

Abstract
The cellular and molecular actions of BW534U87 were studied using intracellular and extracellular recordings from the CA1 region of rat hippocampal slices and whole-cell voltage-clamp recordings of recombinant human brain type IIA Na+ channels expressed in Chinese hamster ovary (CHO) cells.
Normal excitatory and inhibitory postsynaptic potentials evoked in hippocampal slices were unaffected by BW534U87 or the adenosine deaminase inhibitor EHNA. However, epileptiform activity was depressed by BW534U87 (50 μM) and this inhibition was reversed by the adenosine receptor antagonist 8-phenyl theophylline (8-PT, 30 μM). EHNA (10 μM) mimicked the effects of BW534U87. Furthermore, BW534U87 enhanced the inhibitory effects of exogenous adenosine on evoked synaptic potentials. BW534U87 (50 μM) also voltage- and use-dependently inhibited action potentials elicited by current injection, independent of the adenosine system, since it was not affected by 8-PT.
In CHO cells expressing the recombinant human brain Na+ channel, BW534U87 produced a concentration- and voltage-dependent inhibition of Na+ currents with a half-maximal inhibitory concentration of 10 μM at a Vh of −60 mV. Use-dependent inhibition was evident at high-frequencies (20×20 ms pulse train at 10 Hz).
In conclusion, BW534U87 blocks hippocampal epileptiform activity by a dual mechanism. The first action is similar to that produced by EHNA and is dependent on endogenous adenosine probably by inhibition of adenosine deaminase. Secondly, BW534U87 directly inhibits voltage-gated Na+ channels in a voltage- and frequency-dependent manner. Both actions of BW534U87 are activity-dependent and may synergistically contribute to its overall anticonvulsant effects in animal models of epilepsy.
Keywords: Anticonvulsant, BW534U87, EHNA, lamotrigine, adenosine, adenosine deaminase, Na+ channels, intracellular recording, patch-clamp recording, hippocampal slices
Introduction
The novel anticonvulsant compound BW534U87, (1-[(2,6-difluorophenyl)-methyl]-1H-1,2,3-triazolo[4,5-c]) pyridine-4-amine mono hydrochloride) is structurally different from all currently used antiepileptic drugs (AEDs). The compound has been shown to have potent activity against seizures induced by maximal electroshock in rats (a model of generalized tonic-clonic epilepsy, Kelly et al., 1995) and was later shown to be active in a range of acute and chronic epilepsy models (Stratton et al., 1998). However, the cellular and molecular mechanisms underlying the drug's anticonvulsant actions are unknown. Interestingly, the compound was found to cause a reversible competitive inhibition of rat brain adenosine deaminase (ADA) with a Ki of 7 μM in biochemical studies (Dr E. Southam, personal communication; Dupere et al., 1999). Adenosine exerts potent inhibitory modulation on synaptic activity in both peripheral and central nervous systems (for reviews see Greene & Haas, 1991; Brundege & Dunwiddie, 1997). Moreover, adenosine produces differential modulation on evoked excitatory and inhibitory postsynaptic potentials in rat hippocampus (EPSP-IPSPs; Thompson et al., 1992). The inhibition of excitatory postsynaptic potentials and population spikes in the hippocampus is mediated by the adenosine A1 receptor (Schubert & Mitzdorf, 1979; Brundege & Dunwiddie, 1997). Anticonvulsant effects in vivo have been reported after activation of the adenosine system through the use of an adenosine A1 receptor agonist (Dunwiddie & Worth, 1982; Murray et al., 1985; Concas et al., 1993; Malhotra & Gupta, 1997). Similarly, anticonvulsant effects have been observed following inhibition of adenosine metabolic enzymes, mainly adenosine kinase or ADA (Shimuzu et al., 1972; Arch & Newsholme, 1978; Zhang et al., 1993; He et al., 1993), which would result in an elevation and accumulation of endogenous adenosine (Haas & Greene, 1988; Pak et al., 1994).
Adenosine is formed from the metabolism of adenosine triphosphate (ATP). The main source of extracellular adenosine in the brain is from intracellular adenosine via a bidirectional nucleoside transporter or evoked release (Bender et al., 1980; Nimit et al., 1981; Wojcik & Neff, 1982). An additional source of extracellular adenosine is from the breakdown of released ATP (Burger & Lowenstein, 1970; Ribeiro & Sebastiao, 1987; Hamilton & Smith, 1991). The basal extracellular adenosine concentration was estimated to be 1–2 μM (Newman & McIlwain, 1977; Van Wylen et al., 1986) and can be dramatically increased up to approximately 50 μM under certain pathological conditions, such as seizure activity (Winn et al., 1980; Lewin & Bleck, 1981). Adenosine kinase has a Km for the breakdown of adenosine of approximately 2 μM (Arch & Newsholme, 1978), which is close to baseline levels of adenosine. Therefore, the breakdown of adenosine by adenosine kinase is likely to be the main metabolic pathway under normal conditions. ADA deaminates adenosine to inosine with a Km of approximately 50 μM (Arch & Newholme, 1978), which is much higher than the Km of adenosine kinase. Consequently, the rise in extracellular adenosine concentrations during neuronal hyperexcitability or seizures (Winn et al., 1980; Lewin & Bleck, 1981; During & Spencer, 1992) would rapidly saturate adenosine kinase and make ADA the most likely route for metabolism.
In the present study, we have studied the functional significance of inhibition of ADA by this novel anticonvulsant BW534U87 in comparison to the adenosine deaminase inhibitor erythro - 9 - (2 - hydroxy - 3 - nonyl)adenine (EHNA). Other possible mechanisms underlying the anticonvulsant effects of the compound in animal models were also examined using intracellular and extracellular recordings from rat hippocampal slices and whole-cell voltage-clamp recordings of recombinant human brain type IIA Na+ channels expressed in CHO cells. Preliminary data has been presented in abstract form (Dupere et al., 1998; 1999).
Methods
Brain slice preparation
Hippocampal slices were prepared as previously described (Xie & Smart, 1993). Briefly, Sprague Dawley rats (either sex, aged 32–56 days) were killed by cervical dislocation and decapitated. The brain was rapidly removed and placed in ice-cold Krebs-Henseleit medium containing (mM): NaCl 119, NaHCO3 26, KCl 3, MgCl2 2, CaCl2 2, D-Glucose 11, maintained at pH 7.4 by bubbling with 95% O2 5% CO2. Coronal slices (400 μm thick) were prepared using a Vibroslice (Campden Instruments). Slices were stored at room temperature (21–23°C in a holding container for at least 1 h and up to 8 h prior to recording.
Intracellular current-clamp recordings
A single slice was transferred to the recording chamber (1 ml) and continually superfused with Krebs-Henseleit solution at 1 ml min−1 at 30°C. Intracellular recording electrodes were fabricated on a Sutter P87 electrode puller and filled with filtered 3 M KCl solution (resistance 50∼80 MΩ) or 4 M K-acetate (resistance 80∼150 MΩ). Electrodes were advanced into the pyramidal layer of the CA1 region of the hippocampus. Impaled neurones were accepted if the resting membrane potential was at least −55 mV and with an over-shoot of action potential amplitude of 20∼30 mV. The bridge balance in the amplifier (Axoclamp 2B, Axon instruments) was monitored by applying brief hyperpolarizing current injections throughout the experiment and adjusted before each measurement. Orthodromic stimulation (0.2–3 mA, 0.1 ms, 0.1 Hz) to CA1 neurones was applied with bipolar tungsten stimulating electrodes with a separation <50 μm. These electrodes were placed in the stratum lucidum stimulating the Schaffer collateral/commisural afferents to the CA1 subfield. Most intracellular recordings were stable for 1–3 h.
Extracellular recordings
Slices were transferred to the recording chamber as for intracellular recordings. Extracellular recording electrodes were 2–5 MΩ when filled with 4 M NaCl. Electrodes were advanced into the CA1 region. Field potentials were amplified ×1000 through a Neurolog A.C. amplifier (Digitimer), 0.5 Hz–1 kHz bandpass filtered and digitized at 10 kHz using a CED1401 with Spike 2 software (Cambridge Electronic Design).
Cell culture
CHO cells were stably transfected with cDNA encoding the human brain type IIA Na+ channel α-subunit (Xie et al., 1997). These cells were cultured in Iscove's modified Dulbecco's medium (Gibco), containing 10% dialysed foetal calf serum (Gibco), non-essential amino acids (Sigma), H-T supplement (hypoxanthine, thymidine, Gibco) and penicillin/streptomycin (10,000 units ml−1/10 mg ml−1, Gibco). To amplify expression and to prevent cell toxicity resulting from excessive Na+ influx, the media also contained 50 nM methatrexate (Sigma) and 200 nM tetrodotoxin (TTX, Sigma). Cells were grown in a 5% CO2 atmosphere with 95% humidity at 37°C. One to three days prior to electrophysiological recordings the cells were plated onto poly-DL-lysine coated glass coverslips.
Whole-cell voltage-clamp recordings
Cells growing on a glass coverslip were placed into the recording chamber (0.5 ml) and superfused with an extracellular solution at a rate of 2 ml min−1. The extracellular solution contained (mM): NaCl 140, KCl 4.7, MgCl2 1.2, CaCl2 1, glucose 11 and 4-(2-hydroxethyl)-1-piperazineethanesulphonic acid (HEPES, 5). The pH was adjusted to 7.4 using NaOH and the osmolarity ranged from 290–310 mOsm. Patch pipettes (borosilicate glass) were pulled using a Sutter P-97 electrode puller. The pipette was filled with an internal solution consisting of (mM): CsF 120, NaCl 15, ethylene glycol-bis(β-aminoethyl ether) N,N,N′,N -tetra acetic acid Cs salt (Cs-EGTA) 10 and HEPES 10. The pH was adjusted to 7.25 using CsOH and the osmolarity ranged from 275–290 mOsm. When filled with this internal solution, patch electrodes had resistances of 2–6 MΩ. Currents were recorded using standard whole-cell voltage clamp recording techniques (Hamill et al., 1981) at room temperature (21–23°C) using an Axopatch 200A amplifier. Signals were sampled at 25–50 kHz after low pass Bessel filtering at 5 kHz. The majority of series resistance errors (80–85%) were minimized with compensation circuitry. The leak current was subtracted from active currents using the P/4 protocol supplied with the pClamp6 software. Membrane potentials were not corrected for junction potentials (<4 mV). The cells were maintained at a holding potential (Vh) of −90 mV and the currents, evoked by depolarizations to 0 mV, progressively increased over the first 10–15 min.
Drug application
Drugs were applied via the perfusion system at a rate of 1 ml min−1, allowing at least 2 min to incubate the slices or cultured cells. Drugs were applied only after membrane potentials, population spikes or currents had stabilized. BW534U87 (GlaxoWellcome) and EHNA (RBI) were dissolved in 100% dimethyl sulphoxide (DMSO), adenosine (Sigma) in distilled water, bicuculline (Sigma) in 50% NaOH, 50% ethanol and 8-PT (Sigma) in 50% DMSO, 50% distilled water to produce each stock solution of 10 mM. Subsequent dilutions to produce the final concentrations required were made in the external solution. The final concentration of DMSO up to 0.5% had no obvious effects on Na+ currents, membrane excitability or excitatory synaptic transmission.
Data acquisition and analysis
Square wave current or voltage pulses and data collection were performed on-line using pClamp 6 (Axon Instruments) interfaced with amplifiers. Data were analysed off-line using pClamp 6, Spike 3 and Origin (MicroCal) software. Data are presented as either arithmetic mean±s.e.mean or geometric mean [95% confidence limits], where appropriate; n refers to the number of cells or slices in each experiment. Statistic significance levels (P<0.05) were determined with the paired Student's t-test.
In current-clamp recordings, EPSP areas were calculated by integration under the curve using Origin software after removal of spikes if necessary. In voltage-clamp recordings, construction of activation-conductance plots required the calculation of Na+ conductance (gNa) from the peak current (INa) according to the equation gNa=INa/(V–ENa), where V is the test pulse potential and ENa the reversal potential. The activation-conductance plot was fitted using the Boltzmann function: g/gmax=1/[1+exp(V½–V/k], where g is the normalized conductance relative to the maximum conductance (gmax), V½ is the membrane potential at which half the channels are activated and k is the slope of the curve. In the construction of inactivation curves the current was normalized (I) to that produced by a depolarization from −120 to 0 mV (Imax) and plotted against the conditioning pulse potentials. The inactivation curves were fitted according to the Boltzmann function: I/Imax=1/[1+exp(V–V½/k], where V is the conditioning pulse potential, V½ is the membrane potential at which half the channels are inactivated and k is the slope. The concentration-response curves were fitted, where possible, according to an independent binding site receptor model as given by the following equation: y/ymax={1-[D/(D+IC50)]n}, where y is the response in the presence of drug, ymax is the maximal response in the absence of drug, D is the drug concentration, IC50 is the concentration of drug producing a half maximal inhibition of the response and n is the Hill coefficient. Fit estimates were calculated using Marquardt non-linear least squares algorithms.
Results
Preferential inhibition of epileptiform activity by BW534U87 via enhancement of the action of endogenous adenosine
In normal conditions, the average resting membrane potential of hippocampal CA1 pyramidal neurones was −60.4±0.9 mV, with an input resistance of 40.7±1.8 MΩ (n=32 cells). Cells held at −70 mV were virtually devoid of spontaneous synaptic activity. The resting potential and input resistances were unchanged after application of 50 μM BW534U87 (−61.0±1.6 mV and 43.5±2.7 MΩ, n=11 cells) or 10 μM EHNA (−61.4±1.4 mV and 41.2±2.5 MΩ, n=6 cells). Normal EPSP-IPSPs in CA1 region evoked by stimulation of Schaffer collateral/commisural fibres in standard external solution with an EPSP area (excluding potentials below the holding potential) of 297±59 mV ms (n=7 cells). EPSP-IPSPs were not affected by 50 μM BW534U87 (n=7) as shown in the example in Figure 1A. Epileptiform activity was induced by removing extracellular Mg2+, elevating K+ (from 3 to 7 mM) and adding 20 μM bicuculline to the external solution (this buffer is referred to hereafter as ‘Mg2+-free media'). About 15–30 min after application of this media, cells were depolarized by 7.3±2.1 mV without change to the input resistance, which was 40.2±1.8 MΩ prior to application and 41.0±2.2 MΩ after (32 cells). Under these conditions the same stimulation intensity as used in normal solutions, evoked a larger EPSP (without IPSP) with an average EPSP area of 2186±285 mV ms (n=32 cells) and a burst of action potentials superimposed on the EPSP. Evoked synaptic EPSPs were mediated by glutamate both before and after application of the Mg2+-free media since under both conditions EPSPs were completely inhibited by a combination of the N-methyl D-aspartate (NMDA) receptor antagonist D-APV (D-2-amino-5-phosphovanilate, 20 μM) and the α-amino-3-hydroxy- 5-methyl-isoxazole (AMPA) antagonist CNQX (6-cyano-7-nitro-quinoxaline-2,3-dione, 20 μM, six cells, data not shown). It is unlikely that voltage-gated Na+ channels contribute to the EPSPs in control or in Mg2+-free media, since under identical conditions the Na+ channel blocker lamotrigine (50 μM) inhibits action potential firing without affecting the EPSP area (Xie et al., 1995).
Figure 1.
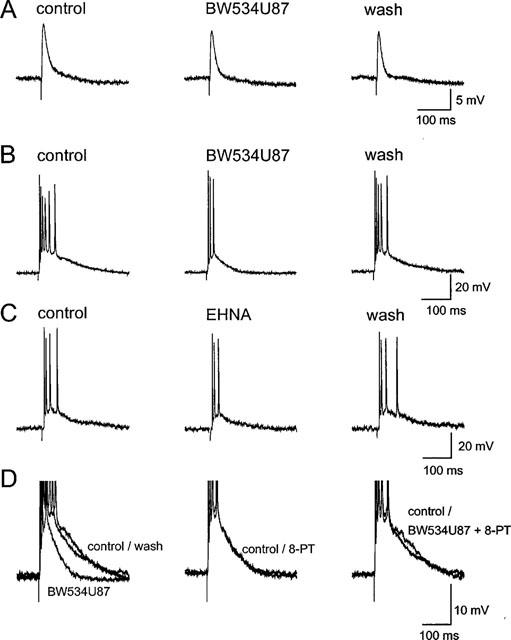
BW534U87 reduced epileptiform activity in hippocampal slices. (A) Normal excitatory and inhibitory postsynaptic potentials (EPSP–IPSPs) recorded in a CA1 pyramidal cell using a sharp microelectrode containing 4 M K-acetate, evoked by stimulation of the Schaffer collateral/commisural fibres, were not affected by BW534U87 (50 μM). (B) In the same neurone, 50 μM BW534U87 depressed epileptiform activity induced by perfusing the slice with a Mg2+-free solution containing 7 mM K+ and 20 μM bicuculline. (C) Depression of epileptiform activity recorded with an electrode containing 3 M KCl in a different neurone by 10 μM of the adenosine deaminase inhibitor EHNA. (D) In another neurone, the inhibition of the epileptiform EPSP area was reversed by co-application with the adenosine receptor antagonist 8-phenyl theophylline (8-PT, 30 μM). The membrane potentials of all cells were held at −70 mV.
In Mg2+-free media, application of 50 μM BW534U87 produced a small 4.2±0.7 mV hyperpolarization without significantly affecting the input resistance (n=6 cells). In the same conditions, the evoked EPSP area was inhibited by 46.3±7.2% and the number of spikes was significantly reduced from 5.2±0.4 to 2.5±0.2 spikes (Figure 1B; n=6 cells). The inhibition of the EPSP area by BW534U87 in Mg2+-free media was mimicked by the EHNA. Following application of 10 μM EHNA the EPSP area was significantly reduced by 52.0±10.7% (Figure 1C, n=5 cells). Similar to BW534U87, EHNA (10 μM) hyperpolarized cells by 5.4±1.1 mV (n=5 cells) without affecting the input resistance (41.2±1.3 MΩ). The depression of epileptiform activity by BW534U87 was reversed by the co-application with the adenosine receptor antagonist 8-PT (30 μM; n=6 cells, Figure 1D), while 8-PT alone produced little effect on the EPSP area or bursts.
Application of Mg2+-free media to hippocampal slices during intracellular recordings produced spontaneous firing of action potentials or bursting in 17/32 slices after 15–30 min. However, spontaneous regular bursts occurred in only 7/32 slices (22%). To examine the effect of BW534U87 on spontaneous burst firing, extracellular electrodes were used and the population burst firing was more stable. After application of the Mg2+-free media for 15 min, spontaneous population bursts were recorded in the CA1 region, with average amplitude of 0.63±0.22 mV at an average frequency of 0.91±0.13 Hz (n=4 slices). In normal media spontaneous bursts were not observed. BW534U87 (50 μM) significantly inhibited the population burst amplitude by 37.3±11.1% and non-significantly inhibited the burst frequency by 31.7±24.4% (Figure 2, n=4 slices). The effects of BW534U87 were fully reversible within 30 min following washout of the drug.
Figure 2.
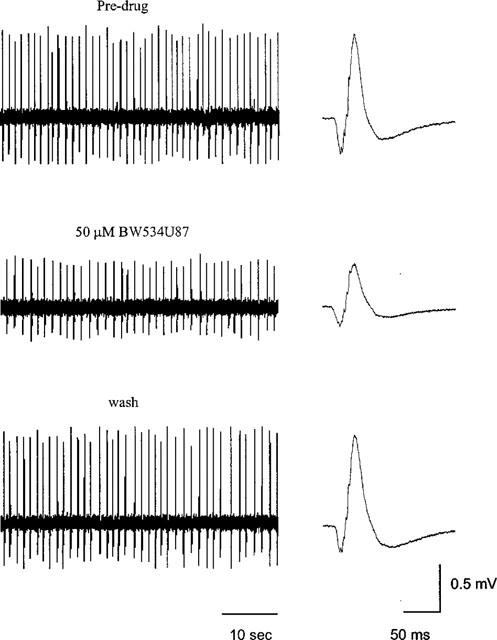
BW534U87 inhibits spontaneous epileptiform activity in hippocampal slices. Extracellular recording using a glass microelectrode from the CA1 region in a hippocampal slice. Spontaneous population bursts were recorded within 15 min following application of a Mg2+-free solution containing 7 mM K+ and 20 μM bicuculline. The top trace shows spontaneous population bursts in the absence of drug (left trace) and the average of 20 population bursts (right trace). Middle traces show the reduction in burst amplitude 15 min following application of 50 μM BW534U87. Bottom traces show the complete reversal of the effects of the drug within 30 min following washout.
Using intracellular recordings, to confirm that inhibition of epileptiform activity by BW534U87 is mediated by an enhancement of adenosine levels, concentration-response relationships for exogenously applied adenosine were constructed in the presence and absence of BW534U87. Application of adenosine (300 μM) produced an 8.6±1.5 mV (n=11 cells) hyperpolarization, which was corrected for by DC current injection to maintain the membrane at −70 mV. Figure 3A shows the concentration-related inhibition of the EPSP area by adenosine and the enhancement of this inhibition by 50 μM BW534U87. The adenosine deaminase inhibitor EHNA (10 μM) mimicked this effect of BW534U87 (Figure 3B). The relationship between the adenosine concentration and the EPSP area in the absence of drug (n=11 cells) and presence of BW534U87 (n=5 cells) or EHNA (n=4 cells) is shown in Figure 3C. The average IC50 value for the inhibition of the EPSP area by adenosine was estimated at 41.0 μM [29.8–53.9 μM] and the Hill coefficient at 1.8±0.1. In the presence of 50 μM BW534U87 the IC50 was reduced to 15.9 μM [7.3–25.8 μM] and the Hill coefficient to 1.5±0.1. In the presence of 10 μM EHNA the IC50 value was 18.9 μM [11.7–30.6 μM] and the Hill coefficient was 1.5±0.2. The IC50 values and the Hill coefficients in the presence of BW534U87 and EHNA were significantly different from those in control.
Figure 3.
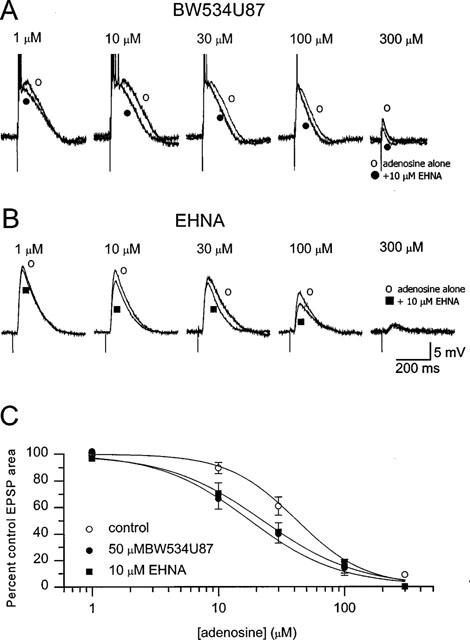
BW534U87 and EHNA enhanced the inhibitory effects of exogenous adenosine on evoked synaptic potentials. (A) Inhibition of the EPSP area in a CA1 pryramidal neurone by increasing concentration of exogenously applied adenosine (1–300 μM alone or in the presence of 50 μM BW534U87. (B) Inhibition of the EPSP by adenosine alone or in the presence of 10 μM EHNA. (C) Concentration-response curves for adenosine in the absence of drug (pooled data from nine cells) and in presence of BW534U87 (n= 5 cells) or 10 μM EHNA (n=4 cells). The smooth curves were obtained by fitting the data to an independent-binding-site receptor model. The IC50 for inhibition of the EPSP area by adenosine was estimated at 41.0 μM [29.8–53.9 μM) and a Hill coefficient was 1.8±0.1. In the presence of BW534U87 (50 μM), the IC50 for adenosine was 15.9 μM [7.3–25.8 μM] and the Hill coefficient was 1.5±0.1. In the presence of EHNA (10 μM) the IC50 for adenosine was 18.9 μM [11.7–30.6 μM] and the Hill coefficient was 1.5±0.2.
Inhibition of action potentials during the latter part of a spike train by BW534U87
Post-synaptic membrane excitability was monitored by electrotonic membrane potentials. Brief injections of depolarizing current (0.8 nA, 600 ms) into cells perfused in the normal control solution produced a train of action potentials which varied from 6 to 28 spikes (n= 11 cells). At a holding potential of −70 mV, a clear reduction in the number of spikes during the latter part of the train was found during perfusion of 50 μM BW534U87 (Figure 4A). On average, the number of spikes in control conditions was 15.1±2.1 spikes, which reduced to 6.9±0.8 in presence of 50 μM BW534U87 (n=11 cells). Virtually complete recovery to 14.0±2.0 spikes was observed after 10 min following washout of the drug. This property of BW534U87 was not shared by EHNA, where cells injected with depolarizing current (0.8 nA, 600 ms, n=6 cells) fired an average of 17.3±2.6 action potentials 10 min after application of 10 μM EHNA similar to the average of 17.0±2.0 prior to application (n=6 cells). The reduction in spike frequency by BW534U87 was ameliorated at the more negative holding potentials of −80 mV and −90 mV (Figure 4A). In the same 5 cells, at −70 mV BW534U87 (50 μM) produced a 49±10% reduction in spike number compared to 18±14% reduction at −80 mV and 14±11% at −90 mV. The reduction in the number of spikes in the train by BW534U87 was not affected by 30 μM 8-PT (Figure 4B). The reduction in spike number by BW534U87 was 54±9% and in the same cells after co-application of 30 μM 8-PT for 10 min, the reduction in spike number was 47±6% (resting potential=−61.9 ±2.3 mV, n=5 cells). The effects of 8-PT alone and in combination with BW534U87 are summarized on Figure 4C (n=5 cells).
Figure 4.
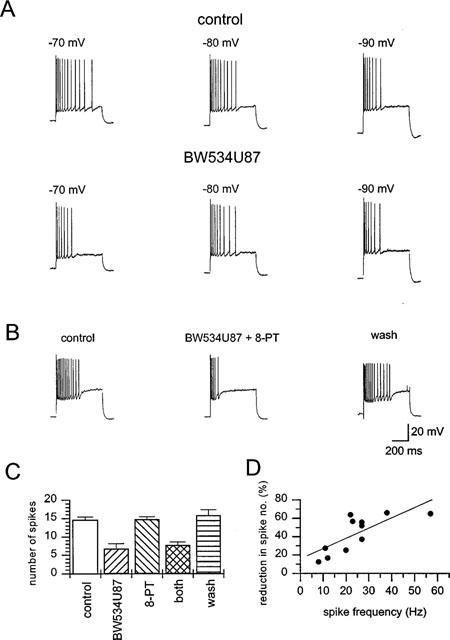
BW534U87 inhibited the latter action potentials in a train in hippocampal CA1 pyramidal neurones. (A) Trains of action potentials were elicited by a depolarizing current injection (0.8 nA, 600 ms) in normal solution (resting potential −63 mV). At a holding potential of −70 mV (left) BW534U87 (50 μM) inhibited the latter action potentials in the train. At the more hyperpolarized potentials of −80 mV (centre) and −90 mV (right) the effect of BW534U87 was less marked. (B) In another neurone, the inhibition of action potentials by BW534U87 was not affected by co-application with 8-phenyl theophylline (8-PT, 30 μM). (C) Summary of the average number of spikes elicited by depolarizing current injection (0.8 nA, 600 ms) into CA1 pyramidal neurones (n=5 cells) in control, in the presence of 50 μM BW534U87, 30 μM 8-PT or combination of both BW534U87 and 8-PT and after washout of drugs (horizontal hatched column). Values are the mean±s.e. mean. (D) Reduction in the number of action potentials in a train by BW534U87 (50 μM) was dependent on the initial adapted spike frequency prior to drug addition. The Spearman's product moment coefficient was 0.76.
The reduction in spike number by BW534U87 was spike frequency-dependent. Figure 4D shows the high correlation between spike frequency and the degree of reduction in spike number by 50 μM BW534U87 (n=11 cells). The correlation was significantly different from random with a Spearman's product moment coefficient (r) of 0.76. The higher the initial spike frequency the greater the reduction in spike number produced by the compound.
Voltage- and use-dependent inhibition of human brain type IIA Na+ channels by BW534U87
To determine whether the adenosine-independent inhibition of action potentials was mediated through a direct interaction with voltage-gated Na+ channels, we used recombinant Na+ channels expressed in CHO cells; a system without the complication of endogenous adenosine. Previous studies have shown that these currents are sensitive to inhibition by tetrodotoxin (Xie et al., 1997). Under voltage-clamp conditions, fast transient Na+ currents (−0.5 nA to −3.5 nA) were elicited by depolarizing cells to a range of test potentials between −80 and +70 mV (8 ms) from a holding potential (Vh) of −90 mV (Figure 5A). These fast Na+ currents were unaffected by 10 μM BW534U87 (n=5 cells). To examine whether the compound interacts with the inactivated channel, the voltage-dependence of inactivation was examined with conditioning pulses of 1 s duration prior to a test pulse to 0 mV (3 ms). BW534U87 (10 μM) produced a significant hyperpolarizing shift of 8.5 mV in the voltage-dependence of inactivation compared to the half-inactivation voltage (V½) in control conditions (control V½=−62.1±1.8 mV, Figure 5B, six cells). The voltage-dependence of inactivation by short conditioning pulses (10 ms) was more hyperpolarized (V½=−39.7±1.7 mV, n=6 cells) than with longer conditioning pulses. BW534U87 (50 μM) produced a non-significant hyperpolarizing shift of 2.6 mV in the V½ following these short conditioning pulses (n=6, data not shown). Since the inhibition of Na+ currents by BW534U87 was dependent on Vh, the concentration-dependence of block was examined at Vhs of −90 mV and −60 mV (pre-conditioned at −60 mV for 1 s from a Vh of −90 mV) and stepping to a test potential of 0 mV. Figure 5C shows the greater effect of 10 μM BW534U87 on currents evoked from a Vh of −60 mV compared to those evoked from a Vh of −90 mV. The onset of the inhibition was rapid (<60 s) and was readily reversible. The concentration-dependence of the inhibition of Na+ currents by BW534U87 at a Vh of −90 mV and −60 mV are shown in Figure 5C (n=5 cells). As BW534U87 comes out of solution at concentration above 50 μM it was not possible to complete a full concentration-response curve. Nevertheless, little inhibition of the current was observed up to 30 μM at a Vh of −90 mV, whereas at a Vh of −60 mV, BW534U87 produced a concentration-dependent inhibition of the current with an estimated IC50 of 10 μM and a Hill coefficient not significantly different from unity.
Figure 5.
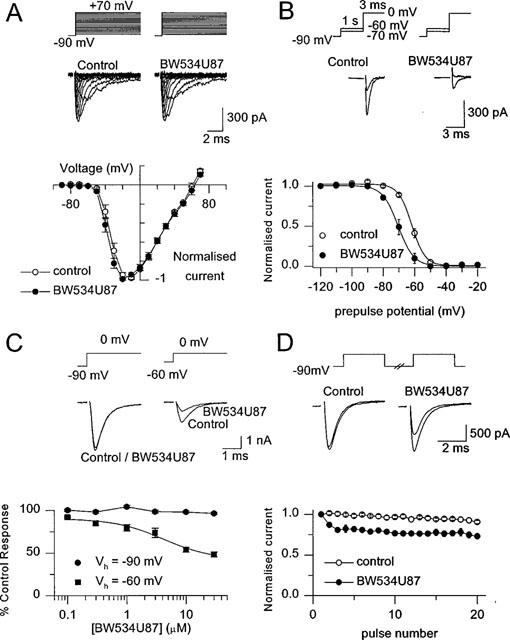
BW534U87 produced a state-dependent inhibition of recombinant human brain Na+ channels expressed in Chinese hamster ovary cells under voltage-clamp conditions. (A, upper traces) Currents evoked from a holding potential (Vh) of −90 mV to a range of potentials between −80 and +70 mV in control and following application of 10 μM BW534U87. (Lower traces) Peak current-voltage relationship in control and after application of BW534U87 (five cells). (B, Upper traces) Currents evoked at a test potential of 0 mV (3 ms) following a conditioning pulse of −70 or −60 mV (1 s) in control and in 10 μM BW534U87. (Lower traces) Effect of BW534U87 on the voltage-dependence of inactivation. Channels were inactivated by a range of conditioning pulses between −120 to −120 mV (1 s) prior to stepping to a test potential of 0 mV. Data are normalized to the current generated following a −120 mV conditioning pulse. Data were fitted with a Boltzmann function, where in control, the V½; was −62.1±1.8 mV and k was 4.4±0.2 mV/e-fold change. In the presence of 10 μM BW534U87, the V½; was −70.3±1.9 mV and k was 4.6±0.2 mV/e-fold change. (C, Upper traces) Currents evoked by a test pulse to 0 mV (8 ms) from a Vh of −90 or −60 mV (conditioning pulse to −60 mV for 1 s) in control and in 10 μM BW534U87. (Lower traces) Concentration-dependence of inhibition of currents by BW534U87 evoked from a Vh of −90 or −60 mV. The data are normalized with respect to the current amplitudes in the absence of the compound. Each point is the mean±s.e. mean (n=4∼7 cells). The smooth curve was obtained by fitting the data to an independent-binding-site receptor model, from which a half-maximal inhibitory concentration of 10 μM and a Hill coefficient of approximately 1 were estimated. (D) Frequency-dependent inhibition of Na+ currents. Currents were elicited by a train of 20 pulses (3.5 ms duration; 10 Hz) from a Vh of −90 to 0 mV. (Upper traces) Currents evoked by the 1st and 20th pulse in the absence of drug and in presence of 10 μM BW534U87. (Lower traces) Average current amplitude (normalized to the first pulse) during a train of 20 pulses in control and in the presence of 10 μM BW534U87. Data points are the mean±s.e. mean (n=6 cells). Leak currents were not subtracted in these experiments.
As the inhibition of action potential frequency in hippocampal neurones by BW534U87 was dependent on the spike frequency prior to drug application, the frequency- or use-dependency was further examined on recombinant channels in CHO cells. Trains of depolarizing pulses (3.5 ms) from a Vh of −90 mV to a test potential of 0 mV were applied to the cell at a frequency of 10 Hz. In control conditions, the current amplitude was sustained for the duration of the train (20 pulses). In the presence of 10 μM BW534U87 the current amplitude diminished during the train indicating a frequency-dependent block (Figure 5D, n=6 cells). In contrast depolarizing trains (20 pulses at 10 Hz) of shorter pulses (0.7 ms), which fully activated channels, but produced less inactivated channels were unaffected by 10 μM BW534U87 (n=5 cells, data not shown).
Discussion
The main findings of the study were that BW534U87 possesses a unique ability to both enhance endogenous adenosine concentrations and inhibit voltage-gated Na+ channels. Under normal conditions, BW534U87 had no effect on resting potential or input resistance. Evoked normal synaptic activity (e.g. EPSP-IPSPs) was not affected by BW534U87, indicating that the compound does not interfere with glutamate or GABA release or their action at postsynaptic receptors per se. In contrast, BW534U87 depressed epileptiform activity induced by exposure of the slice to a Mg2+-free solution containing elevated K+ and bicuculline. The depression of abnormal synaptic activity was reversed by co-application with the adenosine receptor antagonist 8-PT, indicating an involvement of the adenosine system. The selective ADA inhibitor EHNA mimicked these effects of BW534U87. The depression of large synaptic events by BW534U87 is likely to result solely from an inhibition of ADA rather than inhibition of voltage gated Na+ channels. This is supported by the similarity between the effect of BW534U87 and the pure ADA inhibitor EHNA on large synaptic events. In addition lamotrigine, a Na+ channel inhibitor with similar voltage- and frequency-dependent properties to BW534U87, had no effect on large synaptic events induced under identical conditions (Xie et al., 1995).
The differential effect of BW534U87 under normal physiological and hyper-excitable conditions reflects differences in the concentrations of extracellular adenosine and supports an activity-dependent action of the drug. This is likely to be due to the differential effect on the two main adenosine metabolic enzymes, adenosine kinase and ADA. Resting extracellular adenosine levels are estimated to be close to the Km of adenosine kinase (approximately 2 μM; Arch & Newsholme, 1978; Van Wylen et al., 1986) and therefore under normal conditions most endogenous adenosine will be phosphorylated. The inhibition of ADA by BW534U87 would be expected to have little effect on the basal adenosine level. During seizures, brain concentrations of adenosine in patients suffering from temporal lobe epilepsy can substantially increase up to 30 fold of basal levels (During & Spencer, 1992) so that it reaches the Km of ADA (approximately 50 μM; Arch & Newsholme, 1978). BW534U87, which inhibits rat brain ADA with a Ki of 7 μM (Dupere et al., 1999), would be expected to cause accumulation and further elevation of endogenous adenosine. Enhancement of the inhibitory effect of exogenously applied adenosine on EPSPs by BW534U87 confirms the interaction of the compound with the adenosine system. Adenosine inhibited the EPSP with an IC50 of 41.0 μM and a Hill coefficient of 1.8. These values are similar to the IC50 of 29 μM and the Hill coefficient of 2.0 for the inhibition of field EPSP responses by adenosine in rat hippocampus (Dunwiddie & Fredholm, 1984). In the presence of 50 μM BW534U87 the IC50 was reduced to 15.9 μM and the Hill coefficient to 1.5. Similarly in the presence of 10 μM EHNA the IC50 was reduced to 18.9 μM and the Hill coefficient to 1.5. The reductions in the values of both the IC50s and the Hill coefficients by BW534U87 and EHNA are likely to reflect the composite nature of the response. Only a small proportion of the exogenously applied adenosine would be expected to reach A1 receptors at the synapse. A more accurate estimate of the potency of adenosine for A1 receptors at the synapse is obtained when adenosine uptake and metabolism are blocked. Under these conditions the IC50 for the inhibition of field EPSPs by adenosine was estimated to be 630 nM (Mitchell et al., 1993). The IC50s for the inhibition of EPSPs by non metabolized analogues of adenosine such as N6-phenylisopropyl-adenosine, N6-cyclohexyl-adenosine and adenosine 5′-ethyl-carboxamide, are in the nanomolar range and reflect the binding affinities of these compounds for adenosine A1 receptors without the complication of metabolizing enzymes (Dunwiddie & Fredhom, 1984). The IC50s are lower and the Hill coefficients for the inhibition of the field EPSPs are close to unity indicating the binding of one drug molecule to one binding site. During epileptic-like activity, BW534U87 inhibited the EPSP area by 46% and EHNA by 52%. This effect was mimicked by the application of 50 μM exogenous adenosine, which is equivalent to ∼500 nM at the synapse. In other words, during epileptiform activity both BW534U87 and EHNA are likely to elevate the synaptic concentration of adenosine up to at least 500 nM.
Activity-dependent depression of excitatory synaptic transmission in rat hippocampal slices in vitro has been observed previously for EHNA (Mitchell et al., 1993). Interestingly administration of other ADA inhibitors SC1001-sodium or 2′deoxycoformycin produced anticonvulsant effects in animal models in vivo (He et al., 1993; Zhang et al., 1993) supporting blockade of this enzyme as a mechanism for anti-convulsant action.
BW534U87 not only depressed epileptiform synaptic activity, but also reduced the firing of action potentials elicited by a depolarizing current injection a property which is not shared by EHNA. Adenosine itself has been previously shown to reduce neuronal firing in several brain areas including the hippocampus, the laterodorsal tegmental nucleus and the pedunculopontine tegmental nucleus (Dunwiddie, 1980, 1985; Haas & Greene, 1988; Rainnie et al., 1994). However, the reduction of spike firing in hippocampal neurones by BW534U87 was not affected by 8-PT at 30 μM, a concentration which would be expected to virtually abolish any adenosine-mediated responses. Furthermore, the inhibition of spike firing by the drug was dependent on the membrane potential and the firing frequency of the cell prior to drug application. This characteristic and the adenosine-independent modulation of spike firing by the compound suggest that BW534U87 may also directly interact with voltage-gated Na+ channels.
Voltage- and use-dependent inhibition of Na+ channels has been proposed as the main mechanism of action for a series of AEDs including phenytoin, carbamazepine and lamotrigine (Ragsdale et al., 1991;MacDonald & Kelly, 1993; Xie et al., 1995). The main functional component of the Na+ channel is the α-subunit which contains the ionic pore and the voltage-sensing apparatus of the channel. There are at least four different α-subunits found in the rat or human brain (for review see Catterall, 1992). In the rat hippocampal pyramidal layer over 80% of total Na+ channel mRNA are type IIA (Westenbroek et al., 1989; Catterall, 1992). Recently, human brain type IIA Na+ channels have been stably expressed in CHO cells and generate TTX-sensitive Na+ currents with comparable kinetics to those in native neurones (Xie et al., 1997). This cell line therefore provides a convenient model system to investigate a direct interaction of BW534U87 with human brain Na+ channels. Under voltage-clamp conditions, there was very little tonic block of the type IIA channel-conducted currents at a Vh of −90 mV, whereas at a briefly (1 s) depolarized potential of −60 mV, BW534U87 produced a potent inhibition of the currents with an estimated IC50 of 10 μM. The inhibitory effect of BW534U87 at low frequencies of activation (<0.5 Hz) was enhanced at higher frequencies. A number of mechanisms could be responsible for the voltage- and use-dependent effects of BW534U87. The drug could bind to the open state of the channel in a manner similar to the local anaesthetics QX-314 (Strichartz 1973) and GEA968 (Courtney, 1975). However, the use-dependent action of BW534U87 was negligible with brief (0.7 ms) pulses which produced channels mainly in the open state. The use-dependence was most prominent with longer pulses (20 ms), consistent with a voltage-dependent action. Both protocols produce channels in the slow inactivated state. A selective interaction with the slow inactivated state is supported by the experimental data which show that the compound caused a substantial hyperpolarizing shift in the slow inactivation curve. However, an alternative interpretation is that the drug interacts with the fast inactivated or even open state of the channel, but the binding kinetics of the drug are so slow that the drug would not have sufficient time to bind to the channel during the test or short conditioning pulses. This mode of action has been suggested for phenytoin and lamotrigine in native Na+ channels of acutely dissociated hippocampal neurones (Kuo & Bean, 1994; Kuo & Lu, 1997).
In conclusion, BW534U87 preferentially depresses epileptiform activity in hippocampal brain slices in vitro by a dual mechanism. The drug suppresses synaptic epileptiform activity via an augmentation of the endogenous adenosine action, presumably resulting from an inhibition of ADA. In addition, BW534U87 directly inhibits voltage-gated Na+ channels in a voltage- and use-dependent manner. These two activity-dependent mechanisms may synergistically contribute to the selective inhibition of neuronal hyperexcitability observed in the hippocampus in vitro. The dual mode of action of BW534U87 may underlie its anticonvulsant effects in a variety of epilepsy models.
Acknowledgments
We are grateful to our colleagues Drs Eric Southam for kindly sharing his unpublished data; Russ Hagan, Julie Barnes and Luigi Giorgi for reading and making comments on the manuscript.
Abbreviations
- ADA
adenosine deaminase
- AEDs
antiepileptic drugs
- CHO
Chinese hamster ovary cells
- DMSO
dimethyl sulphoxide
- EGTA
ethylene glycol-bis(β-aminoethyl ether) N,N,N′ -tetra acetic acid
- EHNA
erythro-9-(2-hydroxy-3-nonyl)adenine
- EPSP-IPSP
excitatory and inhibitory postsynaptic potentials
- HEPES
4-(2-hydroxethyl)-1-piperazineethanesulphonic acid
- 8-PT
8-phenyl theophylline
- TTX
tetrodotoxin V½, half-maximal activation or inactivation voltage
- Vh
holding potential
References
- ARCH J.R.S., NEWSHOLME E.A. Activities and some properties of 5′-nucleotidase, adenosine kinase and adenosine deaminase in tissues from vertebrates and invertebrates in relation to the control of the concentration and the physiological role of adenosine. Biochem. J. 1978;174:965–977. doi: 10.1042/bj1740965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENDER A.S., WU P.H., PHILLIS J.W. The characterization of [3H] adenosine uptake into rat cerebral cortical synaptosomes. J. Neurochem. 1980;35:629–640. doi: 10.1111/j.1471-4159.1980.tb03702.x. [DOI] [PubMed] [Google Scholar]
- BRUNDEGE J.M., DUNWIDDIE T.V. Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv. Pharmacol. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- BURGER R.M., LOWENSTEIN J.M. Preparation and properties of 5′-nucleotidase from smooth muscle of small intestine. J. Biol. Chem. 1970;245:6274–6280. [PubMed] [Google Scholar]
- CATTERALL W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992;72:S15–48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- CONCAS A., SANTORO G., MASCIA M.P., MACIOCCO E., DAZZI L., ONGINI E., BIGGIO G. Anticonvulsant doses of 2-chloro-N6-cyclopentyladenosine, an adenosine A1 receptor agonist, reduce GABAergic transmission in different areas of the mouse brain. J. Pharmacol. Exp. Ther. 1993;267:844–851. [PubMed] [Google Scholar]
- COURTNEY K.R. Mechanism of frequency-dependent inhibition of Na+ currents in frog myelinated nerve by the lidocaine derivative GEA 968. Pharmacol. Exp. Ther. 1975;195:225–236. [PubMed] [Google Scholar]
- DUNWIDDIE T.V. Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia. 1980;21:541–548. doi: 10.1111/j.1528-1157.1980.tb04305.x. [DOI] [PubMed] [Google Scholar]
- DUNWIDDIE T.V. The physiological role of adenosine in the central nervous system. Int. Rev. Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- DUNWIDDIE T.V., FREDHOLM B.B. Adenosine receptors mediating inhibitory electrophysiological responses in rat hippocampus are different from receptors mediating cyclic AMP formation. Naunyn Schmiedebergs Arch. Pharmacol. 1984;326:294–301. doi: 10.1007/BF00501433. [DOI] [PubMed] [Google Scholar]
- DUNWIDDIE T.V., WORTH T. Sedative and anticonvulsant effects of adenosine analogs in mouse and rat. J. Pharmacol. Exp. Ther. 1982;220:70–76. [PubMed] [Google Scholar]
- DUPERE J.R.B., DALE T.J., SOUTHHAM E., HAGAN R.M., XIE X.M.Depression of epileptiform activity in rat hippocampal slices via inhibition of adenosine deaminase and Na+ channels by the novel anticonvulsant BW534U87 Soc. Neurosci. Abstr. 1999(in press) [DOI] [PMC free article] [PubMed]
- DUPERE J.R.B., SOUTHHAM E., DUFFY C., XIE X.M. Activity-dependent depression of excitatory synaptic transmission in rat hippocampal slices by an inhibitor of adenosine deaminase. Eur. J. Neurosci. 1998;10 Suppl 10:19. 15. [Google Scholar]
- DURING M.J., SPENCER D.D. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- GREENER W., HAASH L. The electrophysiology of adenosine in the mammalian central nervous system. Progress in Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- HAAS H.L., GREENE R.W. Endogenous adenosine inhibits hippocampal CA1 neurones: further evidence from extra- and intracellular recording. Naunyn-Schmiedebergs Arch. Pharmacol. 1988;337:561–565. doi: 10.1007/BF00182732. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P. , MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HAMILTON B.R., SMITH D.O. Autoreceptor-mediated purinergic and cholinergic inhibition of motor nerve terminal calcium currents in the rat. J. Physiol. 1991;432:327–341. doi: 10.1113/jphysiol.1991.sp018387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HE J., CHAI H., CHEN Q., LI Y. Effect of SC1001-sodium on ADA activity of the thymus, spleen and brain in repeated seizure rats. J. West China Univ. School Med. Sci. 1993;24:395–397. [PubMed] [Google Scholar]
- KELLY J.L., KOBLE C.S., DAVIS R.G. , MCLEAN E.W. , SOROKO F.E., COOPERB R. 1(Fluorobenzyl)4-amino-1H-1,2,3-triazolo[4,5-c]pyridines: Synthesis and anticonvulsant activity. J. Med. Chem. 1995;38:4131–4134. doi: 10.1021/jm00020a030. [DOI] [PubMed] [Google Scholar]
- KUO C.-C., BEAN B.P. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 1994;46:716–725. [PubMed] [Google Scholar]
- KUO C.-C., LU L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br. J. Pharmacol. 1997;121:1231–1238. doi: 10.1038/sj.bjp.0701221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIN E., BLECK V. Electroshock seizures in mice: effect on brain adenosine and its metabolites. Epilepsia. 1981;22:577–581. doi: 10.1111/j.1528-1157.1981.tb04129.x. [DOI] [PubMed] [Google Scholar]
- MACDONALD R.L., KELLY K.M. Antiepileptic drug mechanisms of action. Epilepsia. 1993;34 Suppl. 5:S1–8. doi: 10.1111/j.1528-1157.1993.tb05918.x. [DOI] [PubMed] [Google Scholar]
- MALHOTRA J., GUPTA Y.K. Effect of adenosine receptor modulation on pentylenetetrazole-induced seizures in rats. Br. J. Pharmacol. 1997;120:282–288. doi: 10.1038/sj.bjp.0700869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.B., LUPICA C.R., DUNWIDDIE T.V. Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J. Neurosci. 1993;13:3439–3447. doi: 10.1523/JNEUROSCI.13-08-03439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY T.F., SYLVESTER D., SCHULTZ C.S., SZOT P. Purinergic modulation of the seizure threshold for pentylenetetrazol in the rat. Neuropharmacol. 1985;24:761–766. doi: 10.1016/0028-3908(85)90010-3. [DOI] [PubMed] [Google Scholar]
- NEWMAN M.E., MCILWAIN H. Adenosine 3′ : 5′-cyclic monophosphate in nerve-terminal fractions from neocortical tissues: its augmentation by a post-stimulation process. Biochem. Soc. Trans. 1977;5:1074–1075. doi: 10.1042/bst0051074. [DOI] [PubMed] [Google Scholar]
- NIMIT Y., SKOLNICK P., DALY J.W. Adenosine and cyclic AMP in rat cerebral cortical slices: effects of adenosine uptake inhibitors and adenosine deaminase inhibitors. J. Neurochem. 1981;36:908–912. doi: 10.1111/j.1471-4159.1981.tb01680.x. [DOI] [PubMed] [Google Scholar]
- PAK M.A., HAAS H.L., DECKING U.K., SCHRADER J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacol. 1994;33:1049–1053. doi: 10.1016/0028-3908(94)90142-2. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., SCHEUER T., CATTERALL W.A. Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anaesthetic, antiarhythmic and anticonvulsant drugs. Mol. Pharmacol. 1991;40:756–765. [PubMed] [Google Scholar]
- RAINNIE D.G., GRUNZE H.C.R., MCCARLEY R.W., GREENER W. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIBEIRO J.A., SEBASTIAO A.M. On the role, inactivation and origin of endogenous adenosine at the frog neuromuscular junction. J. Physiol. 1987;384:571–585. doi: 10.1113/jphysiol.1987.sp016470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUBERT P., MITZDORF U. Analysis and quantitative evaluation of the depressant effect of adenosine on evoked potentials in hippocampal slices. Brain Res. 1979;172:186–190. doi: 10.1016/0006-8993(79)90910-7. [DOI] [PubMed] [Google Scholar]
- SHIMUZU H., TANAKA S., KODAMA T. Adenosine kinase of mammalian brain: partial purification and its role for the uptake of adenosine. J. Neurochem. 1972;19:687–698. doi: 10.1111/j.1471-4159.1972.tb01384.x. [DOI] [PubMed] [Google Scholar]
- STRATTON S.C., BRACKENBOROUGH K.T., DUFFY C., SARGENT B., PRATT G.D., HAGAN R.M. BW534U87: a novel anticonvulsant, active in a range of rodent seizure models. Epilepsia. 1998;39 Suppl. 6:45. [Google Scholar]
- STRICHARTZ G.R. The inhibition of Na+ currents in myelinated nerve by quaternary derivatives of lidocaine. J. Gen. Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON S.M., HAAS H.L., GÄHWILER B.H. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol. 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN WYLEN D.G.L., PARK T.S., RUBIO R., BERNE R.M. Increases in cerebral interstitial fluid adenosine concentration during hypoxia, local potassium infusion, and ischemia. J. Cereb. Blood Flow Metab. 1986;6:522–528. doi: 10.1038/jcbfm.1986.97. [DOI] [PubMed] [Google Scholar]
- WESTENBROEK R.E., MERRICK D.K., CATTERALL W.A. Differential subcellular localization of RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- WINN H.R., WELSH J.E., RUBIO R., BERNE R.M. Changes in brain adenosine during bicuculline-induced seizures in rats. Effects of hypoxia and altered systemic blood flow. Circ. Res. 1980;47:868–877. doi: 10.1161/01.res.47.4.568. [DOI] [PubMed] [Google Scholar]
- WOJCIK W.J., NEFF N.H. Adenosine measurement by rapid HPLC-fluorimetric method: induced changes of adenosine content in regions of rate brain. J. Neurochem. 1982;39:280–282. doi: 10.1111/j.1471-4159.1982.tb04736.x. [DOI] [PubMed] [Google Scholar]
- XIE X.M., DALE T., PEAKMAN T., CLARE J. Electrophysiological characterization of human brain type IIA Na+ channel α subunits stably expressed in a mammalian cell line. J. Physiol. 1997;504 Suppl:154P. [Google Scholar]
- XIE X.M., LANCASTER B., PEAKMAN T., GARTHWAITE J. Interactions of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native NaP+ channels in rat hippocampal neurones. Pflugers Arch. 1995;430:437–446. doi: 10.1007/BF00373920. [DOI] [PubMed] [Google Scholar]
- XIE X.M., SMART T.G. Properties of GABA-mediated synaptic potentials induced by zinc in adult rat hippocampal pyramidal neurones. J. Physiol. 1993;460:503–523. doi: 10.1113/jphysiol.1993.sp019484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG G., FRANKLIN P.H., MURRAY T.F. Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J. Pharmacol. Exp. Ther. 1993;264:1415–1424. [PubMed] [Google Scholar]