

Abstract
Glibenclamide is a widely used sulphonylurea for the treatment of non-insulin-dependant diabetes mellitus (NIDDM). This agent has been reported to inhibit the activities of various ion channels and transporters. In the present study, we examined the effects of glibenclamide on the function of the H+/peptide cotransporters PEPT1 and PEPT2 by using stable transfectants.
Uptake of [14C]-glycylsarcosine, a typical substrate for peptide transporters, by PEPT1- or PEPT2-expressing transfectant was inhibited by glibenclamide as well as other sulphonylureas including tolbutamide. 3 Kinetic analysis revealed that the inhibition by glibenclamide was noncompetitive. Dixon plot analyses showed that the Ki values of this agent were 25 and 7.8 μM for PEPT1 and PEPT2, respectively.
Glibenclamide did not inhibit Na+-coupled alanine and α-methyl-D-glucoside transport, suggesting that the inhibitory effects of glibenclamide on peptide transporters were not due to nonspecific interactions.
There was little uptake of [3H]-glibenclamide by PEPT-expressing transfectants as compared to mock-transfected cells, suggesting that glibenclamide was not a substrate for these peptide transporters.
In summary, glibenclamide inhibited the [14C]-glycylsarcosine transport by PEPT1 and PEPT2 in a noncompetitive fashion, although glibenclamide per se was not transported through these transporters. These findings would provide important information for clinical, physiological and biochemical aspects of peptide transporters.
Keywords: Glibenclamide, peptide transporters, sulphonylureas, drug interaction, noncompetitive inhibition
Introduction
Glibenclamide is a second-generation sulphonylurea which has been widely used in the management of non-insulin-dependent diabetes mellitus (NIDDM). Glibenclamide induces insulin secretion by blocking ATP-sensitive potassium channels (KATP channel) in pancreatic β-cells (Panten et al., 1996; Babenko et al., 1998). This agent is also known to inhibit the activities of several ion channels and transporters, such as the cystic fibrosis transmembrane conductance regulator (CFTR) and the hepatic bile acid transporter (Fückel & Petzinger, 1992; Sheppard & Welsh, 1992; Kirk et al., 1993; Sheppard & Robinson, 1997). Therefore, it is assumed that glibenclamide not only exhibits its own pharmacological action but also causes unexpected effects by modulating these membrane proteins.
H+-coupled peptide transporters, which are localized at brush-border membranes of epithelial cells, mediate the absorption of various peptide-like drugs, as well as that of di- and tripeptides. As peptide transporters are primarily expressed in the small intestine and kidney, the pharmacokinetic profiles of peptide-like drugs, i.e. their absorption rates and biological half-lives, can be affected by the transport activities of peptide transporters. Two distinct peptide transporters, intestinal PEPT1 and renal PEPT2, have been cloned and characterized (Leibach & Ganapathy, 1996; Daniel & Herget, 1997; Inui & Terada, 1999). Functional studies have confirmed that a wide variety of pharmacologically active peptide-like drugs such as β-lactam antibiotics (Saito et al., 1995; Terada et al., 1997a) and the anticancer agent bestatin (Saito et al., 1996) are substrates of PEPT1 and PEPT2. Furthermore, the structural requirements of substrates for PEPT1 and PEPT2 were recently re-evaluated, and it was shown that PEPT1 and PEPT2 recognize various nonpeptidic substrates (Börner et al., 1998; Döring et al., 1998a,1998b; Ganapathy et al., 1998; Han et al., 1998; Temple et al., 1998).
As glibenclamide is administered orally, we hypothesized that the functions of some intestinal transporters might be affected by this agent crossing the intestinal epithelial cells. The intestinal peptide transporter, PEPT1, attracted our attention because of its important role in the therapeutic efficacy of peptide-like drugs. Recent findings of the broad substrate specificity of PEPT1 also led us to examine the interaction of glibenclamide with this transporter, although glibenclamide is a nonpeptidic compound. Furthermore, comparison of the interaction between PEPT1 and PEPT2 may be important in characterizing the biological aspects of peptide transporters. Therefore, in the present study, we examined the interactions of glibenclamide with peptide transporters using stable transfectants expressing rat PEPT1 or rat PEPT2.
Methods
Materials
[14C]-glycylsarcosine (1.78 GBq mmol−1) was obtained from Daiichi Pure Chemicals Co., Ltd. (Ibaraki, Japan). [3H]-glibenclamide (1.85 Tbq mmol−1) and [3H]-alanine (2.07 Tbq mmol−1) were purchased from Amersham International plc. (Buckinghamshire, U.K.). Methyl-α-D-[14C(U)]-glucopyranoside (α-methyl-D-glucoside (αMG)) (9.66 Gbq mmol−1) was supplied by Moravek Biochemicals Inc. (Brea, CA, U.S.A.). Glycylsarcosine, tolbutamide and chlorpropamide were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Glibenclamide was purchased from Wako Chemical Co. (Kyoto, Japan). All other chemicals used were of the highest purity available. Figure 1 shows the chemical structures of the sulphonylureas used.
Figure 1.
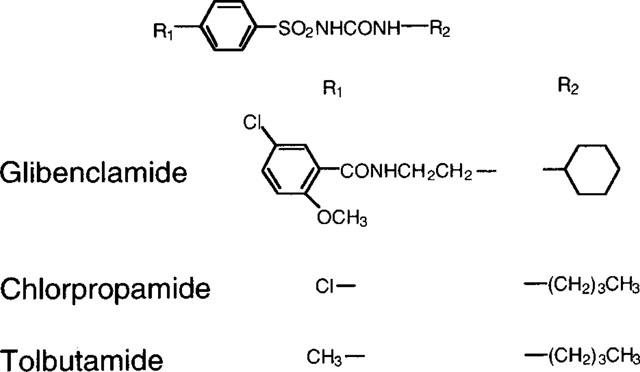
Chemical structures of sulphonylureas.
Cell culture
Parental LLC-PK1 cells were obtained from the American Type Culture Collection (ATCC CRL-1392). The LLC-PK1 cells transfected with rat PEPT1 cDNA (LLC-rPEPT1), with rat PEPT2 cDNA (LLC-rPEPT2) and with empty vector (LLC-pBK) were prepared and cultured as described previously (Terada et al., 1997a,1997b).
Uptake studies by cell monolayers
Cellular uptake of [14C]-glycylsarcosine was measured by monolayers grown in 35-mm plastic dishes as described previously (Terada et al., 1997b). The incubation medium contained (mM): NaCl, 145; KCl, 3; CaCl2, 1; MgCl2, 0.5; D-glucose, 5, and 2-(N-morpholino)ethanesulphonic acid (MES), 5 (pH 6.0) or HEPES, 5 (pH 7.4). Sulphonylureas were dissolved in dimethyl sulphoxide (DMSO) and the final concentration of DMSO in the incubation medium was 1.0%. The protein contents of cell monolayers solubilized in 1 M NaOH were determined by the method of Bradford (1976) using a Bio-Rad Protein Assay Kit (Bio-Rad, Richmond, CA, U.S.A.) with bovine γ-globulin as the standard.
Statistical analysis
Data were analysed statistically by non-paired t-test or one-way analysis of variance followed by Scheffé's test when multiple comparisons were needed.
Results
First, we examined the effects of various sulphonylureas on the uptake of [14C]-glycylsarcosine, a typical substrate for peptide transporters, by rat PEPT1- or rat PEPT2-expressing transfectants (LLC-rPEPT1 or LLC-rPEPT2 cells). As shown in Figure 2, [14C]-glycylsarcosine uptake by LLC-rPEPT1 and LLC-rPEPT2 cells was markedly inhibited by glibenclamide at 50 μM. Other sulphonylureas, i.e. tolbutamide and chlorpropamide, also significantly inhibited [14C]-glycylsarcosine uptake by both transfectants at the concentration of 1 mM. The solvent used for sulphonylureas, DMSO, showed no effect on [14C]-glycylsarcosine uptake (data not shown).
Figure 2.
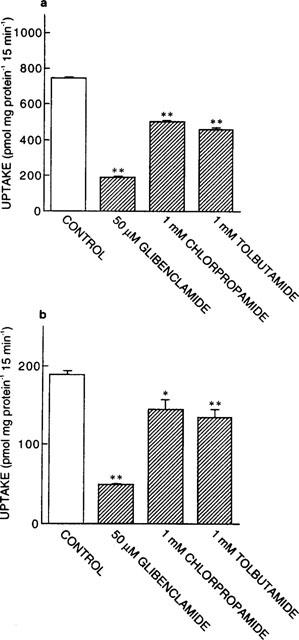
Effects of various sulphonylureas on [14C]-glycylsarcosine uptake by LLC-rPEPT1 (a) and LLC-rPEPT2 cells (b). The cell monolayers were incubated with incubation medium containing [14C]-glycylsarcosine (20 μM, pH 6.0) for 15 min at 37°C in the absence or presence of each inhibitor. Each column represents the mean±s.e.mean of six monolayers from two separate experiments. *P<0.05, **P<0.01, significantly different from control.
To investigate the kinetics of the inhibitory effects of glibenclamide on [14C]-glycylsarcosine uptake, the concentration dependence of [14C]-glycylsarcosine uptake was examined in the absence or presence of this agent (Figure 3). Specific uptake was calculated by subtracting the nonspecific uptake, which was estimated in the presence of excess unlabelled dipeptide, from the total uptake. Kinetic parameters were calculated according to the Michaelis-Menten equation using nonlinear least squares regression analysis. The presence of glibenclamide markedly decreased the maximal velocities (Vmax values for control versus those in the presence of glibenclamide: 74 versus 27 for PEPT1; 1.0 versus 0.24 for PEPT2, nmol mg protein−1 15 min−1). On the other hand, the apparent affinities were comparable (KM values for control versus those in the presence of glibenclamide: 1.8 versus 1.6 for PEPT1; 0.096 versus 0.078 for PEPT2, mM). These results suggested that glibenclamide noncompetitively inhibited [14C]-glycylsarcosine uptake by both PEPT1 and PEPT2.
Figure 3.
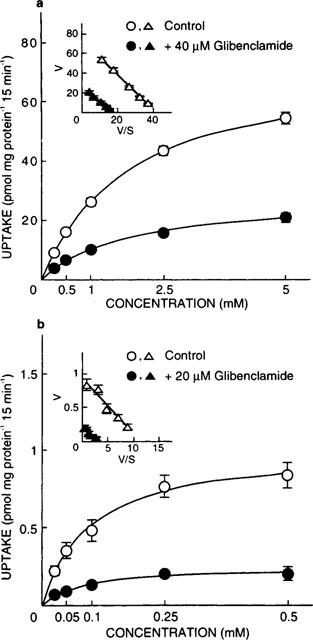
Concentration dependence of [14C]-glycylsarcosine uptake in the absence or presence of glibenclamide in LLC-rPEPT1 (a) and LLC-rPEPT2 cells (b). The cell monolayers were incubated with incubation medium containing various concentrations of [14C]-glycylsarcosine (pH 6.0) for 15 min at 37°C. Nonspecific uptake was evaluated by measuring [14C]-glycylsarcosine uptake in the presence of 50 mM glycylleucine, and the results are shown after correction for the nonsaturable component. Inset, Eadie-Hofstee plots of glycylsarcosine uptake after correction for the nonsaturable component. Each point represents the mean±s.e.mean of six monolayers from two separate experiments. When the error bars are not shown, they are smaller than the symbol.
To determine the inhibition constant (Ki) values of glibenclamide, Dixon plot analyses were performed. As illustrated in Figure 4, the plots were linear and crossed on the abscissa in both transfectants, suggesting that glibenclamide noncompetitively inhibited [14C]-glycylsarcosine uptake. The Ki values calculated from the points of intersection were as follows: 25 μM for PEPT1 and 7.8 μM for PEPT2.
Figure 4.
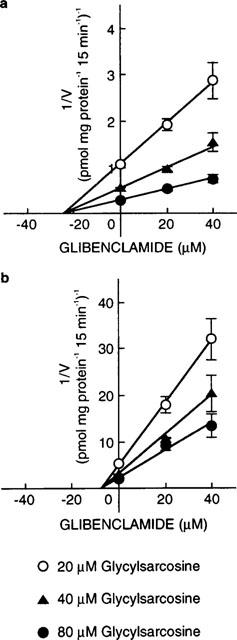
Dixon plots showing the inhibitory effects of glibenclamide on [14C]-glycylsarcosine uptake by LLC-rPEPT1 (a) and LLC-rPEPT2 cells (b). The cell monolayers were incubated for 15 min at 37°C with incubation medium containing [14C]-glycylsarcosine at pH 6.0. Data were corrected for the nonsaturable component by measuring [14C]-glycylsarcosine uptake in the presence of 20 mM glycylleucine. Each point represents the mean±s.e.mean of six monolayers from two separate experiments. When the error bars are not shown, they are smaller than the symbol.
Next, we examined whether glibenclamide affected the H+ gradient as the driving force of PEPT1 and PEPT2. As shown in Figure 5, [14C]-glycylsarcosine uptake at both pH 6.0 (with a H+ gradient) and pH 7.4 (without a H+ gradient) was inhibited by glibenclamide in LLC-rPEPT1 and LLC-rPEPT2 cells.
Figure 5.
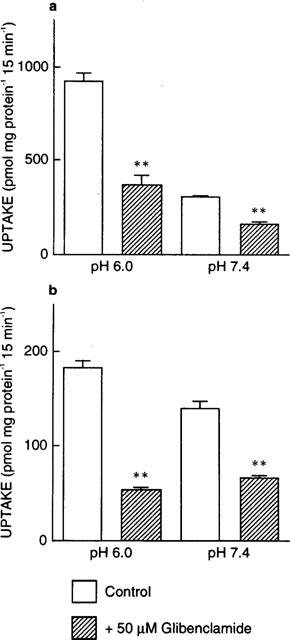
Effects of glibenclamide on [14C]-glycylsarcosine uptake at pH 6.0 or pH 7.4 by LLC-rPEPT1 (a) and LLC-rPEPT2 cells (b). The cell monolayers were incubated with [14C]-glycylsarcosine (20 μM) at pH 6.0 or pH 7.4 in the absence or presence of 50 μM glibenclamide for 15 min at 37°C. Each column represents the mean±s.e.mean of six monolayers from two separate experiments. **P<0.01, significantly different from control.
Parental LLC-PK1 cells were reported to show Na+-dependent and carrier-mediated transport for alanine (Kimmich et al., 1994) and α-methyl-D-glucoside (αMG) (Bennett & Kimmich, 1996). We found that uptake of [3H]-alanine and [14C]-αMG by both transfectants was decreased under Na+-free conditions, and was inhibited in the presence of either unlabelled excess alanine or αMG (data not shown). We subsequently examined the effect of glibenclamide on [3H]-alanine and [14C]-αMG uptake. As shown in Figure 6, glibenclamide did not inhibit [3H]-alanine or [14C]-αMG uptake by either transfectant.
Figure 6.

Effects of glibenclamide on [3H]-alanine (a, b) and [14C]-αMG (c, d) uptake by LLC-rPEPT1 and LLC-rPEPT2 cells. The cell monolayers were incubated with incubation medium containing [3H]-alanine (20 μM, pH 6.0) or [14C]-αMG (100 μM, pH 6.0) in the absence or presence of 50 μM glibenclamide for 15 min at 37°C. Each column represents the mean±s.e.mean of six monolayers from two separate experiments. **P<0.01, significantly different from control.
Finally, we examined whether glibenclamide was transported by peptide transporters. In a preliminary study, we found that the rate of [3H]-glibenclamide uptake by LLC-rPEPT1 cells was linear during the first 1 min of incubation. Therefore, we examined [3H]-glibenclamide uptake by LLC-pBK, LLC-rPEPT1 and LLC-rPEPT2 cells during a 30-s incubation period. As shown in Table 1, there was no increase in [3H]-glibenclamide uptake by LLC-rPEPT1 or LLC-rPEPT2 cells compared to LLC-pBK cells. In addition, 20 mM glycylsarcosine did not show an inhibitory effect on [3H]-glibenclamide uptake. These results suggested that glibenclamide was not transported via peptide transporters.
Table 1.
[3H]-glibenclamide uptake by LLC-pBK, LLC-rPEPT1 and LLC-rPEPT2 cells

Discussion
In the present study, we clearly demonstrated that glibenclamide is a potent inhibitor of the peptide transporters, PEPT1 and PEPT2, although glibenclamide itself was not a substrate for these transporters. Other sulphonylureas, tolbutamide and chlorpropamide, also inhibited the transport activities of PEPT1 and PEPT2. The calculated Ki value of glibenclamide was 25 μM for intestinal PEPT1, one of the highest affinities to this transporter reported to date (Terada et al., 1997b; Ganapathy et al., 1998). These findings have not only significant nutritional but also clinical implications, i.e. drug–drug interactions at the intestinal absorption level are possible. When glibenclamide is coadministered with peptide-like drugs such as oral β-lactam antibiotics to NIDDM patients, there is a possibility that the oral bioavailabilities of peptide-like drugs may be reduced. Therefore, our findings provide important insight into the therapeutic efficacy of such regimens as coadministration of sulphonylureas and peptide-like drugs.
The primary physiological role of peptide transporters is to mediate the transport of small peptides derived from dietary protein. However, recent investigations have indicated the novel physiological and pharmacological aspects of peptide transporters. For example, Döring et al. (1998a) demonstrated that delta-aminolevulinic acid, a precursor of porphyrin synthesis, was transported via PEPT1 and PEPT2 and suggested that it may be useful for photodynamic therapy. Merlin et al. (1998) showed that the intestinal PEPT1 transported formyl-Met-Leu-Phe, a major peptide neutrophil chemotactic factor, and suggested that modulating PEPT1 activity could influence states of intestinal inflammation. It is noted that glibenclamide is not administered transiently because NIDDM is a chronic disease. Therefore, the long-term administration of glibenclamide may affect physiological functions such as porphyrin metabolism and intestinal inflammation. Furthermore, PEPT2 was demonstrated to be expressed in a variety of tissues such as the lung, brain and mammary gland and it was suggested that this transporter may participate in as yet unknown physiological processes (Saito et al., 1996; Döring et al., 1998a). Therefore, it is important to evaluate the effects of long-term administration of glibenclamide on the functions of peptide transporters.
The inhibitory effect of glibenclamide was characterized kinetically as noncompetitive. Several mechanisms of this inhibition were considered. First, the noncompetitive inhibition might occur due to lipophilic nonspecific binding to membrane proteins because glibenclamide was reported to show a high serum protein binding ratio (99%) (Panten et al., 1989). However, these possibilities were excluded because glibenclamide did not show an inhibitory effect on Na+-coupled alanine and αMG transport. The data suggested that the inhibitory effect of glibenclamide on peptide transporters was not evoked by a nonspecific interaction. In fact, glibenclamide showed a stimulatory rather than an inhibitory effect on alanine uptake, although the reasons for this are unclear. Second, glibenclamide could affect the H+ gradient or the membrane potential, which are the driving forces for peptide transporters. This was also unlikely, because glibenclamide inhibited [14C]-glycylsarcosine uptake in the absence of a H+ gradient, and Na+-coupled αMG transport, which was clearly dependent on the membrane potential (Bennett & Kimmich, 1996), was not inhibited by glibenclamide. These results suggested that glibenclamide did not affect the driving force of peptide transporters. Third, it is possible that glibenclamide may bind to the transporter proteins at sites other than the substrate binding sites. Because glibenclamide has a sulphonylurea structure and an amide bond, these structures may be possible interaction sites with PEPTs. Recently, Akarawut et al. (1998) demonstrated that quinapril, an angiotensin converting enzyme inhibitor, noncompetitively inhibited glycylsarcosine uptake by PEPT2 using rabbit renal brush-border membrane vesicles, and suggested that it bound to a site distinct from that of the glycylsarcosine binding site. Although it is unclear whether glibenclamide and quinapril share a common biding site, these agents will be useful compounds to investigate the transport mechanisms of peptide transporters.
In conclusion, we found that the glibenclamide was a noncompetitive inhibitor of peptide transporters, although this agent itself is not a substrate for these transporters. To our knowledge, this is the first demonstration that sulphonylureas, especially glibenclamide, can inhibit the transport activities of peptide transporters. This agent should be a useful compound to examine the transport mechanism of peptide transporters. In addition, investigation of the interaction of sulphonylureas with peptide transporters may provide appropriate information for clinical usage of these drugs.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (B) and a Grant-in-Aid for Scientific Research on Priority Areas (No. 296) from The Ministry of Education, Science, Sports and Culture of Japan, and by a Grant-in-Aid from Japan Research Foundation for Clinical Pharmacology.
Abbreviations
- αMG
α-methyl-D-glucoside
- CFTR
the cystic fibrosis transmembrane conductance regulator
- DMSO
dimethyl sulphoxide
- MES
2-(N-morpholino)ethanesulphonic acid
- NIDDM
non-insulin-dependent diabetes mellitus
References
- AKARAWUT W., LIN C.-J., SMITH D.E. Noncompetitive inhibition of glycylsarcosine transport by quinapril in rabbit renal brush border membrane vesicles: Effect on high-affinity peptide transporter. J. Pharmacol. Exp. Ther. 1998;287:684–690. [PubMed] [Google Scholar]
- BABENKO A.P., AGUILAR-BRYAN L., BRYAN J. A view of SUR/KIR6.X, KATP channels. Ann. Rev. Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- BENNETT E., KIMMICH G.A. The molecular mechanism and potential dependence of the Na+/glucose cotransporter. Biophys. J. 1996;70:1676–1688. doi: 10.1016/S0006-3495(96)79730-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BÖRNER V., FEI Y.-J., HARTRODT B., GANAPATHY V., LEIBACH F.H., NEUBERT K., BRANDSCH M. Transport of amino acid aryl amides by the intestinal H+/peptide cotransport system, PEPT1. Eur. J. Biochem. 1998;255:698–702. doi: 10.1046/j.1432-1327.1998.2550698.x. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- DANIEL H., HERGET M. Cellular and molecular mechanisms of renal peptide transport. Am. J. Physiol. 1997;273:F1–F8. doi: 10.1152/ajprenal.1997.273.1.F1. [DOI] [PubMed] [Google Scholar]
- DÖRING F., WALTER J., WILL J., FÖCKING M., BOLL M., AMASHEH S., CLAUSS W., DANIEL H. Delta-aminolevulinic acid transport by intestinal and renal peptide transporters and its physiological and clinical implications. J. Clin. Invest. 1998a;101:2761–2767. doi: 10.1172/JCI1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DÖRING F., WILL J., AMASHEH S., CLAUSS W., AHLBRECHT H., DANIEL H. Minimal molecular determinants of substrates for recognition by the intestinal peptide transporter. J. Biol. Chem. 1998b;273:23211–23218. doi: 10.1074/jbc.273.36.23211. [DOI] [PubMed] [Google Scholar]
- FÜCKEL D., PETZINGER E. Interaction of sulphonylureas with the transport of bile acids into hepatocytes. Eur. J. Pharmacol. 1992;213:393–404. doi: 10.1016/0014-2999(92)90628-h. [DOI] [PubMed] [Google Scholar]
- GANAPATHY M.E., HUANG W., WANG H., GANAPATHY V., LEIBACH F.H. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem. Biophys. Res. Commun. 1998;246:470–475. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- HAN H-.K., DE VRUEH R.L.A., RHIE J.K., COVITZ K.-M.Y., SMITH P.L., LEE C.-P., OH D.-M., SADÉE W., AMIDON G.L. 5′-amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998;15:1154–1159. doi: 10.1023/a:1011919319810. [DOI] [PubMed] [Google Scholar]
- INUI K., TERADA T.Dipeptide transporters Membrane Transporters as Drug Targets 1999New York: Kluwer Academic/Plenum Publishers; 269–288.Amidon, G.L. & Sadée, W. (eds.) [Google Scholar]
- KIMMICH G.A., RANDLES J., WILSON J. Na+-coupled alanine transport in LLC-PK1 cells. Am. J. Physiol. 1994;267:C1119–C1129. doi: 10.1152/ajpcell.1994.267.4.C1119. [DOI] [PubMed] [Google Scholar]
- KIRK K., HORNER H.A., SPILLETT D.J., ELFORD B.C. Glibenclamide and meglitinide block the transport of low molecular weight solutes into malaria-infected erythrocytes. FEBS Lett. 1993;323:123–128. doi: 10.1016/0014-5793(93)81462-9. [DOI] [PubMed] [Google Scholar]
- LEIBACH F.H., GANAPATHY V. Peptide transporters in the intestine and the kidney. Ann. Rev. Nutr. 1996;16:99–119. doi: 10.1146/annurev.nu.16.070196.000531. [DOI] [PubMed] [Google Scholar]
- MERLIN D., STEEL A., GEWIRTZ A.T., SI-TAHAR M., HEDIGER M.A., MADARA J.L. hPepT1-mediated epithelial transport of bacteria-derived chemotactic peptides enhances neutrophil-epithelial interactions. J. Clin. Invest. 1998;102:2011–2018. doi: 10.1172/JCI4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANTEN U., BURGFELD J., GOERKE F., RENNICKE M., SCHWANSTECHER M., WALLASCH A., ZÜNKLER B.J., LENZEN S. Control of insulin secretion by sulphonylureas, meglitinide and diazoxide in relation to their binding to the sulfonylurea receptor in pancreatic islets. Biochem. Pharmacol. 1989;38:1217–1229. doi: 10.1016/0006-2952(89)90327-4. [DOI] [PubMed] [Google Scholar]
- PANTEN U., SCHWANSTECHER M., SCHWANSTECHER C. Sulfonylurea receptors and mechanism of sulfonylurea action. Exp. Clin. Endocrinol. 1996;104:1–9. doi: 10.1055/s-0029-1211414. [DOI] [PubMed] [Google Scholar]
- SAITO H., OKUDA M., TERADA T., SASAKI S., INUI K. Cloning and characterization of a rat H+/peptide cotransporter mediating absorption of β-lactam antibiotics in the intestine and kidney. J. Pharmacol. Exp. Ther. 1995;275:1631–1637. [PubMed] [Google Scholar]
- SAITO H., TERADA T., OKUDA M., SASAKI S., INUI K. Molecular cloning and tissue distribution of rat peptide transporter PEPT2. Biochim. Biophys. Acta. 1996;1280:173–177. doi: 10.1016/0005-2736(96)00024-7. [DOI] [PubMed] [Google Scholar]
- SHEPPARD D.N., ROBINSON K.A. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J. Physiol. (Lond.) 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEPPARD D.N., WELSH M.J. Effect of ATP-sensitive K+channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J. Gen. Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEMPLE C.S., STEWART A.K., MEREDITH D., LISTER N.A., MORGAN K.M., COLLIER I.D., VAUGHAN-JONES R.D., BOYD C.A.R., BAILEY P.D., BRONK J.R. Peptide mimics as substrates for the intestinal peptide transporter. J. Biol. Chem. 1998;273:20–22. doi: 10.1074/jbc.273.1.20. [DOI] [PubMed] [Google Scholar]
- TERADA T., SAITO H., MUKAI M., INUI K. Characterization of stably transfected kidney epithelial cell line expressing rat H+/peptide cotransporter PEPT1: Localization of PEPT1 and transport of β-lactam antibiotics. J. Pharmacol. Exp. Ther. 1997a;281:1415–1421. [PubMed] [Google Scholar]
- TERADA T., SAITO H., MUKAI M., INUI K. Recognition of β-lactam antibiotics by rat peptide transporters, PEPT1 and PEPT2, in LLC-PK1 cells. Am. J. Physiol. 1997b;273:F706–F711. doi: 10.1152/ajprenal.1997.273.5.F706. [DOI] [PubMed] [Google Scholar]