

Abstract
The effects of 4-Chloro-m-Cresol (4-CmC) were examined on heterologously expressed wild type (WT), Paramyotonia Congenita (R1448H) and Hyperkalemic Periodic Paralysis (M1360V) mutant α-subunits of human muscle sodium channels.
Block of rested sodium channels caused by 4-CmC was concentration-dependent with an ECR50 of 0.40 mM in WT, 0.45 mM in R1448H and 0.49 mM in M1360V.
Inactivation significantly promoted 4-CmC-induced sodium channel block in all clones indicated by 4-CmC-induced shifts of steady-state availability curves, reflecting a higher proportion of channel block at depolarized membrane potentials. Channel block was almost complete (>90%) at concentrations close to the ECR50 (0.5 mM) on application of an inactivating prepulse before the test pulse.
4-CmC accelerated the current decay following depolarization and prolonged recovery from inactivation in all clones. Of these, R1448H, the mutant which displayed severely impaired inactivation in the controls, responded to 4-CmC with the most pronounced acceleration of inactivation. Control experiments revealed enhanced recovery from inactivation in the mutants, which was restored to normal in 0.1 mM 4-CmC.
4-CmC induced no additional frequency-dependent block.
Our results clearly demonstrate that 4-CmC is as effective as lidocaine (Fan et al., 1996) in blocking muscle sodium channels. Low concentrations of the compound (⩽ECR50) were able to restore pathologically accelerated recovery from inactivation and impaired inactivation in the mutants to the WT value.
Keywords: Preservative, 4-Chloro-m-Cresol, muscle sodium channels, Paramyotonia Congenita, Hyperkalemic Periodic Paralysis
Introduction
4-Chloro-m-Cresol (4-CmC) is a preservative added to a wide variety of drugs. It has been shown that 4-CmC was capable of inducing contractures in excised muscle fibre bundles in vitro, with a threshold concentration of 70–100 μmol l−1 (Tegazzin et al., 1996). Experimental evidence suggests that altered function, as well as blockade of muscle sodium channels, is one of the mechanisms modulating the contracture response to various triggering agents in vitro (Fletcher et al., 1997). Therefore, we examined the effects of 4-CmC on wild type (WT) and 2 mutant muscle sodium channels. The mutants cause Paramyotonia Congenita and Hyperkalemic Periodic Paralysis, respectively. These muscle diseases belong to a group of sodium channelopathies which are caused by the exchange of single amino acids within the channel protein, resulting in altered function (Lehmann-Horn & Rüdel, 1995). In the Paramyotonia Congenita (PC) mutant R1448H a positively charged and extracellularly positioned arginine is replaced by a histidine, in the Hyperkalemic Periodic Paralysis (HPP) mutant M1360V an intracellularly positioned methionine is replaced by a valine.
Methods
Molecular biology
WT, R1448H and M1360V mutant α-subunits of human muscle sodium channels were heterologously expressed in human embryonic kidney (HEK293) cells, a stable cell line since 1962 (American Tissue Culture Collection CRL 1573). Site-directed mutagenesis was performed using the pSELECT mutagenesis system (Promega Corporation, Madison, WI, U.S.A.), as described by Chahine et al. (1994) and Wagner et al. (1997). Full-length R1448H and M1360V mutant α-subunits were reassembled in the mammalian expression vector pRc/CMV (Invitrogen, San Diego, CA, U.S.A.). Plasmids containing either WT α-subunits or the mutant sequence along with other regions from the WT channel were transfected into HEK293 cells, employing the calcium phosphate precipitation method, as described by Graham & Van der Eb (1973). Permanent expression was achieved by selection for resistance to the aminoglycoside antibiotic geneticin G418 (Life Technology, Eggenstein, Germany) (Mitrovic et al., 1994).
Experimental set-up
Standard whole-cell and inside-out voltage clamp experiments were performed at 20°C (Hamill et al., 1981). Each experiment consisted of test recordings with the drug present at only one concentration, and of drug-free control recordings before and after the test. At least three experiments were performed for each concentration. Wash-out was successful for all depicted results.
Source of reagents
Test solutions containing 4-CmC (FLUKA, D-82041, Deisenhofen, Germany) in different concentrations were derived from a stock solution of 10 mM 4-CmC in bath solution. Patch electrodes contained [mM]: CsCl2 130, MgCl2 2, EGTA 5, HEPES 10; bath solution contained [mM]: NaCl 140, MgCl2 1, KCl 4, CaCl2 2, HEPES 5, Dextrose 5. For inside-out patch recordings, the solutions were reversed. All solutions were adjusted to 290 mosm and to pH 7.4 by addition of CsOH.
Current recordings and analysis
For data acquisition and further analysis, we used the EPC9 digitally controlled amplifier in combination with Pulse and Pulse Fit software (HEKA Electronics, Lambrecht, Germany). The EPC9 provides automatic subtraction of capacitive and leakage currents by means of a prepulse protocol. Data were filtered at 10 kHz and digitized at 20 μs per point. We used only small cells with capacities between 9 and 15 pF; residual series resistance (after 50% compensation) ranged from 1.2 to 2.5 MΩ; experiments with a rise in series resistance were rejected. The time constant of voltage settling within the membrane (residual series resistance×cell capacitance) was <35 μs.
Resting state affinity
Block of rested channels was defined as the reduction of the maximum current elicited by families of depolarizing pulses going from −120 mV to test potentials ranging from −45 mV to +45 mV, 60 s after start of perfusion with drug-containing solution. Currents were normalized with respect to the current response in the control experiment. Concentration-response curves for drug effects on rested channels were obtained by plotting the fractional maximum currents (INa+) against the applied concentration of 4-CmC. Hill fits (INa+=[1+([C]/ECR50)n]−1) to the data yielded the concentration for half-maximum channel block (ECR50); the Hill-coefficient nH described the stoichiometry of drug binding to the channel. INa+ represents the fractional sodium current in the presence of the drug; [C] represents the applied concentration of 4-CmC.
Inside-out patch recordings
In order to evaluate drug effects on the intracellular side of the sodium channel, excised inside-out patches were exposed to 0.5 mM 4-CmC, and the amount of channel block achieved was compared to the amount of suppression of whole-cell current in the corresponding concentration, applying the pulse protocol described above.
Conductance-voltage plots
The voltage-dependence of Na+-channel conductance (g) was obtained using g=Imax/(V−Vrev), where V is the test potential and Vrev is the reversal potential. Boltzmann fits (g/gmax=(1+ exp(−zaF(Vtest−V0.5)/RT))−1) to the resulting normalized conductance-voltage plots yielded the voltage at half-maximum conductance (V0.5) and the slope factor za.
Frequency-dependent block
The frequency-dependent effects of 4-CmC were studied by applying either 10 or 100 Hz 10 ms depolarizing pulses from a holding potential of −100 mV to 0 mV. The current amplitude during each pulse was normalized to the 1st pulse.
Steady-state availability
The voltage-dependence of Na+-channel availability was obtained from a double-pulse protocol, where the cell membrane was first conditioned by a 20 ms prepulse starting at −150 mV up to prepulse potentials ranging from −150 to −5 mV, immediately followed by a 4 ms test pulse to 0 mV. Peak INa in response to this step was normalized to INa elicited by the test pulse at the most negative prepulse potential (−150 mV). Boltzmann fits to the resulting current-voltage plots yielded the membrane potential at half-maximum channel availability (V0.5) and the slope factor zi: I/Imax=(1+exp(−ziF(Vtest−V0.5)/RT))−1.
Affinity to the slow-inactivated state
In order to estimate drug binding to the slow-inactivated state compared to the resting state, the effects of 0.5 mM 4-CmC on the current response, elicited by 40 ms test pulses from −100 mV to 0 mV, were compared to the effects on the current response when introducing a 2.5 s inactivating prepulse to −35 mV before the test pulse.
Time course of channel inactivation
Time constants of inactivation τh were obtained from single or bi-exponential fits to the decaying currents, following 40 ms voltage jumps from −100 to 0 mV: I(t)=a0+a1exp(−t/τh1)+a2exp(−t/τh2).
Recovery from inactivation
Recovery from inactivation was assessed by a two-pulse recovery protocol: A 36 ms inactivating prepulse to 0 mV was followed by a variable recovery interval at −100 mV. Thereafter, a 25 ms test depolarization to 0 mV was used to elicit a peak current to assess the fraction of channels that had recovered during the preceding recovery interval. The currents elicited by the test pulse were normalized to the peak current elicited by the corresponding prepulse and plotted against the recovery interval. A single or double exponential was fitted to the data yielding the time constant of recovery from inactivation τrec: I(t)=a0+a1exp(−t/τrec1)+a2exp(−t/τrec2).
Statistics
All data are expressed as mean±s.d. Inter-group differences concerning the side of drug application, the applied pulse protocol or differences in shifts of steady-state activation plots with respect to a series of control experiments in bath solution were compared using an unpaired Student's t-test, according to the method of a priori ordered hypotheses (Maurer, 1995; P<0.05 was considered significant.
Results
Tonic inhibition of Na+-current by 4-CmC–resting state affinity
To assess resting affinity, we examined the concentration-dependence of block of maximum Na+-currents evoked by families of depolarizing pulses from a holding potential of −120 mV to the indicated test potentials. 4-CmC effectively blocked whole-cell Na+-currents in WT, Paramyotonia Congenita mutant R1448H and Hyperkalemic Periodic Paralysis mutant M1360V (Figure 1a–c). Data were obtained from different series of experiments in WT (n=6), R1448H (n⩾4) and M1360V (n⩾4) for each concentration of 4-CmC. Hill fits to the concentration-response plots (Figure 1d) yielded an ECR50 value of 0.40 mM in WT, 0.45 mM in R1448H and 0.49 mM in M1360V; nH was 2.6 in WT, 3.4 in R1448H and 3.0 in M1360V. When depolarizing pulses started from a holding potential of −100 mV instead of −120 mV, peak channel block achieved by 0.5 mM 4-CmC was 84±3% in R1448H (n=9) compared to 65.1±4% in WT (n=10)P<0.001 and 62.7±5.6% in M1360V (n=7).
Figure 1.
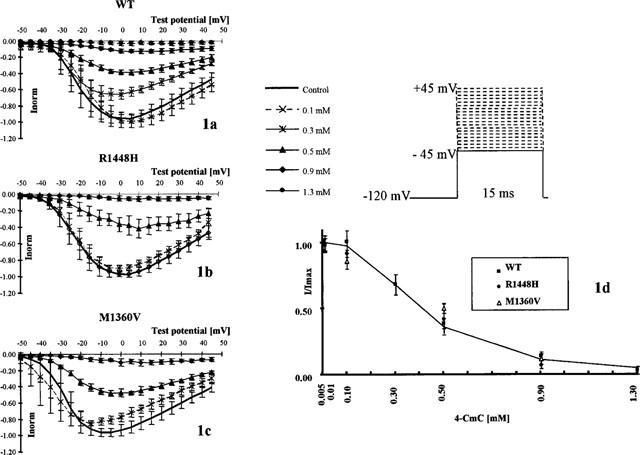
Resting state-dependent block of peak Na+-currents achieved by different concentrations of 4-CmC (n=6 in WT, n⩾4 in R1448H and M1360V for each concentration). (a–c) Normalized peak current-voltage plots following families of 15 ms depolarizing pulses to different test potentials. Each symbol on the curves represents the mean fractional peak current (with respect to the corresponding peak current in the control), elicited by depolarizations from −120 mV to the indicated test potentials [mV]. Error bars are standard deviations (±s.d.). The controls depicted here represent the starting values for all test experiments, each cell was only exposed to one test concentration. 4-CmC blocked sodium currents in a concentration-dependent manner in all clones. This figure does not allow exact interpretation of voltage shifts in the different concentrations of 4-CmC, as the starting values were pooled for all experiments and differed in the test potential at which maximum current was observed. (d) Maximum residual current (with respect to control) elicited in the presence of 4-CmC, applying the protocol of different depolarizing pulses depicted in (a–c) (mean±s.d.). The figure does not depict data measured at a fixed voltage to ensure that the concentration-response curves were unaffected by the choice of the test potential. The abscissa scale represents the concentration [mM] of 4-CmC and the ordinate scale the peak current normalized to the peak current obtained in the absence of the drug. The Hill fit to the WT data is depicted as a solid line yielding an ECR50 for peak current suppression of 0.40 mM in WT.
Effect of 4-CmC on the voltage-dependence of activation
The normalized conductance (gnorm) provides a better estimate of the relative amount of channels opening during each depolarizing pulse than the normalized current-voltage plots taking into account the decrease in driving force for Na+-ions through the channel, when the test potential is at more positive voltages. Figure 2 shows conductance transforms of the peak current-voltage relationships described above for two representative concentrations of 4-CmC: [0.5 mM], near the ECR50, and [0.9 mM] which produced >86% block in all clones. Open symbols represent the conductance in the presence of drug normalized to maximum conductance in the controls (mean±s.d.). When the curves for conductance were normalized to maximum conductance in the presence of the drug (closed symbols), it became evident that 4-CmC shifted the midpoints in the direction of more positive test potentials and reduced the slope in all clones. This means that, in addition to the total reduction in channel availability, depolarizations to more positive test potentials are required in the presence of 4-CmC to activate a corresponding fraction of the available sodium channels compared to control.
Figure 2.
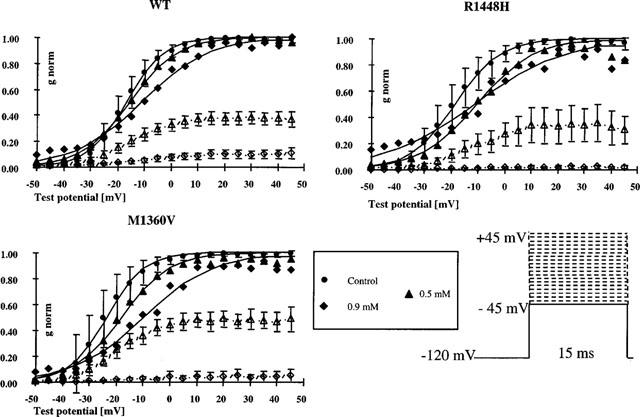
Conductance transforms (mean±s.d.) of the peak current-voltage plots depicted in Figure 1 for two representative concentrations of 4-CmC [0.5 and 0.9 mM], normalized to control (empty symbols) or to the peak conductance in the presence of drug (filled symbols). 4-CmC shifted the midpoints of the curves in the direction of less negative membrane potentials and reduced the slope of the curves indicating that voltage-dependence of activation is slightly reduced in the presence of the drug and the relative amount of channels that open during a depolarization is reduced at a given test potential.
The test potential at half-maximum activation (V0.5) obtained from a Boltzmann fit to the data represents the position of the curve along the voltage axis. The amount of depolarizing shift (dV0.5 [mV]) induced by different concentrations of 4-CmC in the three clones is given in Table 1.
Table 1.
4-CmC-induced shift of steady-state activation plots into depolarizing direction

Drug application to the intracellular side of the channel
The intracellular side of 13 excised inside-out macro patches (8 WT, 5 R1448H) was exposed to 0.5 mM 4-CmC. Maximum currents recorded during families of depolarizing pulses from −120 mV to test potentials ranging from −45 to +45 mV were 71.2±45 pA in the controls and 45.2±24 pA in 0.5 mM 4-CmC. Wash-out was possible and positive in eight cases, in five cases, the lifetime of a stable patch was too short to allow wash-out. This means that the amount of channel block achieved by 0.5 mM 4-CmC applied to the intracellular side of a total of 13 excised inside-out patches was only 33±12% (WT) and 34±12% (R1448H) of current in the controls compared to 62±4% (WT)P<0.01 and 58±12% (R1448H)P<0.01 block of whole-cell currents. This indicates that channel blocking properties of 4-CmC were more pronounced when applying the substance to the extracellular side of the channel of an intact whole-cell patch.
Affinity to the fast-inactivated state estimated by steady-state availability shifts
Drug binding to rested channels does not reflect all aspects of drug effects, especially in pathological conditions, where membrane potential might be more depolarized. After brief depolarizations, Na+-channels enter a fast-inactivated state from which they cannot readily reopen. Time constants for inactivation range from 0.59 to 1.80 ms, dependent on the clone. Thus, 20 ms inactivating pulses are sufficient to reach a steady-state level of inactivated channels at a given depolarized membrane potential. Steady-state inactivation plots assessed by a double-pulse protocol represent the relative fraction of channels that have not been inactivated by the 20 ms prepulse starting from −150 mV to the given prepulse potential, and thus are available on subsequent depolarization to 0 mV (test pulse). Currents elicited by each test pulse were normalized to the current at the most negative prepulse potential (−150 mV) of the same series and plotted against the prepulse potentials. The prepulse potential at half-maximum inactivation (V0.5in) obtained from Boltzmann fits reflects the position of the curve along the voltage axis, which, in control conditions, represents the voltage-dependent distribution between resting state and inactivated state at a given membrane potential. Summarized data for the three channel types (n=3 for each concentration (0.1, 0.5 and 0.9 mM) and each clone) showed that the midpoints (V0.5in) in the controls were more negative in R1448H (V0.5in −69.9±4.6 mV) and M1360V (V0.5in −65.4±4.8 mV) compared to WT (V0.5in −56.4±2.9 mV). These control data confirm INa+-kinetics previously described for those channels (Chahine et al., 1994; Wagner et al., 1997). 4-CmC shifted V0.5in considerably in the direction of more negative test potentials, with the degree of alteration being dependent upon drug concentration and clone (see Figure 3a–c). As virtually no time-dependent shift (<1 mV) of the inactivation curve was observed in control solution, the concentration-dependent left-shifts of the steady-state inactivation curves illustrated in Figure 3a–d reflect the additional reduction of channel availability induced by 4-CmC in the potential range from −20 to −120 mV. This indicates that the drug has a higher affinity to the channel when it is in the inactivated state. To estimate the dissociation constant (KD) of 4-CmC for the fast-inactivated state of the channel in the three clones, we fitted the concentration-dependence of V0.5in to the equation of Bean et al. (1983) (see Figure 3d): V0.5in=kln(1/(1+[C]/KD))+V0.5co where V0.5in is the midpoint in 4-CmC (mean, n=3), V0.5co is the midpoint in control solution, k the slope factor for the availability curve in the controls (k=−RT/Fz), [C] the applied concentration of 4-CmC and KD the dissociation constant for 4-CmC from the inactivated state. Resting state affinity was neglected for this fit. The estimated KD for WT, R1448H and M1360V were 43, 74 and 29 μM, respectively.
Figure 3.
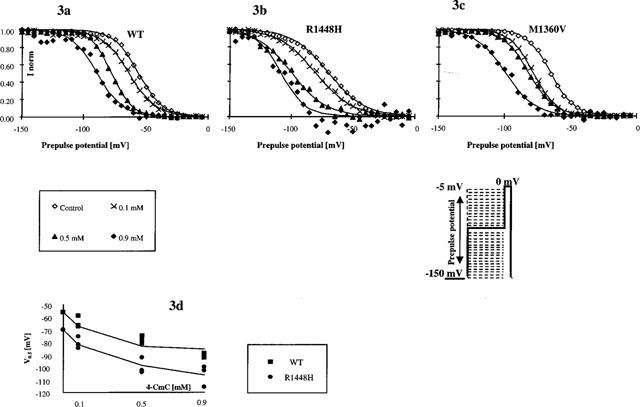
Affinity of 4-CmC to the fast-inactivated state of the channel. (a–c) Steady-state availability curves assessed by a 2-pulse protocol in the absence (control) and presence of 0.1, 0.5 or 0.9 mM 4-CmC. Each symbol represents the mean fractional current (n=3 for each concentration and each clone) elicited by a 4 ms test pulse to 0 mV, following a 20 ms inactivating prepulse from −150 mV to the indicated prepulse potential. Currents were normalized to maximum value (in each series at −150 mV prepotential); solid lines represent the best Boltzmann fit to the data. 4-CmC shifted the midpoints of the curves in the direction of more negative potentials (>−30 mV in 0.9 mM 4-CmC in all clones), indicating that the amount of channel block achieved by the different concentrations was enhanced at depolarized membrane potentials (compared to the block achieved at −150 mV). (d) Midpoints (V0.5) of steady-state availability plots in the absence (control) and presence of different concentrations of 4-CmC in WT and R1448H. Solid lines are least-squares fits to the equation V0.5in=kln(1/(1+[C]/KD))+V0.5co. Estimates of the dissociation constant (KD) of 4-CmC from the fast-inactivated state of the channel were 74 μM in R1448H compared to 43 μM in WT.
Affinity of 4-CmC to the slow-inactivated state of the channel
Prolonged depolarization induces a slow-inactivated state that requires much longer periods for recovery (>1 s). Therefore, the slow-inactivated state assumes particular importance in pathological conditions, such as myotonia or ischemia, in which tissues are depolarized for longer periods. To examine drug binding to the slow-inactivated channel induced by prolonged depolarization, we stepped up the holding potential from −100 to −35 mV for 2.5 s. The membrane potential was then returned to −100 mV for 10 ms allowing recovery from fast inactivation. The availability of unblocked resting channels was then assessed by a 40 ms test pulse to 0 mV. In control solution, the inactivating prepulse caused a reduction of the current elicited by the test pulse by 32±6% in WT, 43±7% in R1448H and 29±3% in M1360V (compare trace 1 and 2 in Figure 4). This is attributed to slow inactivation during the long conditioning prepulse, from which channels did not recover during the 10 ms repolarization (Ragsdale et al., 1994). In the presence of 0.5 mM 4-CmC, the reduction without slow inactivation was 64±1% in WT, 64±4% in M1360V and 82±1% in R1448H (compare trace 1 and 3 in Figure 4), whereas with slow inactivation it was 92±2% in WT, 92±3% in R1448H and 94±1% in M1360V, relative to the amplitude of current obtained with slow activation in the absence of drug (compare trace 2 and 4 in Figure 4). These results indicate a higher affinity of the slow-inactivated state of the channel to 4-CmC, compared to the resting state.
Figure 4.
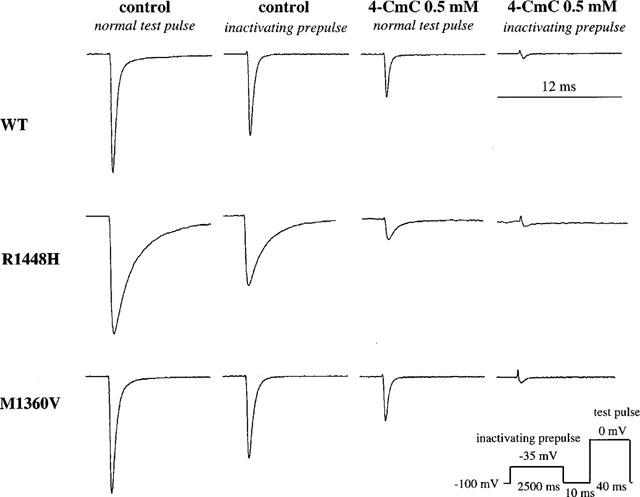
Affinity of 4-CmC to the slow-inactivated state of the channel. Representative current traces (in control solution and in 0.5 mM 4-CmC) evoked by 40 ms test pulses from −100 to 0 mV in the absence (trace 1 and 3) and presence (trace 2 and 4) of a 2.5 s inactivating prepulse introduced before the test pulse. In control solution, the inactivating prepulse caused a reduction of the current elicited by the test pulse due to slow inactivation, from which channels did not recover during the 10 ms repolarization (compare trace 1 and 2). Note that channel block achieved by 0.5 mM 4-CmC was >90% in all clones with slow inactivation (compare trace 2 and 4), whereas without slow inactivation it was only 64±1% in WT, 64±4% in M1360V and 82±1% in R1448H (compare trace 1 and 3).
Acceleration of the Na+-current decay phase by 4-CmC
To examine the time course of Na+-channel inactivation following a depolarization, series of 40 ms voltage steps (n⩾4 for each concentration (0.1, 0.5 and 0.9 mM) and each clone) from a holding potential of −100 mV were performed. The INa+-decay time course was assessed by fitting a single exponential to the decay of currents following depolarizations to 0 mV obtained with WT and M1360V channels and a double exponential in case of R1448H. For M1360V, the thus obtained time constant was not significantly different from WT (τh 0.60±0.1 ms vs τh 0.59±0.1 ms in WT). For R1448H, the decay was markedly slower (τh 1.80±0.4 ms) and obviously contained a second slower component that made up >10%. 4-CmC accelerated the decay of whole-cell currents in a concentration-dependent manner in all clones. R1448H, the mutant displaying severe inactivation deficiency in the controls, responded to 4-CmC with the most pronounced acceleration of the current decay (Figure 5): At a concentration of 0.1 mM, τh was accelerated to 1.44±0.1 ms and current decay became mono-exponential, at a concentration >0.5 mM, 4-CmC restored WT inactivation to R1448H whole-cell currents (τh 0.80±0.09 ms in [0.5 mM], τh 0.47±0.17 ms in [0.9 mM]). In WT, time constants (τh) in the presence of the drug were 0.38±0.07 ms in 0.1 mM, 0.29±0.06 ms in 0.5 mM and 0.26±0.08 ms in 0.9 mM 4-CmC. In M1360V, τh was accelerated to 0.41±0.1 ms in 0.1 mM 4-CmC, higher concentrations of the substance did not exert stronger effects on the current decay in this mutant.
Figure 5.
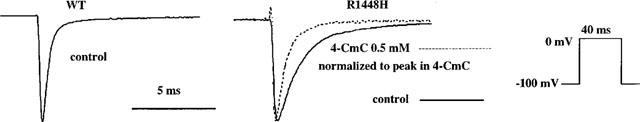
Acceleration of sodium channel inactivation of 4-CmC in R1448H. Representative current traces following a depolarization to 0 mV in WT (control) and in R1448H in the absence (solid line) and presence (dotted line) of 0.5 mM 4-CmC. All currents were scaled to the same size for better comparison of the decay phase. Time course was slowed with R1448H in control solution and could be accelerated to nearly normal by the application of 0.5 mM 4-CmC.
Effects of 4-CmC on recovery from inactivation
Following inactivation, channel reopenings are impossible until channels recover from inactivation, a process requiring several ms after membrane repolarization. Further information about drug effects on the stability of the inactivated state can be derived from the rate of channels recovering from inactivation in the presence of the drug. The time of membrane repolarization required to remove fast inactivation was assessed at −100 mV by a double-pulse protocol with varying time intervals between inactivating prepulse and test pulse (n=3 experiments for each concentration (0.1, 0.5 and 0.9 mM) and each clone). Time constants of recovery τrec, derived from mono-exponential fits to the fractional current having recovered from inactivation, plotted against the time interval between inactivating prepulse and test pulse (see Figure 6), revealed that recovery from inactivation was enhanced in both mutants in controls (τrec 1.42±0.15 ms in M1360V, τrec 1.46±0.18 ms in R1448H) compared to WT (τrec 1.94±0.2 ms). This finding is consistent with former reports (Chahine et al., 1994; Wagner et al., 1997). 4-CmC delayed recovery from inactivation in all clones in a concentration-dependent manner. The low concentration [0.1 mM] restored WT recovery to the mutants, the time course of recovery from inactivation remained mono-exponential in all clones. In the higher concentration [0.5 mM], the time course of recovery contained a second slower component τrec2 that made up 7±2% (WT), 21±7% (M1360V) and 25±16% (R1448H) of the current amplitude. The fast component τrec1 was 3.19±0.15 ms in WT, 2.48±0.5 ms in M1360V and 1.71±0.27 ms in R1448H, the slow component τrec2 was 31±16 ms in WT, 90±63 ms in M1360V and 62±72 ms in R1448H. In 0.9 mM 4-CmC, only very little residual current remained when applying this protocol. Therefore, a fit was not possible for those data.
Figure 6.
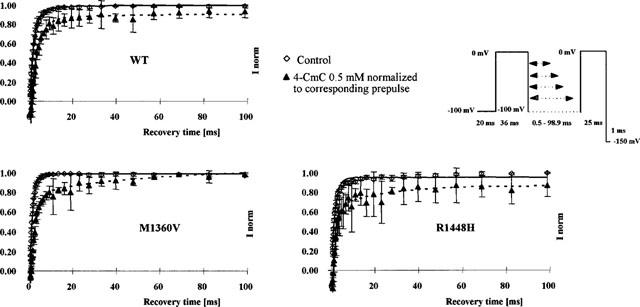
Recovery from inactivation assessed by a 2-pulse protocol in the absence (control) and presence of 0.5 mM 4-CmC. The abscissa represents the recovery time interval between pre- and test pulse [ms]; the ordinate the fractional current (mean±s.d.) elicited by the test pulse with respect to the prepulse in the same series. Solid lines are single exponential fits to the control data. In the presence of 0.5 mM 4-CmC, recovery was markedly delayed in all clones, the best fit to the recovery time was achieved with a double exponential (dotted line).
Frequency-dependent block
The interaction between blocking drugs and the sodium channel is modulated by different binding affinities to active or inactivated states compared to the resting state. When affinity to the resting state is low compared to active or inactivated states, high rates of stimulation may lead to an additional decrease in Na+-current relative to the 1st pulse when the interpulse interval becomes too short to allow complete recovery from block. To get an estimate of the kinetic of drug binding and unbinding during the interpulse interval, we applied a series of depolarizing pulses at high frequencies (10 and 100 Hz). Frequency-dependent block was defined as the additional INa+-decrease for the last pulse relative to the first pulse in a test train in the presence of 4-CmC. No frequency-dependent block could be detected in response to trains of 10-ms depolarizing pulses applied at 10 and 100 Hz in 0.5 mM 4-CmC. Applying the 10 Hz protocol, current amplitudes of the 10th pulse in 0.5 mM 4-CmC were 98±5% (WT), 101±14% (R1448H) and 110±2% (M1360V) of pulse number 1. With the 100 Hz protocol, current amplitudes of the 10th pulse relative to the 1st pulse were 82±8% (WT), 89±4% (R1448H) and 93±1% (M1360V) in bath solution and 66±21% (WT)P=0.3, 90±13% (R1448H)P=0.96 and 68±13% (M1360V)P=0.08 in 0.5 mM 4-CmC. P-values refer to the amplitude reduction relative to the 1st pulse in bath solution, applying this protocol. This means that 4-CmC is able to unbind from closed channels in less than 10 ms during membrane repolarization.
Discussion
Our results clearly suggest that 4-CmC is as potent as lidocaine and twice as potent as benzocaine, causing rest block in normal and mutant muscle sodium channels. The ECR50 for rest block by 4-CmC ranged from 0.40 to 0.49 mM (dependent on the clone), compared to 0.46 mM reported for lidocaine effects on the wild type channel (Fan et al., 1996) and 0.9 mM reported for benzocaine effects on an equine HPP channel (Sah et al., 1998). Similar to lidocaine and benzocaine, 4-CmC binds preferentially to the inactivated state, thereby reducing channel availability at membrane potentials between −120 and −20 mV (dependent on the clone). The estimated affinity KD of 4-CmC to the fast-inactivated channel was 29 μM for M1360V, 43 μM for WT and 74 μM for R1448H, compared to 24 μM (Balser et al., 1996b) and 11–63 μM (Fan et al., 1996) reported for lidocaine. Differences between the KD-values we found for 4-CmC and the values reported for lidocaine may result from the applied pulse protocol. Pulse length of the inactivating prepulse was 500 ms in the lidocaine experiments to ensure equilibration of drug binding to the channel, compared to 20 ms in our experiments. The 500 ms prepulse additionally induces slow inactivation (Balser et al., 1996b), thus the relative contribution of fast and slow inactivation to the measurement remains uncertain. The slow-inactivated state, induced by prepulses of 2.5 s duration, equally shows higher affinity to 4-CmC than the resting state. This finding might gain particular importance in pathological conditions, such as myotonia, in which muscle membrane is depolarized for longer periods (Lehmann-Horn & Rüdel, 1995). 4-CmC additionally stabilized the inactivated state by accelerating the time course of inactivation in all clones and delaying recovery from inactivation. Similar to the effect described for benzocaine on recovery from inactivation (Sah et al., 1998), the time course of recovery was delayed with increasing concentrations of the drug and remained mono-exponential during the first 100 ms in the presence of low concentrations of 4-CmC [0.1 mM]. In contrast to permanently charged local anaesthetic derivatives (QX314), which are ineffective when applied extracellularly (Ragsdale et al., 1994), 4-CmC-induced channel block was more pronounced on application of the substance to the extracellular surface of an intact whole-cell patch. This suggests either an extracellular locus for binding of 4-CmC, or easier access from the extracellular side to the binding site. According to current models for channel gating, the inactivation gate docking site is considered to be located on the intracellular surface of the cell (Catterall, 1996). However, the profound influence of externally positioned mutations on channel inactivation (Chahine et al., 1994) suggests that the structural characteristics of the inactivation mechanism are sensitive to remote allosteric effects. This indicates that drug interaction with the inactivation process does not require drug binding to the inactivation gate itself. The ability of 4-CmC to accelerate the current decay following depolarization should be seen rather in the light of a kinetic model developed by Balser et al. (1996a), based on lidocaine effects on inactivation-deficient rat skeletal muscle Na+-channels: The model supports a reinterpretation of local anaesthetic action, whereby lidocaine functions as an allosteric effector to enhance Na+-channel inactivation. In contrast to lidocaine and many other sodium channel blockers, 4-CmC failed to show additional frequency-dependent block, following repetitive depolarizations applied at 10 and 100 Hz. Lack of frequency-dependent block is equally described for benzocaine and its structural homologue ethyl-4-hydroxybenzoate, two molecules with striking structural similarities to 4-CmC. This behaviour is considered to be caused by fast dissociation of the drugs from the channel during interpulses (Quan et al., 1996).
Myotonia and paralysis are both attributed to altered inactivation phenotypes, a common feature in most sodium channel myopathies (Lehmann-Horn & Rüdel, 1995). Compared to WT, both mutants shifted steady-state inactivation plots in the direction of more negative membrane potentials and enhanced recovery from inactivation in the controls. These findings are consistent with former reports (Chahine et al., 1994; Wagner et al., 1997). In addition, the time course of channel inactivation was severely impaired in R1448H (Chahine et al., 1994). Wagner et al. (1997) described that the time course of inactivation following depolarizations to 0 mV was equally impaired in M1360V (τh 0.98±0.05 ms) compared to WT (τh 0.6±0.05 ms). However, our results revealed no delay in channel inactivation in M1360V. Both the slowed rate of inactivation and the rapid recovery from inactivation would cause hyperexcitability, long duration of action potentials or abbreviated refractory periods after action potentials (Chahine et al., 1994). Incomplete channel inactivation following activation of subpopulations of mutant sodium channels is considered sufficient to cause consecutive membrane depolarization leading to long-lasting hyperexcitability, if depolarization is mild, and to hypoexcitability if depolarization is severe enough to cause channel inactivation (Lehmann-Horn & Rüdel, 1995). Although lidocaine and its analogue mexiletine are the drugs of choice for intermittent or long-term therapy of myotonia (Lehmann-Horn & Rüdel, 1995), neither lidocaine nor mexiletine were able to correct altered inactivation properties in the HPP and PC mutants examined (Sah et al., 1998). Our in vitro results suggest a therapeutic effect of 4-CmC on certain inactivation-deficient sodium channels that goes beyond the effects which could be shown for lidocaine and mexiletine (Fan et al., 1996; Sah et al., 1998): 4-CmC restored WT recovery to both mutants and noticeably accelerated the current decay following depolarizations at a concentration of 0.1 mM. However, complete restitution of altered inactivation kinetics in R1448H was achieved only in concentrations >0.5 mM 4-CmC. Equally benzocaine is reported to be able to accelerate the current decay following depolarizations, but concentrations required to restore altered inactivation to normal in mutant channels are higher (Sah et al., 1998). Besides restitution of impaired inactivation and recovery from inactivation in mutant channels, an important element of drug effects might be that mutant channels are more sensitive to 4-CmC-induced channel block than the WT channels equally expressed in these disorders. While resting state affinity for 4-Cmc, estimated by depolarizations starting from −120 mV was equal for the three clones, channel block was markedly enhanced in R1448H compared to WT and M1360V (84 vs 65 and 63%), when the resting potential was −100 mV. This finding is consistent with reportsof Fan et al. (1996) who compared lidocaine sensitivity of the PC mutant R1448C to WT at different resting potentials: At −120 mV, lidocaine blocked sodium current in R1448C with an EC50 of 0.19 mM compared to 0.46 mM in WT, but these differences diminished when the resting potential was −150 mV. As the affinity of 4-Cmc to the inactivated state was not enhanced in R1448H compared to WT, the voltage-dependent higher sensitivity of R1448H to 4-CmC might be due to altered kinetics of distribution between the resting and inactivated states of the channel in the controls revealed by the amount of hyperpolarizing shift in steady-state inactivation plots compared to WT. A possible explanation might be that the rate of entry into the inactivated state is faster in the mutants at membrane potentials more negative than −60 mV (Yang et al., 1994): In M1360V, the deterioration is probably too small to find significant effects at a membrane potential of −100 mV, but R1448H should reveal more rest block induced by either 4-CmC or lidocaine, due to drug binding to the inactivated state at any given membrane potential more positive than −120 mV. Resting potential of muscle is much less hyperpolarized than the holding potential used in our experiments. In addition, muscle fibre membranes expressing inactivation-deficient Na+-channels might be even more depolarized than normal membranes (Lehmann-Horn & Rüdel, 1995). Thus, shifts in steady-state availability induced by the mutants are likely to be important in vivo, making mutant channels much more susceptible to 4-CmC than normal channels.
Conclusion
(1) 4-CmC is as effective as lidocaine in blocking muscle sodium channels. (2) In contrast to lidocaine, 4-CmC restores sodium channel dysfunction in certain myotonias to normal. (3) If its effects, as described here, are reproducible in heart muscle sodium channels, it may also be an effective anti-dysrythmic drug.
Acknowledgments
We are indebted to Professor Lehmann-Horn, Ulm, for providing us with transfected cells.
Abbreviations
- 4-CmC
4-Chloro-m-Cresol
- ECR50
concentration of 4-CmC which achieved half-maximum reduction of the peak current amplitude following families of depolarizing pulses from a prepulse potential of −120 mV
- F
Faraday's constant (9.648×104 C mol−1)
- HPP
Hyperkalemic Periodic Paralysis
- KD
dissociation constant for 4-CmC from the fast-inactivated state of the channel
- M1360V
sodium channel mutant substituting methionine for valine in the channel protein
- nH
Hill-coefficient describing the stoichiometry of drug binding to rested channels
- PC
Paramyotonia Congenita
- R
Gas constant (8.315 J K−1 mol−1)
- R1448H
sodium channel mutant substituting arginine for histidine in the channel protein
- τh
time constant of inactivation
- τrec
time constant of recovery from inactivation
- V0.5
midpoints of steady-state activation or steady-state inactivation curves
- WT
wild type
- z
effective gating charge which determines the slope of steady-state activation (za) or inactivation (zi) plots
References
- BALSER J.R., BRADLEY NUSS H., ORIAS D.W., JOHNS D.C., MARBAN E., TOMASELLI G.F., LAWRENCE J.H. Local anesthetics as effectors of allosteric gating. J. Clin. Invest. 1996a;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALSER J.R., BRADLEY NUSS H., ROMASHKO D.N., MARBAN E., TOMASELLI G.F. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J. Gen. Physiol. 1996b;107:643–658. doi: 10.1085/jgp.107.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN B.P., COHEN C.J., TSIEN R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CATTERALL W.A. Molecular properties of sodium and calcium channels. J. Bioenerg. Biomembr. 1996;28:219–230. doi: 10.1007/BF02110697. [DOI] [PubMed] [Google Scholar]
- CHAHINE M., GEORGE A.L., ZHOU M., JI S., SUN W., BARCHI R.L., HORN R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- FAN Z., GEORGE A.L., KYLE J.W., MAKIELSKI J.C. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. 1996;496:275–286. doi: 10.1113/jphysiol.1996.sp021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLETCHER J.E., WIELAND S.J., KARAN S.M., BEECH J., ROSENBERG H. Sodium channel in human malignant hyperthermia. Anesthesiology. 1997;86:1023–1032. doi: 10.1097/00000542-199705000-00004. [DOI] [PubMed] [Google Scholar]
- GRAHAM F.L., VAN DER EB A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- LEHMANN-HORN F., RÜDEL R. Hereditary nondystrophic myotonias and periodic paralyses. Curr. Opin. Neurol. 1995;8:402–410. doi: 10.1097/00019052-199510000-00014. [DOI] [PubMed] [Google Scholar]
- MAURER W.Multiple comparisons in drug clinical trials and preclinical assays: a priori ordered hypotheses Testing principles in clinical and preclinical trials 1995Fischer Verlag, Stuttgart; 3–8.Vollmar, J. ed [Google Scholar]
- MITROVIC N., GEORGE A.L., HEINE R., WAGNER S., PIKA U., HARTLAUB U., ZHOU M., LERCHE H., FAHLKE C., LEHMANN-HORN F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle sodium channel by the V1589M mutation. J. Physiol. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAN C., MOK W.M., WANG G.K. Use-dependent inhibition of Na+ currents by benzocaine homologs. Biophys. J. 1996;70:194–201. doi: 10.1016/S0006-3495(96)79563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGSDALE D.S., MC PHEE J.C., SCHEUER T., CATTERALL W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- SAH R.L., TSUSHIMA R.G., BACKX P.H. Effects of local anesthetics on Na+ channels containing the equine hyperkalemic periodic paralysis mutation. Am. J. Physiol. 1998;275:C389–C400. doi: 10.1152/ajpcell.1998.275.2.C389. [DOI] [PubMed] [Google Scholar]
- TEGAZZIN V., SCUTARI E., TREVES S., ZORZATO F. Chlorocresol, an additive to commercial succinylcholine, induces contracture of human malignant hyperthermia-susceptible muscles via activation of the ryanodine receptor Ca2+-channel. Anesthesiology. 1996;84:1380–1385. doi: 10.1097/00000542-199606000-00014. [DOI] [PubMed] [Google Scholar]
- WAGNER S., LERCHE H., MITROVIC N., HEINE R., GEORGE A.L., LEHMANN-HORN F. A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity. Neurology. 1997;49:1018–1025. doi: 10.1212/wnl.49.4.1018. [DOI] [PubMed] [Google Scholar]
- YANG N., JI S., ZHOU M., PTACEK L.J., BARCHI R.L., HORN R., GEORGE A.L. Sodium channel mutations exhibit similar biophysical phenotypes in vitro. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]