

Abstract
New thiazolidinediones BM13.1258 and BM15.2054 were studied with regard to their PPARγ-agonistic activities and to their acute and chronic effects on glucose metabolism in soleus muscle strips from lean and genetically obese rats.
Both BM13.1258 and BM15.2054 revealed to be potent PPARγ-activators in transient transfection assays in vitro.
In insulin-resistant obese rats, but not in lean rats, 10 days of oral treatment with either compound increased the stimulatory effect of insulin on muscle glycogen synthesis to a similar extent (insulin-induced increment in μmol glucose incorporated into glycogen g−1 h−1: control, +1.19±0.28; BM13.1258, +2.50±0.20; BM15.2054, +2.55±0.46; P<0.05 vs control each).
In parallel to insulin sensitization, mean glucose oxidation increased insulin-independently in response to BM13.1258 (to 191 and 183% of control in the absence and presence of insulin, respectively; P<0.01 each), which was hardly seen in response to BM15.2054 (to 137 and 124% of control, respectively; ns).
Comparable effects on PPARγ activation and on amelioration of insulin resistance by BM13.1258 and BM15.2054 were therefore opposed by different effects on glucose oxidation.
In contrast to chronic oral treatment, acute exposure of muscles to BM13.1258 or BM15.2054 in vitro elicited a distinct catabolic response of glucose metabolism in specimens from both lean and obese rats.
The results provide evidence that BM13.1258 and BM15.2054 can affect muscle glucose metabolism via more than one mechanism of action.
Further efforts are required to clarify, to what extent other mechanisms besides insulin sensitization via the activation of PPARγ are involved in the antidiabetic actions of thiazolidinediones.
Keywords: Thiazolidinedione, BM13.1258, BM15.2054, insulin sensitivity, glucose transport, glycogen synthesis, glucose oxidation, skeletal muscle
Introduction
Common metabolic diseases like obesity and type 2 diabetes are associated with insulin resistance and derangement of glucose homeostasis (DeFronzo, 1988; Olefsky et al., 1982). During the last decades, major efforts to find new drugs with insulin sensitizing properties resulted in the development of a new class of antidiabetic compounds, the thiazolidinediones (TZD). In various animal models of obesity and type 2 diabetes, chronic oral TZD administration markedly ameliorates insulin resistance in association with an improvement of glucose and lipid metabolism (Bowen et al., 1991; Fujiwara et al., 1988; Kuehnle, 1996; Oakes et al., 1994; Stevenson et al., 1990). The precise mechanism via which TZD exert their metabolic actions is, however, still not completely understood.
TZD are known to bind to and activate the nuclear peroxisome proliferator-activated receptor γ (PPARγ), what leads to the modulation of gene expression rates and triggers adipocyte differentiation (Okuno et al., 1998; Spiegelman, 1998). During the last years, evidence has accumulated that PPARγ-dependent modulation of gene transcription is essential for TZD-induced improvement of metabolic parameters (Berger et al., 1996; 1999; Lehmann et al., 1995; Spiegelman, 1998). In some contrast to that assumption, several studies document acute responses to TZD within a time range too short to suggest that the underlying mechanism involves the modulation of gene transcription rates (Fujiwara et al., 1988; Fürnsinn et al., 1997; Kreutter et al., 1990; Lee & Olefsky, 1995; Okuno et al., 1997). Therefore, it can not be excluded that TZD affect glucose metabolism via more than one biochemical mechanism, and that PPARγ-independent pathways contribute to the various metabolic effects of TZD.
The present study was undertaken to examine the effects of the new TZD compounds BM13.1258 and BM15.2054 on key parameters of glucose metabolism in skeletal muscle, which is the quantitatively most important target tissue of insulin (Baron et al., 1988; DeFronzo, 1988). To provide evidence for or against action via more than one mechanism, PPARγ agonistic activities of BM13.1258 and BM15.2054 were determined and modulation of muscle glucose handling was examined in response to chronic oral treatment in vivo and acute exposure in vitro. To this end, glucose transport rates in parallel with intracellular glucose flux into glycogen synthesis and glycolysis were determined in muscle specimens from lean as well as insulin resistant obese rats. The results suggest that interaction of the employed TZD compounds with muscle glucose metabolism may not exclusively rely on PPARγ-dependent insulin sensitization.
Methods
TZD compounds
The TZD compounds BM13.1258, BM15.2054, troglitazone (CS-045), and rosiglitazone (BRL 49653) were provided by Boehringer Mannheim (Mannheim, Germany). The chemical structures of BM13.1258 and BM15.2054 are depicted in Figure 1 and demonstrate that these compounds belong to the TZD class.
Figure 1.
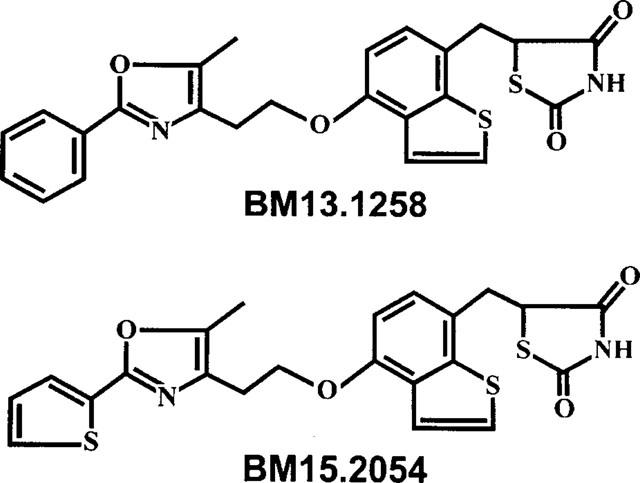
Chemical structures of BM13.1258 and BM15.2054. The structures on the right indicate that the compounds belong to the class of thiazolidinediones.
TZD action on PPARγ
Plasmids
Mouse PPARγ2 cDNA was amplified from a mouse fat cell cDNA-library (Clonetech) by using the primers (5′ - ATATACCCGGGACCATGGGTGAAACTCTGGGA -G-3′ and 5′-GATACGCTAGCCRTAGTACAAGTCCTTGTAGATCTCC-3′). The PCR product was subsequently cleaved with SmaI and NheI and cloned into the SV40 early promotor-driven expression vector pSG7 (a derivate of pSG5; Green et al., 1988). The expression vector pSG5-hRXRa encoding the human retinoid X receptor α cDNA was a gift from H. Stunnenberg (EMBL, Heidelberg, Germany). A PPAR-responsive reporter plasmid was generated by inserting three copies of a peroxisome proliferator responsive element (PPRE) consensus binding element (5′-AAGCTTACGTTGACCTTTGTCCTGGTCCCGTCGAC-3′) upstream of a HSV-thymidine kinase minimal promoter (−109/+38) fused to a luciferase reporter gene. Plasmid p-cytomegalovirus β (pCMVβ; Clonetech, Heidelberg, Germany) contains the E. coli beta-galactosidase coding sequence under control of the CMV immediate early promoter/enhancer. All plasmids were verified for their integrity by DNA sequencing.
Transient transfections
CV-1 cells (ATCC CCL70) were cultivated in Dulbecco's modified Eagle's medium supplemented with 10% Char-coal stripped foetal calf serum at 37°C in a 95% O2:5% CO2 atmosphere. The day prior transfection cells were seed out at a density of 1×105 per 35 mm dish. Cells were transfected using a modified calcium precipitate technique (Chen & Okayama, 1988). Twenty hours later cells were washed and treated for 30–36 h with the indicated amounts of TZD (0.1, 0.5, or 1.0 μmol l−1) or a dimethyl sulphoxide (DMSO; from Sigma, Deisenhofen, Germany) vehicle control. After ligand exposure cells were harvested and assayed for luciferase and β-galactosidase activity. Luciferase activity was normalized to β-galactosidase activity which served as an internal control for transfection efficiency, and is expressed as fold-activation relative to DMSO-treated control cells. The amount of DMSO used did not exceed 0.1% (vol vol−1). All transfections were done in triplicates and repeated at least twice.
Rats
Male Sprague-Dawley (SD) rats were purchased from the breeding facilities of the University of Vienna (Himberg, Austria), genetically obese Zucker rats (fa/fa; HsdOla) were from Harlan (Borchen, Germany). Rats were kept at an artificial 12 h light/dark cycle at constant room temperature, and conventional laboratory diet and tap water were provided ad libitum until the evening before killing, when only food was withdrawn. Rats were killed by cervical dislocation between 08 : 30 and 09 : 30 h. All experiments were performed according to local law and to the principles of good laboratory animal care.
Chronic oral TZD treatment in vivo
BM13.1258 and BM15.2054 were suspended in 5% arabic gum solution (2 mg TZD ml−1) that was mixed with a Polytron homogenizer (Kinematica, Luzern, Switzerland) immediately before use. In the morning, a daily dose of ∼4.4 mg TZD kg body weight−1 was administered by gavage during 10 consecutive days to lean SD rats (363±2 g body weight; daily oral dose of 0.8 ml containing 1.6 mg TZD) and obese Zucker rats (458±5 g body weight; daily oral dose of 1 ml containing 2 mg TZD). Control animals received the same amount of arabic gum solution without TZD. On day 11, i.e. 24 h after the last dose of TZD was administered, overnight fasted animals were killed.
Immediately after killing, two (SD rats) or three (Zucker rats) longitudinal soleus muscle strips per leg were prepared, weighed (approximately 25 mg), and tied under tension on stainless steel clips as previously described (Crettaz et al., 1980). Muscles were immediately put into 25 ml-Erlenmeyer flasks coated with BlueSlick solution (Serva, Heidelberg, Germany) and placed into a shaking waterbath (1 strip/flask; 37°C; 130 cycles min−1). Each flask contained 3 ml Krebs-Ringer buffer solution (KRB, pH 7.35) supplemented with 5.5 mmol l−1 glucose and 1% (wt vol−1) bovine serum albumine (BSA). An atmosphere of 95% O2 : 5% CO2 was continuously provided within the flasks.
The experiments started with a preincubation period of 30 min. After preincubation, muscles were immediately transferred into another set of flasks and incubated in 3 ml of identical buffer solution additionally supplemented with palmitate as dissolved in ethanol to give final concentrations of 300 μmol l−1 palmitate and 0.8% (vol vol−1) ethanol. Furthermore, the solution alternatively contained trace amounts of D-[U-14C]glucose, [U-14C]palmitic acid, or 2-deoxy-D-[2,6-3H]glucose plus [U14C]sucrose (all from Amersham, Amersham, U.K.), and where indicated, human insulin (Actrapid from Novo, Bagsvaerd, Denmark). After incubation for 60 min, muscles were quickly removed, blotted, and frozen in liquid nitrogen. Later, muscle strips were lysed in 1 mol l−1 KOH at 70°C, the lysate was then employed for further analytical procedures as described below.
Acute TZD exposure in vitro
Untreated rats were killed and two (SD rats; ∼140 g body weight) or four (Zucker rats; ∼450 g body weight) soleus muscle strips were immediately prepared. The muscle incubation procedure was as described for chronic treatment experiments, except that KRB was always devoid of BSA, ethanol, and labelled or unlabelled palmitate. Instead, TZD compounds as dissolved and diluted in DMSO were added to the medium during the preincubation as well as during the incubation period to give final TZD concentrations as indicated. To allow for intraindividual comparison, one muscle strip from each rat was incubated in the absence of TZD, while DMSO concentration was identical in all media including TZD-free control media (0.1%, vol vol−1).
Analytical procedures
Net uptake of 2-deoxy-D-[2,6-3H]glucose, a glucose analogue which does not enter glycolysis, was determined employing [14C]sucrose as an extracellular space marker by methods described previously (Fürnsinn et al., 1995). Under the applied experimental conditions, insulin-stimulated 3H-2-deoxy-glucose uptake does not reach saturation within the incubation period of 60 min (data not shown). Net rates of glucose incorporation into glycogen are referred to as glycogen synthesis, and were determined measuring the conversion of [14C]glucose to [14C]glycogen by methods described previously (Crettaz et al., 1980). The rates of CO2 production from glucose and palmitate, respectively, were calculated from the conversion of [14C]glucose or [14C]palmitate into 14CO2, which was trapped with a solution containing methanol and phenethylamine (1 : 1) (Fürnsinn et al., 1995). Rates of lactate release were calculated from lactate accumulated in the incubation buffer during the experiment. Buffer lactate concentration was determined enzymatically employing the lactate dehydrogenase method (Engel & Jones, 1978). For the determination of muscle glycogen content prevailing at the end of the experiment, glycogen in the muscle lysate was completely degraded to glucose with amyloglucosidase (Dimitriadis et al., 1988). Glucose was then measured enzymatically by a commercial kit from Human (Taunusstein, Germany).
Statistics
All data are presented as means±s.e.mean and a P<0.05 was considered significant. Where not stated otherwise, multiple comparisons with a control were performed as described for unpaired data by Dunnett (Dunnett, 1964), or with an analogous test adapted for paired data.
Results
TZD action on PPARγ
Transient transfection studies were performed to test for ligand activities of TZD compounds. To this end, CV-1 cells were transfected with expression plasmids for PPARγ2 and its heterodimerization partner RXRα along with a PPRE-luciferase reporter plasmid. All TZD compounds activated PPARγ2-dependent luciferase expression in a dose-dependent manner (Figure 2). To exclude possible stimulation of RXRα present in the heterodimer, transfection studies were repeated with RXRα alone. In this context, no stimulation by the TZD ligands was observed clearly indicating that the compounds function as PPARγ but not RXRα agonists. In agreement with previous studies, rosiglitazone exhibited significantly higher PPARγ-agonistic activity than troglitazone (Berger et al., 1996; 1999), while activation by both BM13.1258 and BM15.2054 tended to be even superior to rosiglitazone.
Figure 2.
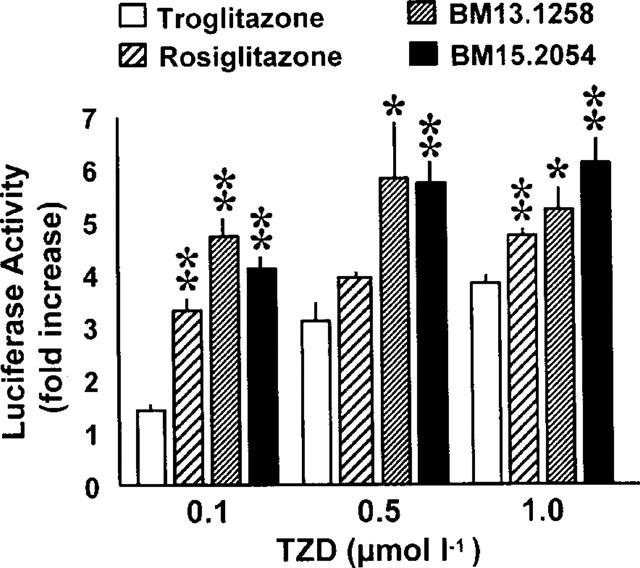
Activation of PPARγ by TZDs. 1×105 CV-1 cells were transfected with 100 ng pSGS-hRXRα, 200 ng pSG7-mPPARγ2, 300 ng of the PPRE luciferase plasmid and 20 ng of pCMVβ as described. Cells were exposed to the indicated amounts of TZDs for 30–36 h and the relative activation of PPAR-mediated gene expression is given as fold stimulation compared to DMSO cells. Means±s.e.mean are indicated from transfections done in triplicates and repeated at least twice. *P<0.05, **P<0.01 vs troglitazone by unpaired Student's t-test.
Chronic oral TZD treatment, body weight gain
No significant influence of oral TZD administration on body weight gain was observed both in lean SD rats (body weight gained during 10 days of treatment: control, +30±3 g; BM13.1258, +34±4 g; BM15.2054, +28±6 g; ns vs control) and in obese Zucker rats (control, +43±5 g; BM13.1258, +56±3 g; BM15.2054, +46±8 g; ns vs control). One obese Zucker rat for unknown reasons lost 53 g during 10 days of treatment and was therefore excluded from the study.
Figure 3.
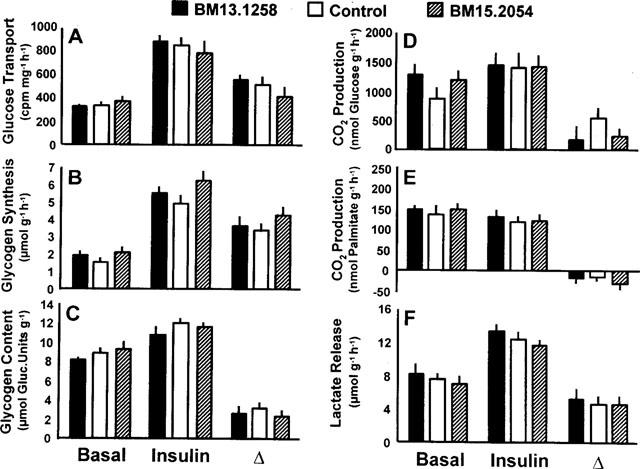
Rates of basal and insulin stimulated (10 nmol l−1) 3H-2-deoxy-glucose transport (A), glycogen synthesis (B), CO2 production from glucose (D) and palmitate (E), and lactate release (F) in soleus muscle strips from healthy Sprague-Dawley rats after oral administration of BM13.1258 or BM15.2054 (4.4 mg kg−1 d−1) for 10 consecutive days. Control rats received placebo; glycogen content as determined after the experiment (C). Means±s.e.mean; n=6 each; no significant differences for treated vs control rats.
Figure 4.
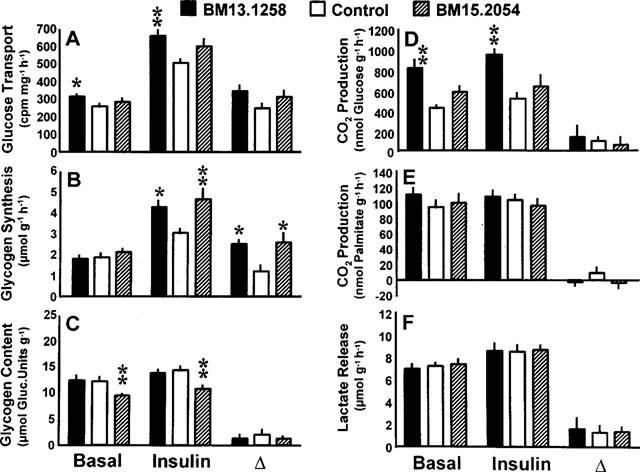
Rates of basal and insulin stimulated (10 nmol l−1) 3H-2-deoxy-glucose transport (A), glycogen synthesis (B), CO2 production from glucose (D) and palmitate (E), and lactate release (F) in soleus muscle strips from genetically obese Zucker rats (fa/fa) after oral administration of BM13.1258 (n=7) or BM15.2054 (n=10) (4.4 mg kg−1 d−1) for 10 consecutive days. Control rats received placebo (n=11); glycogen content as determined after the experiment (C). Means±s.e.mean; *P<0.05, **P<0.01 vs control rats.
Chronic oral TZD treatment, glucose metabolism (Figures 3 and 4)
Lean rats (Figure 3)
In soleus muscle strips prepared from lean SD rats, the various parameters of glucose metabolism were not significantly affected by 10 days of oral TZD administration.
Obese rats (Figure 4)
After 10 days of oral treatment with BM13.1258, 3H-2-deoxy-glucose transport in muscle strips from obese rats was significantly increased in the absence and presence of insulin, while only non-significant trends were observed in response to BM15.2054 (Figure 4A). The rates of glycogen synthesis remained unaffected under basal conditions, but were markedly increased in the presence of insulin with the mean stimulatory (incremental) effect of insulin augmented to a very similar extent by the two compounds (to 210 and 214% of control by BM13.1258 and BM15.2054, respectively; Figure 4B). After termination of the incubation experiment, muscle glycogen content was decreased in muscles from BM15.2054-treated rats only (Figure 4C). CO2 production from glucose nearly doubled in BM13.1258-treated obese rats reaching 191 and 183% of control values in the absence and presence of insulin, respectively, and hence the BM13.1258-induced increase in glucose oxidation was independent of concomitant insulin stimulation (Figure 4D). In parallel to what was observed for glucose transport, only a non-significant stimulatory trend occurred with regard to basal and insulin-stimulated glucose oxidation after treatment with BM15.2054 (to 137 and 124% of control). Rates of lactate release and CO2 production from palmitate were not affected by oral TZD treatment for 10 days (Figure 4E,F).
Figure 5.
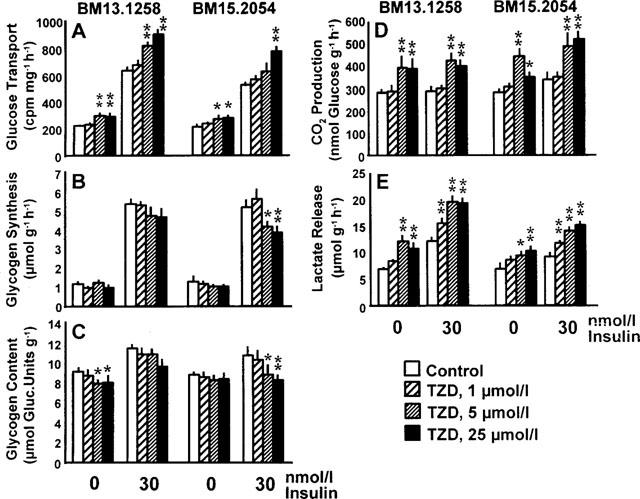
Rates of 3H-2-deoxy-glucose transport (A), glycogen synthesis (B), CO2 production from glucose (D), and lactate release (E) in soleus muscle strips from healthy Sprague-Dawley rats exposed to 0, 1, 5, or 25 μmol l−1 of the thiazolidinediones (TZD) BM13.1258 or BM15.2054 under basal or insulin-stimulated conditions. Glycogen content as determined after the experiment (C). Means±s.e.mean; *P<0.05; **P<0.01 vs intra-individual control.
Figure 6.
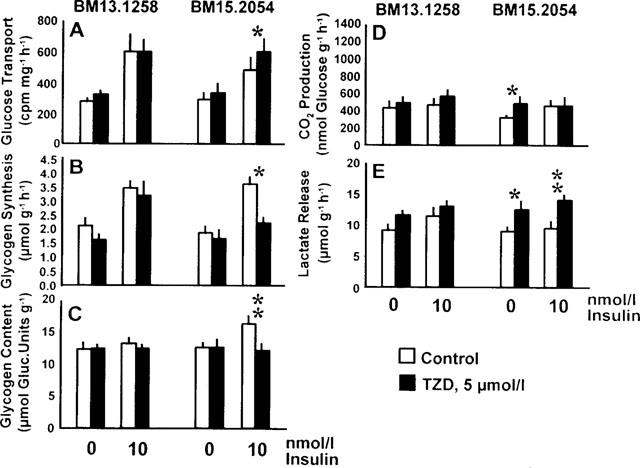
Rates of 3H-2-deoxy-glucose transport (A), glycogen synthesis (B), CO2 production from glucose (D), and lactate release (E) in soleus muscle strips from genetically obese Zucker rats (fa/fa) exposed to 0 or 5 μmol l−1 of the thiazolidinediones (TZD) BM13.1258 or BM15.2054 under basal or insulin-stimulated conditions. Glycogen content as determined after the experiment (C). Means±s.e.mean; *P<0.05, **P<0.01 vs intra-individual control by paired Student's t-test.
Acute TZD exposure, glucose metabolism (Figures 5 and 6)
Lean rats (Figure 5)
Acute TZD exposure of muscle strips from lean SD rats resulted in a distinct and dose-dependent catabolic response of glucose metabolism. Independent of insulin stimulation, both compounds triggered a significant increase in the rates of glucose transport and aerobic as well as anaerobic glycolysis, when used at concentrations of 5 or 25 μmol l−1 (Figure 5A,D,E). Even 1 μmol l−1 TZD sufficed to significantly increase the rates of insulin-stimulated lactate release under the employed experimental conditions (Figure 5E). With regard to the glycogenic pathway, an inhibitory effect was indicated by decreases in insulin-stimulated glycogen synthesis and associated muscle glycogen content at the end of the incubation. Significance was found with BM15.2054 only, what may indicate a somewhat more pronounced catabolic effect of BM15.2054 than BM13.1258 (Figure 5B,C).
Obese rats (Figure 6)
Acute TZD treatment of insulin resistant muscles from obese rats was examined at 5 μmol l−1 TZD only and an insulin concentration of 10 nmol l−1 was selected to ensure that potential changes in insulin sensitivity would become obvious (submaximally effective insulin concentration in isolated soleus muscle from obese rats; data not shown). In parallel to the results from healthy lean rats (Figure 5), BM15.2054 elicited a catabolic response rather than insulin sensitization (i.e. intracellular glucose metabolism was shifted from the glycogenic towards the glycolytic pathway) (Figure 6A–E). With BM13.1258, only non-significant trends were seen.
Discussion
Chronic oral TZD treatment in vivo
Chronic oral treatment with BM13.1258 or BM15.2054 failed to significantly affect glucose handling in skeletal muscle from lean SD rats exhibiting normal insulin sensitivity, what is in line with little if any metabolic effects of other TZD in healthy rodents (Bowen et al., 1991; Fujiwara et al., 1988; Kuehnle, 1996; Oakes et al., 1994). In soleus muscle from obese Zucker rats, we found distinct insulin sensitizing action of oral treatment with BM13.1258 and BM15.2054, whereby the two compounds exhibited a similar potential to counteract prevailing insulin resistance. Insulin sensitizing mode of action is indicated by the rates of glycogen synthesis that were unchanged in the absence of insulin, but exhibited more than doubled responses to insulin stimulation in muscles from TZD-treated obese rats as compared to such from placebo-treated animals. Counteracting insulin resistance, antidiabetic TZD compounds are often referred to as insulin sensitizers, but further findings from the present study indicate that this attribute meets only one out of a variety of actions exerted on skeletal muscle glucose metabolism by BM13.1258 and BM15.2054.
Thus, chronic treatment of obese rats with BM13.1258 led to markedly increased rates of muscle glucose oxidation, which reached a similar extent under basal and insulin-stimulated conditions. Although less distinct in amplitude, a parallel trend was seen with BM15.2054. This finding is in agreement with what has been observed in other studies employing genetically obese rodents treated with the TZD compounds englitazone and troglitazone (Stevenson et al., 1990; and unpublished results from this laboratory). Completely independent of concomitant insulin stimulation, the stimulatory effect on glucose oxidation can not be regarded as a direct consequence of TZD-induced insulin sensitization. In chronically TZD treated rats, a causal relationship to augmented expression of uncoupling protein as described to occur in response to TZD administration (Aubert et al., 1997; Camirand et al., 1998; Shimabukuro et al., 1997) is to be considered. Compared to healthy littermates, soleus muscle from obese Zucker rats is characterized by decreased rates of uncoupling protein-3 expression (Boss et al., 1998) and glucose oxidation (Crettaz et al., 1980; Fürnsinn et al., 1997), and hence, increased CO2 production from glucose may represent another aspect of TZD-induced amelioration of their obesity-associated metabolic syndrome. Following that line of interpretation, improved insulin sensitivity and increased glucose oxidation might be triggered via a single PPARγ-dependent mechanism.
At variance to the established hypothesis that PPARγ-activation may suffice to explain metabolic TZD actions, however, our findings contain relevant evidence that a PPARγ-independent mechanism is also involved in the responses elicited by the employed TZD compounds. Thus, a similar potential of BM13.1258 and BM15.2054 to activate PPARγ and to insulin-sensitize the glycogenic pathway is opposed by an approximately 3 fold difference in their insulin-independent potential to affect oxidative glycolysis as well as by different glycogen content (BM13.1258 vs BM15.2054 by Tukey test: P<0.05 for both glucose oxidation and glycogen content under both basal and insulin-stimulated conditions). While it may be hypothesized that blunted glycogen stores limit intracellular availability of glucosyl units and therefore are in a causal relationship to BM15.2054's relatively lower impact on glucose oxidation, the lack of correlation between chronic oral actions on glycogenic and glycolytic fluxes clearly hints at two independent underlying mechanisms.
One of that two mechanisms is likely to involve activation of PPARγ, because antidiabetic potentials of TZD have been shown to correlate with their respective PPARγ-agonistic activities (Berger et al., 1996; 1999). Accordingly, BM13.1258 and BM15.2054 revealed to be very potent PPARγ-agonists and doubled muscle insulin sensitivity at daily oral doses, which are more than 30 fold below the troglitazone doses described to improve metabolic parameters in obese Zucker rats (Fujiwara et al., 1988; Okuno et al., 1998). Taken together, parallel potentials of BM13.1258 and BM15.2054 for PPARγ-activation and for improvement of insulin-stimulated muscle glycogenesis suggest the involvement of PPARγ in insulin sensitization, while the compounds' differential effects on muscle glucose oxidation hint at an underlying mechanism independent of PPARγ.
The measurement of glucose transport into isolated insulin target cells has been used by others to indicate insulin-mimetic and insulin-sensitizing properties of TZD compounds (Bähr et al., 1996; Kreutter et al., 1990; Park et al., 1998). In this study, the extent to which muscle glucose transport is increased by chronic TZD-treatment of obese Zucker rats obviously reflects the sum of substrate requirements caused by the insulin-dependent increase in substrate flux through the glycogenic pathway plus the insulin-independent increase in glycolytic flux. Thus, the superior potency of BM13.1258 vs BM15.2054 to insulin-independently stimulate glucose oxidation is reflected by significantly increased glucose uptake into muscle of BM13.1258-treated, but not of BM15.2054-treated rats. The obvious dependency of glucose transport rates on insulin-independent processes triggered by BM13.1258 and BM15.2054 corroborates our previously published view that substrate flux through the glycogenic pathway is a much more reliable parameter than glucose transport to quantify insulin-sensitizing and insulin-mimetic actions in isolated muscle (Fürnsinn et al., 1996; 1997). In that context it is of note that in the present study, solely relying on glucose transport rates might have lead to the doubtful conclusion that BM13.1258 is a more powerful insulin sensitizer than BM15.2054.
Acute TZD exposure in vitro
In a preceding study, we have described non-insulin-like stimulation of rat muscle glucose catabolism by acute exposure to the TZD troglitazone (Fürnsinn et al., 1997). That effect is obviously not specific for troglitazone, because BM13.1258 and BM15.2054 elicit comparable catabolic responses that are characterized by markedly increased glycolysis, reduced glycogen synthesis, and accelerated glucose transport. Increased rates of glucose transport in association with a non-insulin-like catabolic response again emphasize the limitations of glucose uptake as an appropriate measure of insulin-mimetic and insulin-sensitizing actions.
Furthermore, different biochemical mechanisms seem to be responsible for the catabolic effect triggered in vitro vs chronic action in vivo, because in contrast to the effects of oral treatment with BM13.1258 and BM15.2054, the catabolic response to acute exposure was associated with augmented rates of lactate release and equally potent in muscle specimens obtained from both lean SD and obese Zucker rats. It must be considered, however, that strong binding of other TZD to serum albumin and plasma proteins has been demonstrated (Shibukawa et al., 1995), what hampers the estimation of biologically available TZD concentrations prevailing in vivo and in vitro. Reliable conclusions from in vitro results to the in vivo situation are therefore difficult, what renders the specificity and the relevance of mechanisms activated by TZD exposure in vitro unclear. Although we do not know whether the mechanisms underlying acute actions are of any relevance in vivo, the results provide additional evidence that the employed TZD carry the potential to affect muscle glucose metabolism independently of insulin stimulation and PPARγ-receptors, because short-term exposure equally affected muscle metabolism in the absence and presence of insulin within 90 min, i.e. more rapidly than should be expected via PPARγ-induced modulation of gene transcription.
Conclusions
Considerable evidence has been provided that the therapeutic effects of TZD are mediated via activation of PPARγ resulting in insulin sensitization and improved glucose metabolism (Berger et al., 1996; 1999; Lehmann et al., 1995; Spiegelman, 1998). The results of this study do not challenge that hypothesis, but suggest that the new TZD compounds BM13.1258 and BM15.2054 may additionally affect muscle glucose metabolism via other biochemical pathways. That conclusion is supported by a lack of correlation between chronic oral actions of BM13.1258 and BM15.2054 on glycogenic and glycolytic fluxes as well as by acute and insulin-independent catabolic stimulation of glucose metabolism in vitro. Based on the findings from the present study, further efforts are required to clarify in detail, to what extent other mechanisms than PPARγ-dependent insulin sensitization may be involved in the antidiabetic action of TZD compounds.
Acknowledgments
We appreciate the help of the staff at the Biomedical Research Centre, University of Vienna, who took care of the rats. This work was supported by the Austrian Science Fund (Grant No. P13049-MED).
Abbreviations
- BSA
bovine serum albumine
- CMV
cytomegalovirus
- DMSO
dimethyl sulphoxide
- KRB
Krebs-Ringer buffer solution
- PPARγ
nuclear peroxisome proliferator-activated receptor γ
- PPRE
PPAR responsive element
- TZD
thiazolidinedione
References
- AUBERT J., CHAMPIGNY O., SAINT-MARC P., NEGREL R., COLLINS S., RICQUIER D., AILHAUD G. Up-regulation of UCP-2 gene expression by PPAR agonists in preadipose and adipose cells. Biochem. Biophys. Res. Commun. 1997;238:606–611. doi: 10.1006/bbrc.1997.7348. [DOI] [PubMed] [Google Scholar]
- BÄHR M., SPELLEKEN M., BOCK M., VON HOLTEY M., KIEHN R., ECKEL J. Acute and chronic effects of troglitazone (CS-045) on isolated rat ventricular cardiomyocytes. Diabetologia. 1996;39:766–774. doi: 10.1007/s001250050509. [DOI] [PubMed] [Google Scholar]
- BARON A.D., BRECHTEL G., WALLACE P., EDELMAN S.V. Rates and tissue sites of non-insulin and insulin-mediated glucose uptake in humans. Am. J. Physiol. 1988;255:E769–E774. doi: 10.1152/ajpendo.1988.255.6.E769. [DOI] [PubMed] [Google Scholar]
- BERGER J., BAILEY P., BISWAS C., CULLINAN C.A., DOEBBER T.W., HAYES N.S., SAPERSTEIN R., SMITH R.G., LEIBOWITZ M.D. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-γ: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 1996;137:4189–4195. doi: 10.1210/endo.137.10.8828476. [DOI] [PubMed] [Google Scholar]
- BERGER J., LEIBOWITZ M.D., DOEBBER T.W., ELBRECHT A., ZHANG B., ZHOU G., BISWAS C., CULLINAN C.A., HAYES N.S., LI Y., TANEN M., VENTRE J., WU M.S., BERGER G.D., MOSLEY R., MARQUIS R., SANTINI C., SAHOO S.P., TOLMAN R.L., SMITH R.G., MOLLER D.E. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARδ ligands produce distinct biological effects. J. Biol. Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- BOSS O., SAMEC S., KUHNE F., BIJLENGA P., ASSIMACOPOULOS-JEANNET F., SEYDOUX J., GIACOBINO J.P., MUZZIN P. Uncoupling protein-3 expression in rodent skeletal muscle is modulated by food intake but not by changes in environmental temperature. J. Biol. Chem. 1998;273:5–8. doi: 10.1074/jbc.273.1.5. [DOI] [PubMed] [Google Scholar]
- BOWEN L., STEIN P.P., STEVENSON R., SHULMAN G.I. The effect of CP 68,722, a thiozolidinedione derivative, on insulin sensitivity in lean and obese Zucker rats. Metabolism. 1991;40:1025–1030. doi: 10.1016/0026-0495(91)90124-f. [DOI] [PubMed] [Google Scholar]
- CAMIRAND A., MARIE V., RABELO R., SILVA J.E. Thiazolidinediones stimulate uncoupling protein-2 expression in cell lines representing white and brown adipose tissue and skeletal muscle. Endocrinology. 1998;139:428–431. doi: 10.1210/endo.139.1.5808. [DOI] [PubMed] [Google Scholar]
- CHEN C.A., OKAYAMA H. Calcium-phosphate-mediated gene transfer: A highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- CRETTAZ M., PRENTKI M., ZANIETTI D., JEANRENAUD B. Insulin resistance in soleus muscle from obese Zucker rats. Involvement of several defective sites. Biochem. J. 1980;186:525–534. doi: 10.1042/bj1860525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEFRONZO R.A. The triumvirate β-cell, muscle, liver: a collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- DIMITRIADIS G.D., LEIGHTON B., VLACHONIKOLIS I.G., PARRY-BILLINGS M., CHALLISS R.A.J., WEST D., NEWSHOLME E.A. Effects of hyperthyroidism on the sensitivity of glycolysis and glycogen synthesis to insulin in the soleus muscle of the rat. Biochem. J. 1988;253:87–92. doi: 10.1042/bj2530087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNNETT C.W. New tables for multiple comparisons with a control. Biometrics. 1964;20:482–491. [Google Scholar]
- ENGEL R.C., JONES J.B. Causes and elimination of erratic blanks in enzymatic metabolic assays involving the use of NAD+ in alkaline hydrazine buffers: improved conditions for the assay of L-glutamate, L-lactate and other metabolites. Anal. Biochem. 1978;88:475–484. doi: 10.1016/0003-2697(78)90447-5. [DOI] [PubMed] [Google Scholar]
- FUJIWARA T., YOSHIOKA S., YOSHIOKA T., USHIYAMA I., HORIKOSHI H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes. 1988;37:1549–1558. doi: 10.2337/diab.37.11.1549. [DOI] [PubMed] [Google Scholar]
- FÜRNSINN C., EBNER K., WALDHÄUSL W. Failure of GLP-1(7-36)amide to affect glycogenesis in rat skeletal muscle. Diabetologia. 1995;38:864–867. doi: 10.1007/s001250050365. [DOI] [PubMed] [Google Scholar]
- FÜRNSINN C., ENGLISCH R., EBNER K., NOWOTNY P., VOGL C., WALDHÄUSL W. Insulin-like vs non-insulin-like stimulation of glucose metabolism by vanadium, tungsten, and selenium compounds in rat muscle. Life Sciences. 1996;59:1989–2000. doi: 10.1016/s0024-3205(96)00550-4. [DOI] [PubMed] [Google Scholar]
- FÜRNSINN C., NESCHEN S., NOE C., BISSCHOP M., RODEN M., VOGL C., SCHNEIDER B., WALDHÄUSL W. Acute non-insulin-like stimulation of rat muscle glucose metabolism by troglitazone in vitro. Br. J. Pharmacol. 1997;122:1367–1374. doi: 10.1038/sj.bjp.0701527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN S., ISSEMANN I., SHEER E. A versatile in vivo and in vitro eukaryotic expression vector for protein engineering. Nucleic Acids Res. 1988;16:369. doi: 10.1093/nar/16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KREUTTER D.K., ANDREWS K.M., GIBBS E.M., HUTSON N.J., STEVENSON R.W. Insulin-like activity of new antidiabetic agent CP 68722 in 3T3-L1 adipocytes. Diabetes. 1990;39:1414–1419. doi: 10.2337/diab.39.11.1414. [DOI] [PubMed] [Google Scholar]
- KUEHNLE H.F. New therapeutic agents for the treatment of NIDDM. Endocrinology & Diabetes. 1996;104:93–101. doi: 10.1055/s-0029-1211429. [DOI] [PubMed] [Google Scholar]
- LEE M.K., OLEFSKY J.M. Acute effects of troglitazone on in vivo insulin action in normal rats. Metabolism. 1995;44:1166–1169. doi: 10.1016/0026-0495(95)90010-1. [DOI] [PubMed] [Google Scholar]
- LEHMANN J.M., MOORE L.B., SMITH-OLIVER T.A., WILKINSON W.O., WILLSON T.M., KLIEWER S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) J. Biol. Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- OAKES N.D., KENNEDY C.J., JENKINS A.B., LAYBUTT D.R., CHISHOLM D.J., KRAEGEN E.W. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes. 1994;43:1203–1210. doi: 10.2337/diab.43.10.1203. [DOI] [PubMed] [Google Scholar]
- OKUNO A., IKEDA K., SHIOTA M., FUJIWARA T., YOSHIOKA S., SUGANO T., HORIKOSHI H. Acute effect of troglitazone on glucose metabolism in the absence or presence of insulin in perfused rat hindlimb. Metabolism. 1997;46:716–721. doi: 10.1016/s0026-0495(97)90019-6. [DOI] [PubMed] [Google Scholar]
- OKUNO A., TAMEMOTI H., TOBE K., UEKI K., MORI Y., IWAMOTO K., UMESONO K., AKANUMA Y., FUJIWARA T., HORIKOSHI H., YAZAKI Y., KADOWAKI T. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Invest. 1998;101:1354–1361. doi: 10.1172/JCI1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLEFSKY J.M., KOLTERMAN O.G., SCARLETT J.A. Insulin action and resistance in obesity and noninsulin-dependent type II diabetes mellitus. Am. J. Physiol. 1982;243:E15–E30. doi: 10.1152/ajpendo.1982.243.1.E15. [DOI] [PubMed] [Google Scholar]
- PARK K.S., CIARALDI T.P., ABRAMS-CARTER L., MUDALIAR S., NIKOULINA S.E., HENRY R.R. Troglitazone regulation of glucose metabolism in human skeletal muscle cultures from obese type II diabetic subjects. J. Clin. Endocrinol. Metab. 1998;83:1636–1643. doi: 10.1210/jcem.83.5.4764. [DOI] [PubMed] [Google Scholar]
- SHIBUKAWA A., SAWADA T., NAKAO C., IZUMI T., NAKAGAWA T. High-performance frontal analysis for the study of protein binding of troglitazone (CS-045) in albumin solution and in human plasma. J. Chromatography A. 1995;697:337–343. doi: 10.1016/0021-9673(94)00929-4. [DOI] [PubMed] [Google Scholar]
- SHIMABUKURO M., ZHOU Y.-T., LEE Y., UNGER R.H. Induction of uncoupling protein-2 mRNA by troglitazone in the pancreatic islets of Zucker diabetic fatty rats. Biochem. Biophys. Res. Commun. 1997;237:359–361. doi: 10.1006/bbrc.1997.7140. [DOI] [PubMed] [Google Scholar]
- SPIEGELMAN B.M. PPARγ: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- STEVENSON R.A., HUTSON N.J., KRUPP M.N., VOLKMANN R.A., HOLLAND G.F., EGGLER J.F., CLARK D.A., MCPHERSON R.K., HALL K.L., DANBURY B.H., GIBBS E.M., KREUTTER D.K. Actions of novel antidiabetic agent englitazone in hyperglycemic hyperinsulinemic ob/ob mice. Diabetes. 1990;39:1218–1227. doi: 10.2337/diab.39.10.1218. [DOI] [PubMed] [Google Scholar]