was confirmed as Ip5I by HPLC detection. Samples were treated with phosphodiesterase (3 mU, at 37°C) from Crotalus durissus (EC.3.1.15.1) (for rationale, see Results) then diluted 1 : 100 with distilled water for HPLC separation and detection of Ip5I breakdown products.
Chromatographic procedures
The chromatographic equipment consisted of a Waters 600E delivery system, a Waters 717+ autosampler and a Waters 2487 dual wavelength absorbance detector, which were managed by Millenium 2010 software. Analyses were performed under reverse-phase chromatography conditions, equilibrating the system with 100 mM KH2PO4, 4% methanol, pH 6.0, at 1.5 ml min−1. The column was a Spherisorb ODS-2 (25 cm length, 0.4 cm diameter; from Waters). Detection was monitored at 260 nm wavelength. For phosphodiesterase measurements, ion-pair chromatography was performed. The mobile phase conditions were 10 mM KH2PO4, 2 mM tetrabutyl ammonium, 15% acetonitrile, pH 7.5, at 2 ml min−1. The column was a Spherisorb ODS-2. Detection was performed as above.
Oocyte preparation
Xenopus laevis frogs were anaesthetized in Tricaine (0.2% w v−1), killed by decapitation, and the ovarian lobes removed surgically. Oocytes (stages V and VI) were defolliculated by a 2-step process involving collagenase treatment (Type IA, 2 mg ml−1 in a Ca2+-free Ringer's solution, for 2–3 h) followed by stripping away the follicular layer with fine forceps. Defolliculated oocytes were stored in Barth's solution (pH 7.5, at 4°C) containing (mM): NaCl, 110; KCl, 1; NaHCO3, 2.4; Tris HCl, 7.5; Ca(NO3)2, 0.33; CaCl2, 0.41; MgSO4, 0.82; gentamycin sulphate, 50 μg l−1. Separate batches of defolliculated oocytes were injected cytosolically (40 nl, 1 μg ml−1) with cRNAs for rat P2X1–4 receptors (see Acknowledgements), incubated for 24–48 h at 18°C in Barth's solution and, thereafter, kept at 4°C for up to 12 days until used in electrophysiological experiments.
Electrophysiology
ATP-evoked membrane currents (IATP) (Vh=−60 to −90 mV) were recorded from cRNA-injected oocytes using a twin-electrode voltage-clamp amplifier (Axoclamp 2B). The voltage-recording and current-recording microelectrodes (1–5 MΩ tip resistance) were filled with 3.0 M KCl. Oocytes were superfused with a Ringer's solution (5 ml min−1, at 18°C) containing (mM): NaCl, 110; KCl, 2.5; HEPES, 5; BaCl2, 1.8, adjusted to pH 7.5. The extracellular pH was maintained at pH 7.5 in all experiments, since the potency of ATP at P2X1–4 receptors is affected by H+ ions (King et al., 1996; Wildman et al., 1999b,1999c).
Solutions and drugs
All solutions were nominally Ca2+-free to avoid the activation of a Ca2+-dependent Cl− current (ICl,Ca) in oocytes. ATP was prepared in Ca2+-free Ringer's solution at the concentrations stated, and superfused via a gravity-feed continuous flow system which allowed rapid addition and washout. IpnI compounds were dissolved in a buffer solution (HEPES 50 mM, pH 6.5 with KOH) to give a 10 mM stock solution, then diluted further using Ringer's solution and readjusted to pH 7.5. For inhibition curves, ATP (at the EC70 value at pH 7.5 (in μM): P2X1, 1; P2X2, 20; P2X3, 3; P2X4, 10) was added to the superfusate for 60–120 s, then washed off with Ringer's solution for 30 min. After obtaining agonist responses of consistent amplitude (Figure 1A), diinosine polyphosphates (IpnI, 0.1–100,000 nM) were added to the superfusate for 30 min before and during re-application of ATP. The blocking activity of IpnI compounds did not improve with pre-incubation periods longer than 10 min (Figure 1B) which suggested that IpnI blockade was not use-dependent, although IpnI compounds were routinely applied throughout the full 30 min of the ATP-washout period. For Schild analyses, the concentration/ response (C/R) relationship was determined for ATP (0.01–300 μM), then cRNA-injected oocytes were incubated with IpnI compounds for 30 min, after which ATP C/R curves constructed again in the continued presence of IpnI. EC50 values and slopes of C/R curves were taken from Hill plots, using the transform log (I/ImaxI) where I is the current evoked by each concentration of ATP. pA2 values were determined by Schild analysis, using the equation {log (DR–1)−log [IpnI]}.
Figure 1.

Consistency of agonist and antagonist activity. (A) histograms of the amplitudes of paired agonist-responses at homomeric rP2X1 and rP2X3 receptors (using ATP: rP2X1, 1 μM; rP2X3, 3 μM). Washout periods of 30 min were used between first and second applications of ATP, a periodicity sufficient to yield IATP responses of consistent amplitude. Thus, IpnI-related antagonism could not be explained in terms of a rundown of ATP-responses at P2X1 and P2X3 receptors. (B) the effect of increasing the pre-incubation period (10, 30 and 60 min) to Ip5I (3 μM) on the level of antagonism of ATP-responses (using 3 μM) at homomeric rP2X3 receptors. The blocking activity of Ip5I neither improved nor waned over 60 min pre-incubation, indicating that blockade was not use-dependent. Data: mean±s.e.mean (n=4) in A and B.
Statistics
Data are presented as means±s.e.mean of 4–6 sets of data from different oocyte batches. Significant differences were determined by either unpaired or paired Student's t-test using commercially-available software (Instat v2.05a, GraphPad).
Materials
All common salts were AnalaR grade (Aldrich Chemicals, U.K.). Adenosine 5′-triphosphate, diadenosine polyphos-phates (Ap3A, Ap4A, Ap5A) and phosphodiesterase (EC.3.1.15.1) (from Crotalus durissus) were purchased from Boehringer (Mannheim, Germany). 5′-adenylic acid deaminase (from Aspergillus sp.) was purchased from Sigma Chemical Co. (St. Louis, U.S.A.).
Results
Production of IpnI
5′-adenylic acid deaminase of Aspergillus sp. efficiently transformed Ap5A into Ip5I, fully converting the substrate into product over 90 min without further transformation of Ip5I into other by-products. During intermediate times, HPLC analysis of the reaction medium revealed an additional peak with a retention time between that of the initial Ap5A peak and final Ip5I peak (Figure 2A). This additional peak gradually disappeared over 90 min and was attributed to the intermediary reaction product (Ip5A), where only one of two adenosine moieties had been deaminated. Like results were obtained when producing Ip4I from Ap4A and Ip3I from Ap3A.
Figure 2.
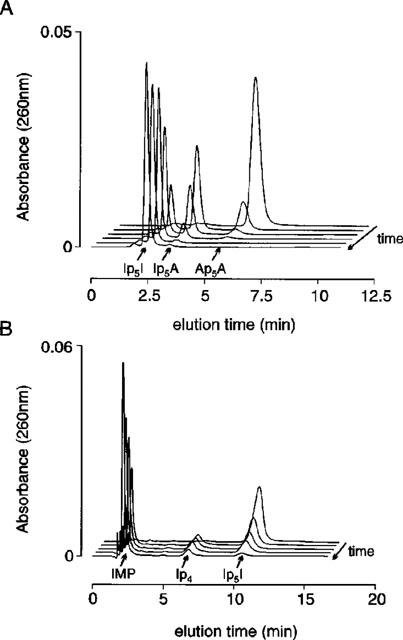
HPLC analysis of Ip5I production. (A) a series of chromatographic profiles for the time-dependent enzymatic conversion of Ap5A into the intermediary product Ip5A and final product Ip5I by adenylate deaminase (Aspergillus sp.) over a period of 90 min (chromatograms at: T=0, 15′, 30′, 45′, 60′ and 90′). (B) a series of chromatographic profiles for the time-dependent production of IMP and Ip4 by phosphodiesterase breakdown of the reaction product Ip5I (chromatograms at: T=0, 5′, 10′, 15′ and 30′). Ordinate scalars (A and B) as AUFS (absorbance units full scale), as defined by Millennium 2010 software (Waters).
The final reaction product was confirmed as Ip5I (as opposed to related breakdown products, i.e. IMP and Ip4) in experiments where phosphodiesterase was used. This enzyme cleaves Ip5I to yield inosine monophosphate (IMP) and the mononucleotide inosine 5′-tetraphosphate (Ip4) (Figure 2B). The retention times for these breakdown products was compared and matched against times for standard solutions of IMP and Ip4. The absence of IMP and Ip4 peaks (prior to enzymatic treatment at zero time) in HPLC analysis indicated Ip5I had not been broken down during its synthesis. Similar results were observed when testing the purity of Ip4I and Ip3I.
Blockade of P2X receptors by Ip5I
The pentaphosphate Ip5I (1 μM) was a potent inhibitor of ATP-responses at rP2X1 receptors, and weak inhibitor of ATP-responses at rP2X3 receptors (Figure 3A,B and Table 1). However, Ip5I failed to block rP2X2 receptors and potentiated ATP-responses at rP2X4 receptors (Figure 3C,D and Table 1). Thus, Ip5I is an antagonist at Group 1 P2X receptors (as defined by Humphrey et al., 1998), yet approximately 900 fold more potent at rP2X1 than rP2X3 receptors. The inhibitory and facilitatory effects of Ip5I were reversed on washout. Micromolar levels of Ip5I had no effect on the holding current of either Xenopus oocytes injected with cRNA for rP2X1–4 receptors or water-injected (control) oocytes. Thus, Ip5I inhibition or facilitation was not due to a partial agonistic effect.
Figure 3.
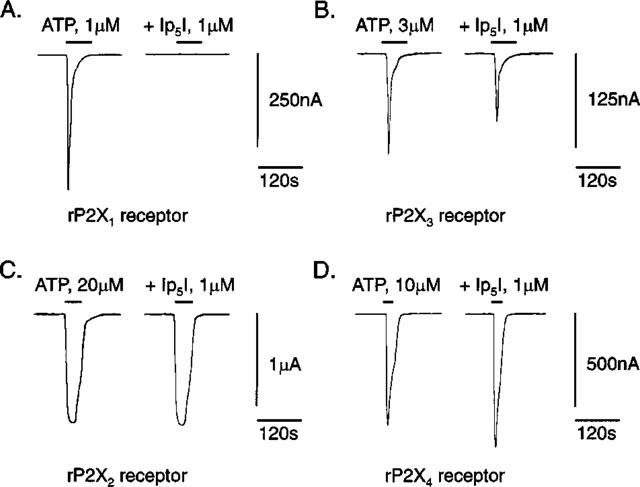
Ip5I antagonism of ATP-responses at rP2X1–4 receptors. ATP-activated whole-cell inward currents (IATP), before (first record) and during (second record) superfusion of micromolar Ip5I (1 μM, 30 min pre-incubation). Paired IATP records were taken from single cRNA-injected oocytes expressing homomeric rP2X1 (in A), rP2X3 (in B), rP2X2 (in C) and rP2X4 receptors (in D). ATP was applied at a concentration equivalent to the EC70 value for each recombinant rP2X1–4 receptor (see Methods). Vh=−60 mV in A–D.
Table 1.
Activity indices of IpnI compounds at rP2X1-4 receptors

Blockade of P2X receptors by IpnI series
The blocking activity Ip4I and Ip3I was also investigated at Group 1 P2X receptors (Figure 4A,B and Table 1). Ip4I was 180 fold less potent than Ip5I as an antagonist of rP2X1 receptors yet, in contrast, 3 fold more potent than Ip5I as an antagonist of P2X3 receptors. The triphosphate Ip3I was a weak antagonist of rP2X1 and rP2X3 receptors, only showing significant levels of blockade at high concentrations (>10μM). The blocking actions of Ip4I and Ip3I were reversed on washout.
Figure 4.

Inhibition curves for IpnI series at Group 1 P2X receptors. (A) Concentration-dependent inhibition of IATP (ATP, 1 μM) at homomeric rP2X1 receptors by Ip5I, Ip4I and Ip3I. (B) Concentration-dependent inhibition of IATP (ATP, 3 μM) at homomeric rP2X3 receptors by the same diinosine polyphosphates. IC50 values and slopes of inhibition curves are given in Table 1. Data: mean±s.e.mean (n=5) in A and B.
The actions Ip4I and Ip3I were also studied at rP2X2 and rP2X4 receptors (Figure 5A,B and Table 1). Both IpnI compounds, like Ip5I, were inactive as either antagonists or potentiators at rP2X2 receptors (Figure 5A). At rP2X4 receptors, Ip4I was over 800 fold less potent than Ip5I at potentiating ATP-responses (an effect reversed on washout), while Ip3I had no effect (Figure 5B and Table 1).
Figure 5.

Activity of IpnI series at rP2X2 and rP2X4 receptors. (A) The IpnI series neither inhibited nor potentiated ATP-responses (using 20 μM) at rP2X2 receptors. (B) Ip5I and Ip4I potentiated ATP-responses (using 10 μM) at rP2X4 receptors. Activity indices for IpnI series given in Table 1. Data: mean±s.e.mean (n=5) in A and B.
Schild analysis of Ip5I blockade
Ip5I (10, 30 and 100 nM) caused a rightwards displacement of the ATP concentration-response (C/R) curve, without altering the maximum agonist effect (Figures 6A and 8 and Table 2). However, higher concentrations of Ip5I (1 and 10 μM) completely blocked the agonist effects of ATP (⩽1 mM) (Figure 8), beyond which ATP exerts non-specific excitatory effects on Xenopus oocytes (Kupitz & Atlas, 1993). Thus, Ip5I blockade was surmountable at low Ip5I concentrations (⩽100 nM), and nonsurmountable at micromolar concentrations. The antagonistic effects of Ip5I (10–100 nM) on ATP C/R curves (EC50 values, Hill coefficients and pA2 values) are summarized in Table 2. Schild analysis of paired C/R curves revealed that pA2 values were not uniform, varying between 7.70–9.23 (range of 12 determinations) for the mean pA2 values listed (Table 2). This variability, in combination with nonsurmountable blockade with micromolar Ip5I, indicated that this dinucleotide did not antagonize ATP-mediated membrane currents at rP2X1 receptors in a simple competitive manner.
Figure 6.
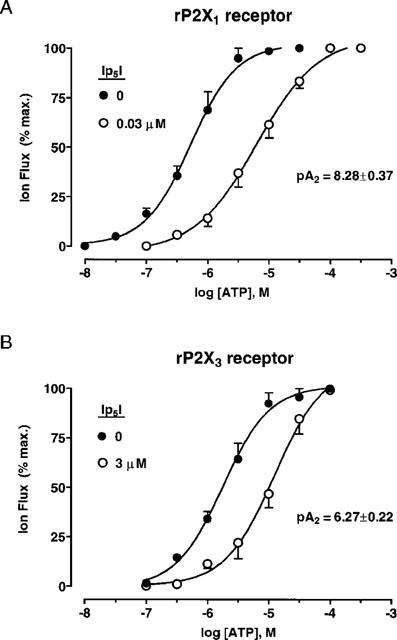
Ip5I antagonism of ATP-responses at Group 1 P2X receptors. (A) Concentration-response curves for ATP (0.01–300 μM) at homomeric rP2X1 receptors, before and during the presence of Ip5I (30 nM). (B) Concentration-response curves for ATP (0.1–100 μM) at homomeric rP2X3 receptors, before and during the presence of Ip5I (3 μM). EC50 values, Hill coefficients and pA2 values given in Table 2. Data: mean±s.e.mean (n=4) for paired C/R curves, in A and B.
Figure 8.
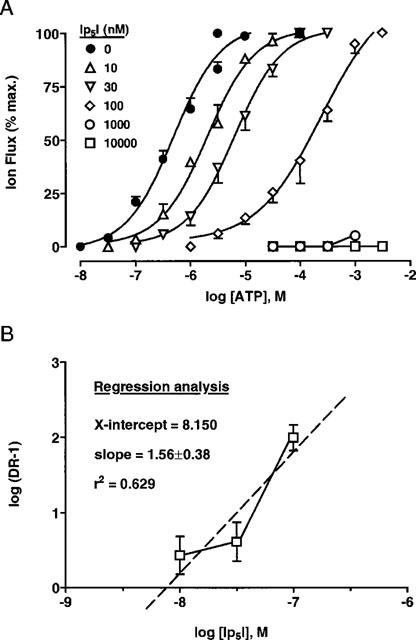
Schild analysis of Ip5I blockade of rP2X1 receptors. (A) Concentration/response (C/R) curves for the ATP-activated rP2X1 receptor, in the absence and presence of Ip5I (10 − 10,000 nM). The control C/R curve represented pooled data of 20 determinations, andtest C/R curves determined for four experiments for each of the five concentrations of Ip5I used (10, 30, 100, 1000 and 10,000 nM). (B) The Schild plot of C/R data according to EC50 values stated in Table 2. Linear regression (Prism v2.0, GraphPad) gave an estimated pA2 of 8.150, mean slope of 1.56 (i.e., greater than unity), and a low correlation coeffcient (r2) of 0.629 for linearity. The Schild plot was clearly biphasic.
Table 2.
Schild analysis of Ip5I displacement of ATP C/R curves at rP2X1 receptors

On the other hand, high concentrations of Ip5I (3 μM, approximate IC50 value) did cause a surmountable antagonism of ATP-responses at rP2X3 receptors (Figure 6B). EC50 values and Hill coefficients for ATP were (for the following Ip5I concentrations): 0 μM, 1.4±0.4 μM (nH, 1.13±0.12); 3 μM, 9.6±2.5 μM (nH: 1.08±0.18) (n=4). The pA2 value for Ip5I antagonism at rP2X3 receptors was 6.27±0.22 (n=4).
Schild analysis of Ip4I blockade
Ip4I (3 μM, approximate IC50 value) caused a nonsurmountable inhibition of ATP-response at rP2X1 receptors, in the manner of non-competitive antagonist (Figure 7A). EC50 values and Hill coefficients for ATP were (for the following Ip4I concentrations) : 0 μM, 0.38±0.06 μM (nHH, 0.84±0.17); 3 μM, 1.91±0.43 μM (nH, 0.63±0.46) (n=4). The maximum ATP effect was reduced by 56±7% (n=4) by the dinucleotide. Thus, Ip4I caused a reduction in both agonist potency and efficacy.
Figure 7.
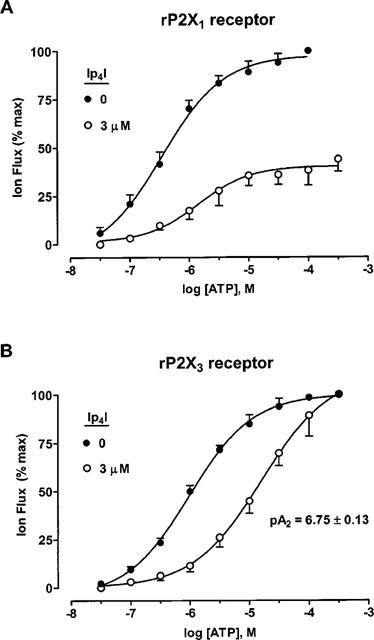
Ip4I antagonism of ATP-responses at Group 1 P2X receptors. (A) Concentration-response curves for ATP (0.03–300 μM) at homomeric rP2X1 receptors, before and during the presence of Ip4I (3 μM). (B) Concentration-response curves for ATP (0.03–300 μM) at homomeric rP2X3 receptors, before and during the presence of Ip4I (3 μM). EC50 values, Hill coefficients and pA2 values given in the text. Data: means±s.e.mean (n=4) for paired C/R curves, in A and B.
At rP2X3 receptors, Ip4I (3 μM, approximate IC75 value) caused a surmountable inhibition of ATP responses (Figure 7B). EC50 values and Hill coefficients for ATP were (for the following Ip4I concentrations): 0 μM, 1.0±0.3 μM (nH, 0.82±0.11); 3 μM, 17.8±5.3 μM (nH, 0.76±0.19) (n=4). The pA2 value for Ip4I antagonism at rP2X3 receptors was 6.75±0.13 (n=4).
Discussion
The present results showed that Ip5I was an effective antagonist of ATP-responses at Group 1 P2X receptors (P2X1 and P2X3), being selective for rP2X1 receptors at low (⩽100 nM) concentrations. This pentaphosphate was 900 fold less potent at P2X3 receptors (comparing IC50 values), at which micromolar concentrations were required to block ATP-activated inward currents. The blocking actions of Ip5I at P2X1 receptors at nanomolar concentrations initially seemed consistent with a competitive antagonism. However, several features suggested that the mechanism of Ip5I blockade was more complex than originally thought. First, micromolar concentrations of Ip5I caused a nonsurmountable inhibition of ATP-responses (Figure 8A). Second, determinations of the pA2 value were dependent on the Ip5I concentration used (see Table 2). Third, a Schild plot of combined Ip5I data (10, 30 and 100 nM) was non-linear and the slope (by regression analysis) was greater than unity (Figure 8B). Taken together, these features suggested that Ip5I is not a simple competitive antagonist at rP2X1 receptors. Also, the parent compound Ap5A is a partial agonist at rP2X1 receptors (Wildman et al., 1999a), suggesting that the binding site for the pentaphosphate (as either Ap5A or Ip5I) might not be the same position as the ATP docking site.
For P2X1 receptors, the blocking activity of the IpnI series decreased as the phosphate chain was reduced in length. Thus, Ip5I was 180 fold more potent than Ip4I and greater than 10,000 fold more potent than Ip3I. This potency order for antagonism clearly contrasted with the agonist potency order of their parent compounds at rP2X1 receptors, where Ap4A (7.4)>Ap5A (6.0)>Ap3A (>4) (pEC50 values) (Wildman et al., 1999a). Like Ip5I, Ip4I was a non-competitive antagonist at rP2X1 receptors and significantly reduced the maximum ATP effect. Its parent compound, Ap4A, is a partial agonist at rP2X1 receptors, suggesting the binding site for the tetraphosphate (as either Ap4A or Ip4I) might again differ from the ATP docking site. Interestingly, neither dinucleotide triphosphate (Ip3I and Ap3A) interacted well with rP2X1 receptors in terms of antagonist and agonist activities, yet rP2X1 receptor is activated by a number of mononucleoside triphosphates.
The rP2X3 receptor showed a slight preference for Ip4I over Ip5I, their pA2 values being 6.75 and 6.27, respectively. Both diinosine compounds appeared to act as competitive antagonists, causing a parallel rightwards shift of the ATP C/R curve without reducing the maximum ATP effect (Figures 6B and 7B). Their parent compounds, Ap4A and Ap5A, are both full agonists at rP2X3 receptors, at which the tetraphosphate (pEC50, 6.10) is slightly more potent than the pentaphosphate (pEC50, 5.88) (Wildman et al., 1999a). As far as the triphosphate is concerned, Ip3I is a weak antagonist and Ap3A a partial agonist at rP2X3 receptors.
The IpnI series lacked activity at rP2X2 receptors at which the parent ApnA compounds otherwise showed interesting effects. Ap4A is a full agonist at rP2X2 receptors, although 4 fold less potent than ATP, while nanomolar Ap5A is a potent potentiator of ATP-responses (Pintor et al., 1996). Thus, the inability of Ip4I and Ip5I to interact with rP2X2 receptors contrasted sharply with our earlier results with their parent compounds (Ap4A and Ap5A), although the lack of activity of the diinosine polyphosphates at least reflects the efficiency of the enzyme degradation process to make IpnI compounds. Ip5I was reported to be inactive against ATP-responses at P2X2-like receptors in neonatal rat cerebellar Purkinje neurons, although Ip3I and Ip4I have not yet been tested in this model (Garcia-Lecea et al., 1999).
For rP2X4 receptors, the potentiating effects of Ip5I and Ip4I struck a chord with similar potentiating effects of other compounds (e.g. suramin, PPADS and Reactive blue 2) tested as P2 receptor antagonists at the rat P2X4 receptor (Bo et al., 1995) and mouse P2X4 receptor (Townsend-Nicholson et al., 1999). The IpnI compounds proved just as ineffective as antagonists at rP2X4 receptors as other compounds tested so far. The parent dinucleotide of Ip4I is a partial agonist at rP2X4 receptors (pEC50, 5.5) (Wildman et al., 1999a). However, the potentiating effect of Ip4I was not accompanied by any change in holding currents and was not believed to be due to Ap4A contamination.
Of the IpnI series, only Ip5I has been tested at native P2X receptors. Ip5I is considerably less potent as an antagonist at the P2X1-like receptor in guinea-pig vas deferens (pA2, 6.5±0.1) (Hoyle et al., 1997), compared to its blocking activity at the recombinant rP2X1 receptor in the present study (pA2 range, 7.70–9.23). This reduction in activity may be due to Ip5I breakdown by ecto-nucleotidases in guinea-pig vas deferens, or perhaps due to a difference between guinea-pig and rat P2X1 receptors. The guinea-pig P2X1 receptor has not yet been cloned and, furthermore, Ip5I has not yet been tested on the human P2X1 receptor. Thus far, the blocking activity of Ip5I has only been characterized at the rat P2X1 receptor. Potential species differences in blocking activity notwithstanding, the observed Ip5I activity at native P2X1 receptors in guinea-pig still compares favourably with the non-competitive blocking activity of suramin (pKb, 5.3±0.2), iso-PPADS (pKb, 6.6±0.2) and Reactive blue 2 (pKb, 5.8±0.2) at P2X1-like receptors in rat vas deferens (Khakh et al., 1994). To this end, Ip5I may yet prove to be a useful pharmacological tool in bioassays of naturally-occurring P2X1 receptors, for example human HL60 cells (Buell et al., 1996) and murine thymocytes (Chavtcho et al., 1996) as well as P2X1-like receptors in vas deferens and vascular smooth muscle of various mammalian species (Humphrey et al., 1998).
Acknowledgments
This work was supported by grants from British Heart Foundation (U.K.), DGCYT (PM-98 0089; Spain), CAM (8.5/18/98; Spain) and BIOMED II (European Union). We are also grateful for financial support from Roche BioScience (Palo Alto, U.S.A.). We thank the following investigators for P2X receptor plasmids: rP2X1, Dr Gary Buell (Glaxo, Geneva, Switzerland); rP2X2, Professor David Julius (UCSF, California, U.S.A.); rP2X3, Professor John Wood (UCL, U.K.); rP2X4, Dr Xuenong Bo (UCL, U.K.). We acknowledge the assistance of Dr Andrea Townsend-Nicholson (UCL, U.K.) who prepared cRNAs for rP2X1–4.
Abbreviations
- ATP
adenosine 5′-triphosphate
- Ap5A
diadenosine pentaphosphate
- D-β, γ-meATP
D isomer of β,γ-methylene ATP
- L-β,γ-meATP
L isomer of β,γ-methylene ATP
- DIDS
4,4′-diisothiocyanostilbene-2,2′-disulphonic acid
- IATP
ATP-activated membrane current
- IC50
concentration causing 50% reduction of agonist response
- Ip3I
diinosine triphosphate
- Ip4I
diinosine tetraphosphate
- Ip5I
diinosine pentaphosphate
- KN-62
1-[N,O-bis(5-isoquinolinesulphonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine
- MRS 2220
cyclic pyroxidine-α4,5-monophos-phate-6-azophenyl-2′,5′-disulphonic acid
- NF023
8,8′-(carbonylbis(imino-3,1-phenylene carbonyliminobis-(1,3,5-napthalenetrisulphonic acid)
- nH Hill coefficient; pA2
negative log of antagonist concentration causing a 2 fold decrease in agonist potency
- pEC50
negative log of agonist concentration causing 50% of maximum effect
- pIC50
negative log of antagonist concentration causing 50% reduction of agonist response
- PPADS
pyridoxal-α5-phosphate-6-azophenyl-2′,4′-disulphonic acid
- rP2X1–4
rat P2X1–4 receptor subtypes, RB-2, Reactive blue 2
- TNP-ATP
2′,3′-O-(2,4,6-trinitrophenol)-ATP
- Vh
holding potential
References
- BO X., ZHANG Y., NASSAR M., BURNSTOCK G., SCHOEPFER R. A P2X purinoceptor cDNA conferring a novel pharmacological profile. FEBS Lett. 1995;375:129–133. doi: 10.1016/0014-5793(95)01203-q. [DOI] [PubMed] [Google Scholar]
- BUELL G., MICHEL A.D., LEWIS C., COLLO G., HUMPHREY P.P., SURPRENANT A. P2X1 receptor activation in HL60 cells. Blood. 1996;87:2659–2664. [PubMed] [Google Scholar]
- CHAVTCHO Y., VALERA S., AUBRY J.P., RENNO T., BUELL G., BONNEFOY J.Y. The involvement of an ATP-gated ion channel, P(2X1), in thymocyte apoptosis. Immunity. 1996;5:275–283. doi: 10.1016/s1074-7613(00)80322-2. [DOI] [PubMed] [Google Scholar]
- COLLO G., NORTH R.A., KAWASHIMA E., MERLO-PICH E., NEIDHART S., SURPRENANT A., BUELL G. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS R.J., LEWIS C., BUELL G., VALERA S., NORTH R.A., SURPRENANT A. Pharmacological characterization of heterolously expressed ATP-gated cation channels (P2X purinoceptors) Mol. Pharmacol. 1995;48:178–183. [PubMed] [Google Scholar]
- EVANS R.J., SURPRENANT A. P2X receptors in autonomic and sensory neurons. Semin. Neurosci. 1996;8:217–223. [Google Scholar]
- EVANS R.J., SURPRENANT A., NORTH R.A.P2X receptors: Cloned and Expressed The P2 Nucleotide Receptors 1998Humana Press, New Jersey, Ch. 2; 43–62.In: Turner J.T, Weisman G.A., Fedan J.S. (eds) [Google Scholar]
- GARCIA-LECEA M., DELICADO E.G., MiRAS-PORTUGAL M.T., CASTRO E. P2X2 characteristics of the ATP receptor coupled to [Ca2+]i increases in cultured Purkinje neurons from neonatal rat cerebellum. Neuropharmacol. 1999;38:699–706. doi: 10.1016/s0028-3908(98)00225-1. [DOI] [PubMed] [Google Scholar]
- GURANOWSKI A.E., STARZYNSKA E., GÜNTHER SILLERO M.A., SILLERO A. Conversion of adenosine(5′)oligophospho(5′)adenosines into inosine(5′)oligophospho(5′)inosines by non-specific adenylate deaminase from the snail Helix pomatia. Biochim. Biophys. Acta. 1995;1243:78–84. doi: 10.1016/0304-4165(94)00110-j. [DOI] [PubMed] [Google Scholar]
- HOYLE C.H.V., PINTOR J., GUALIX J., MIRAS-PORTUGAL M.T. Antagonism of P2X receptors in guinea-pig vas deferens by diinosine pentaphosphate. Eur. J. Pharmacol. 1997;333:R1–R2. doi: 10.1016/s0014-2999(97)01129-1. [DOI] [PubMed] [Google Scholar]
- HUMPHREY P.P.A., KHAKH B.S., KENNEDY C., KING B.F., BURNSTOCK G. IUPHAR Compendium of Receptor Characterization and Classification. IUPHAR Media Publications; 1998. Nucleotide receptors: P2X receptors; pp. 195–208. [Google Scholar]
- HUMPHREYS B.D., VIRGINIO C., SURPRENANT A., RICE J., DUBYAK G.R. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol. Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- JACOBSON K.A., KIM Y.C., WILDMAN S.S., MOHANRAM A., HARDEN K., BOYER J.L., KING B.F., BURNSTOCK G. A pyridoxine cyclic phosphate and its 6-azoaryl-derivative selectively potentiate and antagonize activation of P2X1 receptors. J. Med. Chem. 1998;41:2201–2206. doi: 10.1021/jm980183o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHAKH B.S., MICHEL A., HUMPHREY P.P.A. Estimates of antagonist affinities at P2X purinoceptors in rat vas deferens. Eur. J. Pharmacol. 1994;263:301–309. doi: 10.1016/0014-2999(94)90726-9. [DOI] [PubMed] [Google Scholar]
- KIM M., YOO O.J., CHOE S. Molecular assembly of the extracellular domain of P2X2, an ATP-gated ion channel. Biochem. Biophys. Res. Comm. 1997;240:618–622. doi: 10.1006/bbrc.1997.7713. [DOI] [PubMed] [Google Scholar]
- KING B.F.Molecular Biology of P2X Purinoceptors Cardiovascular Biology of Purines 1998Kluwer Academic Publications, Massachusetts; 159–186.In: Burnstock G., Dobson J.G., Liang B.T., Linden J. (eds)Ch. 10 [Google Scholar]
- KING B.F., ZIGANSHINA L.E., PINTOR J., BUNSTOCK G. Full sensitivity of P2X2 purinoceptor to ATP revealed by changing extracellular pH. Br. J. Pharmacol. 1996;117:1371–1373. doi: 10.1111/j.1476-5381.1996.tb15293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUPITZ Y., ATLAS D. A putative ATP-activated Na+ channel involved in sperm-induced fertilization. Science. 1993;261:484–486. doi: 10.1126/science.8392753. [DOI] [PubMed] [Google Scholar]
- NICKE A., BÄUMERT H.G., RETTINGER J., EICHELE A., LAMBRECHT G., MUTSCHLER E., SCHMALZING G. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J. 1998;17:3016–3028. doi: 10.1093/emboj/17.11.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTOR J., DÍAZ-HERNÁNDEZ M., BUSTAMANTE C., GUALIX J., DE TERREROS F.J., MIRAS-PORTUGAL M.T. Presence of dinucleotide and ATP receptors in human cerebrocortical synaptic terminals. Eur. J. Pharmacol. 1999;366:159–165. doi: 10.1016/s0014-2999(98)00922-4. [DOI] [PubMed] [Google Scholar]
- PINTOR J., GUALIX J., MIRAS-PORTUGAL M.T. Diinosine polyphosphates, a group of dinucleotides with antagonistic effects on diadenosine polyphosphate receptor. Mol. Pharmacol. 1997;51:277–284. doi: 10.1124/mol.51.2.277. [DOI] [PubMed] [Google Scholar]
- PINTOR J., KING B.F., MIRAS-PORTUGAL M.T., BURNSTOCK G. Selectivity and activity of adenine dinucleotides at recombinant P2X2 and P2Y1 receptors. Br. J. Pharmacol. 1996;119:1006–1012. doi: 10.1111/j.1476-5381.1996.tb15771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAE M.G., ROWAN E.G., KENNEDY C. Pharmacological properties of P2X3-receptors present in neurones of the rat dorsal root ganglia. Br. J. Pharmacol. 1998;124:176–180. doi: 10.1038/sj.bjp.0701803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- SOTO F., LAMBRECHT G., NICKEL P., STUHMER W., BUSCH A.E. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacol. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- TORRES G.E., EGAN T.M., VOIGT M.M. Hetero-oligomeric assembly of P2X receptor subunits. J. Biol. Chem. 1999;274:6653–6659. doi: 10.1074/jbc.274.10.6653. [DOI] [PubMed] [Google Scholar]
- TOWNSEND-NICHOLSON A., KING B.F., WILDMAN S.S., BURNSTOCK G. Molecular cloning, functional characterization and possible cooperativity between the murine P2X4 and P2X4a receptors. Mol. Brain. Res. 1999;64:246–254. doi: 10.1016/s0169-328x(98)00328-3. [DOI] [PubMed] [Google Scholar]
- VIRGINIO C., ROBERTSON G., SURPRENANT A., NORTH R.A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X receptors. Mol. Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- WILDMAN S.S., BROWN S.G., KING B.F., BURNSTOCK G. Selectivity of diadenosine polyphosphates for rat P2X receptor subunits. Eur. J. Pharmacol. 1999a;367:119–123. doi: 10.1016/s0014-2999(98)00976-5. [DOI] [PubMed] [Google Scholar]
- WILDMAN S.S., KING B.F., BURNSTOCK G. Modulation of ATP-responses at recombinant rP2X4 receptors by extracellular pH and Zinc. Br. J. Pharmacol. 1999b;126:762–768. doi: 10.1038/sj.bjp.0702325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILDMAN S.S., KING B.F., BURNSTOCK G. Modulatory activity of extracellular H+ and Zn2+ on ATP-responses at rP2X1 and rP2X3 receptors. Br. J. Pharmacol. 1999c;128:486–492. doi: 10.1038/sj.bjp.0702802. [DOI] [PMC free article] [PubMed] [Google Scholar]