

Abstract
The antiarrhythmic drug mexiletine (Mex) is also used against myotonia. Searching for a more efficient drug, a new compound (Me5) was synthesized substituting the methyl group on the chiral carbon atom of Mex by an isopropyl group. Effects of Me5 on Na+ channels were compared to those of Mex in rat skeletal muscle fibres using the cell-attached patch clamp method.
Me5 (10 μM) reduced the maximal sodium current (INa) by 29.7±4.4 % (n=6) at a frequency of stimulation of 0.3 Hz and 65.7±4.4 % (n=6) at 1 Hz. At same concentration (10 μM), Mex was incapable of producing any effect (n=3). Me5 also shifted the steady-state inactivation curves by −7.9±0.9 mV (n=6) at 0.3 Hz and −12.2±1.0 mV (n=6) at 1 Hz.
In the presence of sea anemone toxin II (ATX; 5 μM), INa decayed more slowly and no longer to zero, providing a model of sodium channel myotonia. The effects of Me5 on peak INa were similar whatever ATX was present or not. Interestingly, Me5 did not modify the INa decay time constant nor the steady-state INa to peak INa ratio.
Analysis of ATX-induced late Na+ channel activity shows that Me5 did not affect mean open times and single-channel conductance, thus excluding open channel block property.
These results indicate that increasing hindrance on the chiral atom of Mex increases drug potency on wild-type and ATX-induced noninactivating INa and that Me5 might improve the prophylaxis of myotonia.
Keywords: Na+ channel, rat skeletal muscle, patch clamp, mexiletine derivative, sea anemone toxin, myotonia
Introduction
Molecular interactions of local anaesthetics (LAs) and related antiarrhythmic and anticonvulsant drugs with voltage-gated sodium channels are under intensive study. Biophysical evidences and binding studies first indicated that these drugs bind to the sodium channel protein on a site within the ion-conducting pore and accessible from the cytoplasmic side of the channel (Frazier et al., 1970; Hille, 1977; Postma & Catterall, 1984). Accordingly, heterologous expression of sodium channels and site-directed mutagenesis have recently allowed identification of several amino acids in segment 6 of domain IV of the sodium channel α-subunit as well as residues of the selectivity filter in domains II and III, which may compose part of the binding site (Ragsdale et al., 1994; 1996; Qu et al., 1995; Sunami et al., 1997; Wright et al., 1998). Block of sodium channels by LAs shows complex voltage- and frequency-dependent properties that have been explained by the drug binding dependence on the channel state (Snyders et al., 1992). Such phenomenon may result from a change in receptor affinity according to the modulated receptor hypothesis (Hille, 1977; Hondeghem & Katzung, 1977) or access of the drug to a receptor of fixed affinity, according to the guarded receptor hypothesis (Starmer et al., 1984). The higher the frequency of membrane depolarization, the greater is the blocking action, which provides a general mechanism for the selective action of this class of drugs on the arrhythmic heart (Catterall, 1987; Grant & Wendt, 1992).
In the skeletal muscle, excessive and sustained firing of action potentials results in myotonia, characterized by long-lasting involuntary muscle contractions. Inherited nondystrophic myotonias are due to mutations in the genes encoding the chloride or the sodium channel, which lead to a loss or a gain of function, respectively (for review, see Cannon, 1997; Ptáček, 1998). On the basis of the frequency-dependent blocking mechanism of antiarrhythmic drugs, the orally effective lidocaine derivative mexiletine appears as a drug of choice for treating myotonic syndromes (De Luca et al., 1997b; Ptáčček, 1998). However, in the particular case of sodium channel myotonias, mutations impair the channel inactivation process so important in LAs action. In fact, the drug was shown to be more potent in blocking wild-type than inactivation-deficient channels. (Sah et al., 1998; Fleischhauer et al., 1998; Desaphy et al., 1999), suggesting that its beneficial action is rather symptomatic: blocking wild-type Na+ channels may counteract sarcolemma hyperexcitability due to mutant Na+ channel activity. In a previous screening (Tricarico et al., 1991; De Luca et al., 1997a,1997b), we showed that chemical modification on the chiral carbon atom of mexiletine or the related drug tocainide could modify their blocking properties of muscle sodium channels as well as their antimyotonic efficacy against skeletal muscle hyperexcitability of adr mice, a model of chloride channel myotonia. Here we report the effect of a newly synthesized compound (Me5), in which an isopropyl group substitutes for the methyl group on the chiral carbon atom of mexiletine (Figure 1), on native rat skeletal muscle sodium channels recorded by means of the cell-attached patch clamp method. We also investigate the effect of Me5 on sodium currents modified by the sea anemone toxin II (ATX). This toxin binds to sodium channels and impairs channel inactivation, providing an in vitro pharmacological model of sodium channel myotonia (Cannon & Corey, 1993; Cannon et al., 1993). We found that the chemical modification of mexiletine greatly enhances sodium channel block. This may result from a facilitated access of the mexiletine analogue to the channel from intracellular side and a greater affinity of Me5 for its receptor. Moreover, the finding that Me5, in contrast with mexiletine, reduces wild-type and ATX-induced noninactivating sodium currents to the same extent, suggests that Me5 might improve the prophylaxis of myotonias.
Figure 1.
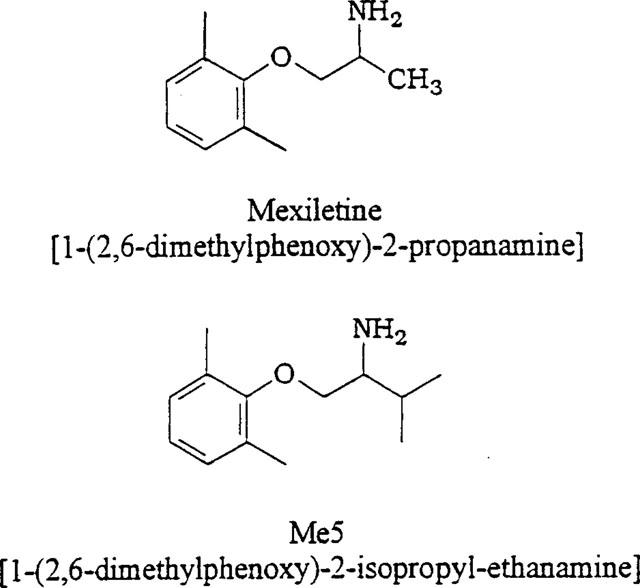
Chemical structure of mexiletine and its newly synthesized analogue Me5.
Methods
Cell preparation
Fibres from flexor digitorum brevis muscles of the hind feet were obtained from adult rats as previously described (Desaphy et al., 1998a). Briefly, animals were killed by an overdose of urethane (i.p. injection). Muscles were promptly removed and placed in Ringer solution supplemented with 2.5 mg ml−1 collagenase (3.3 iu ml−1, type XI-S, Sigma, St Louis, MO U.S.A.). They were shaken at 70 min−1 for 1–2 h at 32°C under a 95% O2/5% CO2 atmosphere. During this incubation, dissociated cells were sampled, rinsed several times with bath solution, and transferred to the RC-11 recording chamber (Warner Instruments, Hamden, CT, U.S.A.).
Cell-attached voltage-clamp recordings
Sodium currents were recorded at room temperature (21±2°C) in the cell-attached configuration of the patch-clamp method (Hamill et al., 1981) with an AxoPatch 1D amplifier and a CV-4-0.1/100U headstage (Axon Instruments, Foster City, CA, U.S.A.). Pipettes were formed from Corning 7052 glass (Garner Glass, Claremont, CA, U.S.A.) with a vertical puller (PP-82, Narishige, Tokyo, Japan). They were coated with sylgard 184 (Dow Corning) and heat polished on a microforge (MF-83, Narishige). Pipettes had resistances ranging from 2–4 MΩ when filled with the recording pipette solution. Voltage-clamp protocols and data acquisition were performed with pCLAMP 6.0 software (Axon Instruments) through a 12-bit A-D/D-A interface (digidata 1200, Axon Instruments). Currents were low-pass filtered at 2 kHz (−3 dB) by the amplifier four-pole Bessel filter and digitized at 10–20 kHz.
Membrane passive responses were controlled during the experiments and the patches in which eventual change might have modified sodium current characteristics were discarded from analysis. Capacitance currents were almost totally cancelled by the compensation circuit of the amplifier. To further eliminate residual capacitance transient and leak currents, the scaled passive ensemble average current recorded on return to the holding potential was subtracted from the current traces elicited by the depolarizing pulse. Particular care was taken to avoid contamination of the scaled passive ensemble average current by late sodium channel activity. This method usually gives satisfactory results, similar to the P/4 subtraction procedure of pCLAMP 6.0 (Desaphy et al., 1998a). The latter was not used in the present study because it might interfere with the effects of mexiletine and its derivative, which are particularly dependent on voltage protocol. After about 10 min of incubation in the CsCl bath solution, the fibres were depolarized to −7.6±1.0 mV (mean±s.e.mean, n=45 fibres), as determined by intracellular microelectrode measurements, and maintained this depolarized membrane potential (Vm) for a long duration (Desaphy et al., 1998a). The values of potential given in the manuscript are not corrected for Vm.
Sodium current analysis
Patches contained a large number of sodium channels, the activation of which resulted in macroscopic-current-like sodium currents. Throughout the experiment, the patch membrane was repetitively depolarized for 30 ms to −20 mV from a holding potential of −100 mV, at a frequency of 0.3 or 1.0 Hz. Such frequencies allowed complete recovery from inactivation between test pulses in the absence of drug. Patches with run-down of sodium current not greater than 15% in 20 min were considered for analysis, as already described (Desaphy et al., 1998a). When steady current levels were reached, both before and after drug application, a specific voltage–clamp protocol was applied to the patch membrane for the measure of the current–voltage relationship and the voltage dependence of activation and steady-state inactivation: the holding potential was −110 mV; a first pulse of 450 ms in duration was applied to potentials ranging from −140 to +80 mV in 10 mV increments and allowed the construction of the current–voltage relationship; a second pulse of 30 ms in duration was applied to −20 mV and allowed the measure of steady-state inactivation as a function of the first-pulse potential. The interpulse duration was set at 1 or 3 s to respect 1 or 0.3 Hz frequencies, respectively. This protocol was repeated 5 times and peak current amplitude values were reported as means±s.e.mean. The activation curve was constructed from the current–voltage relationship by converting current to conductance: the current amplitude was divided by the driving force (V−VNa), where V was the potential applied to the patch and VNa was the equilibrium electrochemical potential for sodium ions. VNa was estimated to be +70 mV from linear extrapolation of single-channel current/voltage curves. Activation curves were fitted to the Boltzmann equation: G/Gmax=1/{1+exp[(V−Vx)/K]} where G is conductance, Gmax is the maximal conductance, K is the slope factor, and Vx is the potential for having half of the channels activated. The steady-state inactivation curves were fitted to the Boltzmann equation: I/Imax=1/{1+exp[(V−Vh)/K]} where I is current, Imax is the maximal current, K is the slope factor, and Vh is the potential for having half of the channels inactivated. Single-channel analysis was performed on late sodium channel openings recorded in the presence of ATX during depolarizing pulses of 150 ms in duration applied to various potentials from a holding potential of −70 mV at 1 Hz frequency. Such a depolarized holding potential was used to reduce the number of overlapping events. Single-channel currents were estimated from all-points amplitude histograms fitted with multi-Gaussian functions by the Levenberg-Marquardt least-squares fitting routine of pSTAT software (pCLAMP 6.0 package). The single-channel conductance was determined as the slope of the single-channel current-voltage linear relationship. Open-time histograms were constructed using the half-amplitude threshold criterion (Colquhoun & Sigworth, 1983), discarding overlapping events as well as closed times shorter than 0.4 ms, considered as burst activity. In a previous study, open-time histograms were well fitted with a mono-exponential function (Desaphy et al., 1998b). Here, in the presence of ATX, the fits of histograms by the Levenberg-Marquardt least-squares fitting routine of pSTAT were significantly improved with a two-terms exponential, as evaluated by the F value (P<0.05), which compares the sum of squared errors of the different fitting models. This resulted in the determination of two mean open times, corresponding to the two exponential time constants of the function. The proportion of the two terms in the fit was not taken into consideration because excluding overlapping events might have introduced a bias, the probability of observing an overlap being greater for the longer mean open time. Open probability (NPo) was calculated for each depolarization as the sum of the open times (at each single-channel current level) occurring during the interval 10–145 ms divided by the total interval duration (135 ms). The mean NPo was further calculated as the mean±s.e.mean of NPo measured on at least 100 consecutive depolarizations. Averaged results are reported as means±s.e.mean for n patches. Statistical analysis was performed with Student's t-test for paired or grouped data, considering P<0.05 as significant.
Solutions and chemicals
Ringer solution contained (mM): NaCl 145, KCl 5, MgCl2 1, CaCl2 1, MOPS 10 and Glucose 5. Bath solution contained (mM): CsCl 145, EGTA 5, MgCl2 1, HEPES 10 and Glucose 5. Pipette solution contained (mM): NaCl 150, MgCl2 1, CaCl2 1 and HEPES 10. All solutions were buffered at pH 7.3. Mexiletine [1-(2,6-dimethylphenoxy)-2-propanamine] and its analogue Me5 [1-(2,6-dimethylphenoxy)-2-isopropyl-ethanamine] were synthesized in our laboratories directly in the R(−)-enantiomeric form as hydrochloride salt according to procedures previously described (Figure 1) (Franchini et al., 1994). The compounds were analysed by spectroscopy to control that the proportions of C, N and H atoms range within ±0.4% of the theorical values. They were first dissolved in bath solution and then added to the recording chamber at the final desired concentration. The sea anemone toxin II isolated from Anemonia sulcata was purchased from Alomone Labs (Jerusalem, Israel). ATX was dissolved in the pipette solution at the concentration of 5 μM.
Results
Comparison of the effects of mexiletine and Me5 on sodium currents
Cell-attached patches of freshly dissociated rat skeletal muscle fibres contained a large number of sodium channels. Thus macroscopic-current-like sodium currents were elicited by depolarizing the patch membrane from a holding potential of −100 mV to −20 mV (Figure 2). Control peak current amplitude remained quite stable during the experiment, indicating complete recovery from inactivation at stimulation frequencies of 0.3 Hz or 1 Hz (Figure 2). Bath application of 10 μM Me5 reduced current amplitude by about 30% at the frequency of 0.3 Hz. Increasing the frequency to 1 Hz further reduced peak current to about 30% of control (Figure 2). In the same experimental conditions, 10 μM mexiletine did not affect significantly the peak sodium current at 0.3 Hz (Figure 2). Indeed, the small reduction of current amplitude observed in the presence of mexiletine (about 5% of control current in 15 min, Table 1) well superimposed the spontaneous run-down usually observed in these conditions (Desaphy et al., 1998a). At 1 Hz, the effect of mexiletine was evident in only one out of three patches, in which the peak sodium current was reduced by 30%. In the two other patches, the reduction of current was not distinguishable from run-down. Thus Me5 appeared significantly more potent than its parent compound in reducing sodium currents in skeletal muscle fibres (Table 1).
Figure 2.
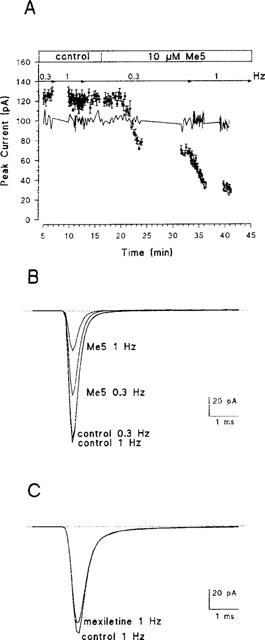
Frequency-dependent effect of Me5 on macroscopic–current-like Na+ currents in a cell-attached patch of rat skeletal muscle fibre. (A) Time course of changes in peak Na+ currents induced by 10 μM Me5 added to the bath solution. Na+ currents were elicited by depolarizing the patch membrane from a holding potential of −100 mV to −20 mV, at a frequency of 0.3 Hz or 1 Hz as indicated. Peak current amplitudes were measured on each trace, averaged every five consecutive traces and reported as means +s.e.mean as a function of the recording time. The blanks in the time course of Me5 effect correspond to the application of voltage protocols for the measure of I–V and steady-state inactivation relationships, maintaining the 1 Hz or the 0.3 Hz stimulation frequencies. The solid line indicates a typical time course of changes in peak Na+ currents recorded in the presence of 10 μM mexiletine. (B) Ensemble average Na+ currents constructed from 30 consecutive traces elicited as in (A) (same patch) at stimulation frequencies of 0.3 or 1 Hz, before (control) and after application of 10 μM Me5. (C) Ensemble average Na+ currents constructed as in (B), before and after application of 10 μM mexiletine. Only currents elicited at 1 Hz are illustrated.
Table 1.
Comparison of the effects of 10 μM of drug on peak sodium current parameters in rat skeletal muscle fibres
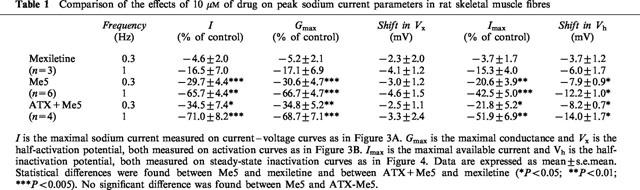
Sodium currents were observed between −80 and +60 mV, and peaked between −40 and −10 mV (−22.2±2.2 mV, n=9) (Figure 3A). Me5 did not modify the pattern of the current–voltage curve, reducing current amplitude at all voltages (Figure 3A). The voltage dependence of the activation curve was not modified by Me5 (Figure 3B). The half-activation potential Vx was −40.3±1.9 (n=9) in control, and the negative shift recorded in the presence of Me5 was similar to that recorded in the presence of mexiletine (Table 1) and to that we systematically observed during long cell-attached recordings (Desaphy et al., 1998a), suggesting that it is an artifact introduced by the methodology we use rather than an effect of drugs.
Figure 3.
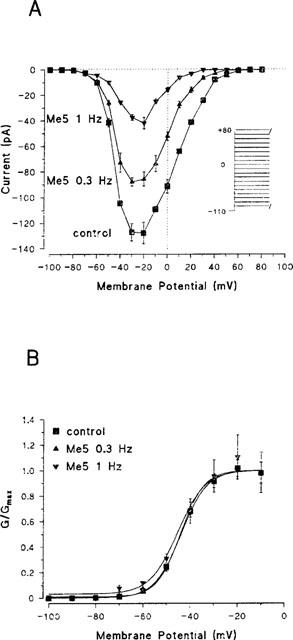
Effect of Me5 on current–voltage relationship and activation curve of peak sodium currents in a cell-attached patch of rat skeletal muscle fibre. (A) Current–voltage relationships were constructed before (control, 1 Hz) and after application of 10 μM Me5 from protocol shown in inset. Same patch as in Figure 2. (B) Normalized activation curves were constructed from current–voltage relationships shown in (A) by converting current to conductance considering a reversal potential of +70 mV for sodium ions (see Methods). Data were fitted to the Boltzmann equation with the following parameters: Vx=−44.0 mV and K=5.5 mV for control; Vx=−44.1 mV and K=5.0 mV for Me5 at 0.3 Hz; Vx=−43.9 mV and K=6.8 mV for Me5 at 1 Hz.
In control conditions, the half-inactivation potential Vh measured on steady-state inactivation curves was −84.7±2.5 mV in average (n=9) (Figure 4). In a previous study, we reported a shift of Vh of about −4 mV occurring spontaneously within 20 min of cell-attached recording (Desaphy et al., 1998a). The negative shift of steady-state inactivation curves recorded in the presence of 10 μM mexiletine was quite similar to this spontaneous shift (Table 1). In contrast, the Vh shift recorded in the presence of Me5 was significantly greater (Figure 4; Table 1). The slope factor of steady-state curves was 5.4±0.2 mV (n=9) in control and 5.6±0.3 mV (n=6) with Me5 at 1 Hz (P=0.84 between paired data). The maximal sodium current Imax, measured on nonnormalized steady-state inactivation curves, was significantly reduced by 10 μM Me5 as compared with the same concentration of mexiletine (Table 1). The effects of Me5 on Vh and Imax were both dependent on stimulation frequency.
Figure 4.
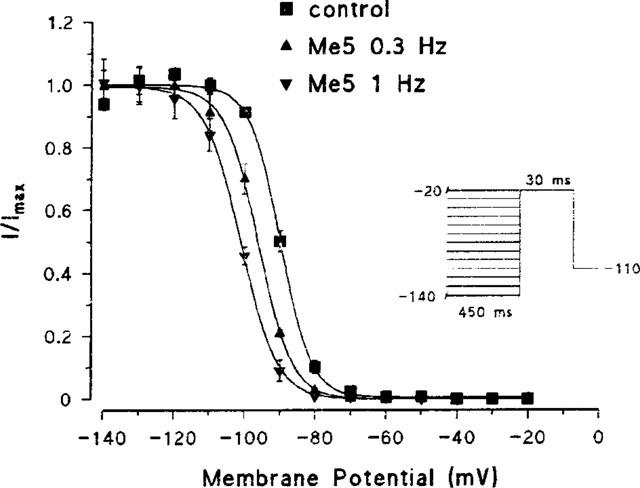
Effect of Me5 on steady-state inactivation curve of peak sodium currents in a cell-attached patch of rat skeletal muscle fibre. Normalized steady-state inactivation curves were constructed from the currents elicited in the same patch as in Figure 2 by the protocol shown in inset, before (control, 1 Hz) and after application of 10 μM Me5. Data were fitted to the Boltzmann equation with the following parameters: Vh=−89.9 mV and K=4.4 mV for control; Vh=−96.1 mV and K=4.8 mV for Me5 at 0.3 Hz; Vh=−101.1 mV and K=5.1 mV for Me5 at 1 Hz.
Effect of Me5 on sodium currents in the presence of ATX
We investigated the effect of Me5 on sodium channels in the presence of 5 μM sea anemone toxin II (ATX), which binds to and impairs the inactivation process of the channels (Rogers et al., 1996). We took two advantages of the sodium current changes induced by the sea anemone toxin II (ATX). First, this provides a model of sodium channel myotonia (Cannon & Corey, 1993; Cannon et al., 1993; Desaphy et al., 1999). Second, this allows the collection of numerous single channel events to perform accurate single channel analysis.
In the presence of ATX, macroscopic-current-like sodium currents decayed about five times more slowly than those recorded in control (compare Figure 5A with Figure 2B). In both cases, the decay was well fitted with an exponential function considering a single inactivation time constant τh, which was 0.27±0.01 ms (n=9) in control and 1.32±0.07 ms (n=4) with ATX (P<0.001 between grouped data). Also, the noninactivating steady-state sodium current measured at the end of the 30-ms long depolarizing pulse was significantly increased in the presence of ATX. The steady-state current to peak current ratio (Iss/Ipk) was, in percentage, 0.26±0.04 (n=9) in control and 2.72±0.39 (n=4) in the presence of the toxin (P<0.01 between grouped data). In contrast, ATX did not modify the voltage-dependence of current–voltage curves, activation curves, and steady-state inactivation curves of peak sodium currents (data not shown). Also the presence of the toxin did not modify significantly the reduction of I, Gmax and Imax and the shift of Vh induced by 10 μM Me5 at 0.3 and 1 Hz (Table 1). Interestingly, the mexiletine analogue had no significant effect on τh and Iss/Ipk (Figure 5B,C). In the presence of Me5 (n=4), τh was 1.33±0.07 ms at 0.3 Hz and 1.25±0.09 ms at 1 Hz (P=0.88 and P=0.41 with paired control data, respectively) and Iss/Ipk was 2.91±0.74% at 0.3 Hz and 2.57±0.19% at 1 Hz (P=0.83 and P=0.71 with paired control data, respectively). Therefore, in contrast to mexiletine which reduces peak current significantly more than steady-state current, Me5 has the same blocking activity on the peak and the steady-state current (Figure 5D).
Figure 5.
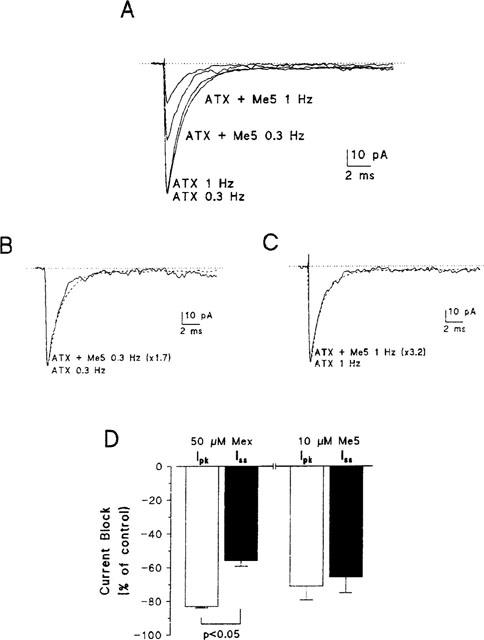
Effect of Me5 on macroscopic-current-like sodium currents recorded in the presence of ATX in a cell-attached patch of rat skeletal muscle fibre and comparison to the effects of mexiletine. (A) Ensemble average sodium currents constructed from 30 consecutive traces elicited by depolarizing the patch membrane from a holding potential of −100 mV to −20 mV, at stimulation frequencies of 0.3 Hz or 1 Hz, in the presence of 5 μM ATX in the pipette solution, before and after application of 10 μM Me5. (B, C) The scaled currents recorded at 0.3 Hz (B) or 1 Hz (C) after effect of Me5 (continuous lines) well superimposed their respective control current (dashed lines). (D) Bar graph showing the reduction of sodium current measured at the peak (open bars) and at the steady state (filled bars) induced by 50 μM mexiletine (n=4) and 10 μM Me5 (n=4), both in the presence of ATX and at a frequency of stimulation of 1 Hz. Me5 reduces peak and steady-state currents to the same extent whereas mexiletine reduces peak current significantly more than steady-state current.
ATX greatly increased late activity of sodium channels during the entire depolarization. Figure 6 shows late activity of sodium channels recorded in the presence of the toxin. The patch contained more than 50 channels as roughly determined by dividing the maximal peak current elicited from −100 to −20 mV by the single channel current. To perform single channel analysis, sodium currents were recorded within the interval 10–145 ms of depolarizing pulses applied to −20 mV for 150 ms at a frequency of 1 Hz. To reduce the number of overlapping events, the holding potential was set to −70 mV. From a total of 200 trials recorded in the presence of ATX only, no blank trace was observed. Numerous single openings and overlapping events (up to five channels opened simultaneously in this patch) of sodium channels occurred throughout the entire depolarization (Figure 6A). The same behaviour was observed after application of Me5 but the number of openings was reduced (Figure 6B). Late activity of sodium channels resulted in persistent ensemble average currents (Figure 6C, D), which were reduced by 65.7±9.3% (n=4) after application of 10 μM Me5. The mean NPo was reduced to a similar extent (Figure 6E).
Figure 6.
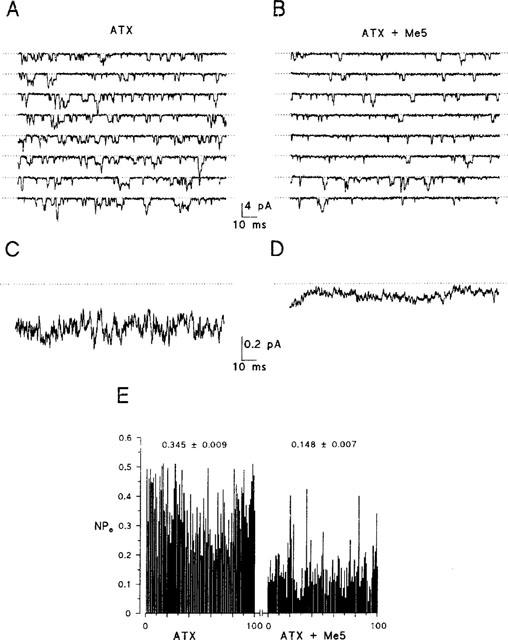
Effect of Me5 on open probability of sodium channels in the presence of ATX in a cell-attached patch of rat skeletal muscle fibres. (A, B) Sodium currents elicited during the time interval 10–145 ms of eight consecutive depolarizing pulses of 150 ms in duration applied to −20 mV from a holding potential of −70 mV, in the presence of 5 μM ATX, before and after application of 10 μM Me5. Stimulation frequency was 1 Hz. (C, D) Ensemble average currents constructed from 200 current traces elicited as in (A) and (B). (E) Diary of sodium channel open probability (NP0) during 100 consecutive depolarizing pulses used for the ensemble average currents shown in (C) and (D). The means±s.e.mean of NP0 calculated before and after application of Me5 are reported.
We looked at possible open channel block by Me5 in the presence of ATX. Open channel block is expected to reduce channel open times if block and channel gating operate on a similar time scale or to reduce single-channel current if block is very fast compared to the acquisition sampling rate. Single-channel current amplitude was measured on all-points amplitude histograms constructed at −20 mV in the presence of 5 μM ATX before and after application of 10 μM Me5 (Figure 7). The higher Gaussian bell corresponds to the closed state of the channels. The lower Gaussian bells correspond to the opening of one or two channels. Single-channel current was not significantly modified by the mexiletine analogue. Single-channel conductance calculated as the slope of single-channel current–voltage relationships was 17.4±1.0 pS and 17.3±0.9 pS (n=4, P=0.96 between paired data) before and after application of Me5, respectively (Figure 7C). To ascertain the lack of single-channel current reduction, current traces were directly scanned; we found no evidence of change in current amplitude within long open channel events. Figure 8 shows representative open-time histograms constructed at −20 mV in the absence or the presence of 5 μM ATX. Without toxin, histograms were best fitted by a mono-exponential function (Figure 8A), as already described (Desaphy et al., 1998b). In the four patches investigated with the toxin (Figure 8B,C), histograms were best fitted by considering two exponential time constants τfast and τslow. On average, τfast was 0.253±0.030 ms and 0.252±0.026 ms (P=0.88 between paired data) and τslow was 1.206±0.127 ms and 1.163±0.127 ms (P=0.42 between paired data), before and after application of Me5, respectively. Thus Me5 did not reduce significantly the mean open times of sodium channels. Nevertheless, because of the non-negligible number of overlapping events, we could not determine whether the drug modifies the relative proportion of τfast and τslow (see Methods).
Figure 7.
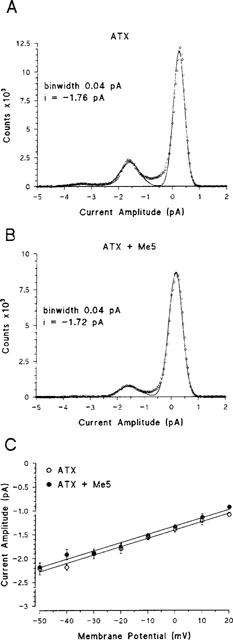
Effect of Me5 on single-channel current amplitude and conductance in the presence of ATX in cell-attached patches of rat skeletal muscle fibres. (A,B) All-points amplitude histograms constructed at −20 mV from current traces elicited as in Figure 6A (same patch), before (A) and after (B) application of 10 μM Me5. Sampled data (open circles) were fitted to a three-Gaussian function in (A) and a two-Gaussian function in (B). (C) Single-channel current–voltage relationships constructed from single-channel current amplitudes determined either by all-points amplitude histograms as in (A) and (B) or by direct measure on square-shaped single-channel events. Each data is the mean±s.e.mean from four patches. Data collected before and after application of 10 μM Me5 were linearly correlated (P<0.001 for both), indicating single-channel conductances of 17.4 and 17.3 pS, respectively.
Figure 8.
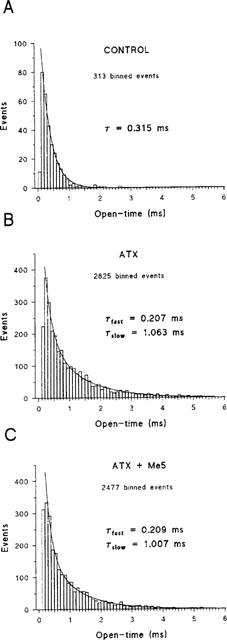
Effect of Me5 on mean open times of sodium channels recorded in the presence of ATX in a cell-attached patch of rat skeletal muscle fibre. (A) Typical open-time histogram constructed using the half-amplitude criterion from current traces elicited at −20 mV in a cell-attached patch in the absence of toxin. (B,C) Open-time histograms were constructed from current traces elicited at −20 mV in a cell-attached patch in the presence of ATX as in Figure 6A, before (B) and after (C) application of 10 μM Me5. Overlapping events and closed-time shorter than 0.4 ms were not included in the histograms. The sampling frequency of currents was 10 kHz and the histogram bin width was 100 μs. Excluding initial low-counts bins, data in (A) were well fitted with a mono-exponential function whereas data in (B) and (C) were best fitted with a two-exponential function. The exponential time constants (τ in A, τfast and τslow in B and C) give values of mean open times.
Discussion
Enhanced sodium channel block by Me5 compared to mexiletine
In a recent study (Desaphy et al., 1999), we reported that 50 μM mexiletine reduces by about 75% the sodium currents elicited from −100 to −20 mV every 3 s (0.3 Hz) in rat skeletal muscle fibres. Here, we show that 10 μM mexiletine has no effect on sodium currents in the same recording conditions. In contrast, significant reduction of sodium currents is obtained in response to 10 μM Me5 exposure. Therefore, isopropyl substitution on the chiral atom of mexiletine increases the apparent affinity between the LA and the rat skeletal muscle sodium channels. This is comparable to previous results (Tricarico et al., 1991; De Luca et al., 1997a) demonstrating that substitutions on the chiral centre of tocainide favour sodium channel block in frog muscle fibres or human myoballs. Altogether, these findings argue for a general law about tertiary amine LAs: sodium channel block is enhanced by increased steric hindrance on the chiral centre. Since the drug reaches its receptor from the intracellular side of the channel and we applied the compounds outside the cell, a difference in apparent affinity may come from difference in drug lipophilicity or pKa. In fact, the introduction of an isopropyl group in place of the methyl group on the chiral carbon atom is expected to increase both lipophilicity and pKa of Me5 compared to mexiletine, therefore facilitating the diffusion of Me5 through the membrane and increasing the amount of drug that has access to its receptor from the cytoplasmic side (Snyders et al., 1992). It should be noted however that the pKa of mexiletine is close to nine, so that mexiletine molecules are essentially permanently charged at physiological pH and, consequently, increasing pKa of mexiletine may have only small influence on the partitioning of the drug between compartments in our experiments. On the other hand, the chemical modification of mexiletine may increase the binding capacity of the drug to its receptor. The aromatic ring of LAs has been proposed to bind to a tyrosine residue (at position 1586 in the rat skeletal muscle sodium channel) while the terminal amine group of the drugs may associate with a neighbouring phenylalanine residue through π electron interaction (position 1579) (Ragsdale et al., 1994; Wright et al., 1998). Earlier studies suggested that additional carbons on the terminal amine group, which increase drug binding affinity, may prolong the life time of the blocked state via interaction between the amino terminal alkyl chains and the hydrophobic phenylalanine residue (Sheldon et al., 1991; Zamponi & French, 1994). Recently, site-specific mutagenesis experiments have added other evidence to the importance of hydrophobicity near or at the LA receptor for drug binding affinity (Sunami et al., 1997; Wang et al., 1998). By analogy, we believe that the presence of an isopropyl group in lieu of the methyl group on the mexiletine chiral carbon atom, which is very close to the amine group, may reduce drug unbinding rate.
Lack of open-channel block by Me5
Mexiletine is a very poor open-channel blocker and mainly binds to inactivated channels (Courtney, 1981; Sunami et al., 1991; Desaphy et al., 1999). Structural requirements for discriminating between open and inactivated channel blockers have not been clarified yet. Hille (1977) first proposed that LAs must be charged to block open channels. Molecular modelling suggested that such property may depend on the size of the tertiary amine; an open channel blocker might have an X dimension longer than 5 Å and an XY cross-sectional area larger than 45 Å2 (Courtney, 1988). Zamponi & French (1993) dissected lidocaine action and proposed that the aromatic ring causes inactivated channel block while the amine moiety is responsible for open channel block. In fact, the blocking property of a LA may result from the melange of all these characteristics. In the present study, we investigated open-channel block by Me5 in the presence of ATX. This toxin binds to the sodium channel α-subunit at the neurotoxin site 3, which lies on the extracellular side of the channel pore (Thomsen & Catterall, 1989; Rogers et al., 1996). The consequence is the impairment of the fast-inactivation process of the channels. Our results indicate that, compared to the toxin-free channels characterized by a unique mean open time, ATX-bound skeletal muscle Na+ channels can undergo a second, longer mean open time (τslow) and that they recover more rapidly from inactivation, since numerous reopenings occur within the entire depolarization duration. Me5 did not reduce the unitary current nor either of the two mean open times, providing no evidence of open channel block by Me5. Thus the increase of pKa, which might have only lightly increased the proportion of charged Me5 (see above), and the increase of drug size, both resulting from the isopropyl substitution, are not sufficient to confer open-channel block characteristic to the mexiletine analogue.
Potential pharmacological interest of Me5 in the treatment of myotonia
ATX-modified sodium channels provide a good model for the inherited myotonic diseases due to mutations in the gene encoding the skeletal muscle sodium channel (Cannon & Corey, 1993; Cannon et al., 1993; Desaphy et al., 1999). Although the use of heterologously expressed recombinant mutant channels in mammalian cell lines may provide interesting information on the interaction between mutant channels and drugs at the molecular level, the use of ATX allows to test drugs on the native tissue in a situation that closely mimics the molecular and cellular natural defect. On this basis, the toxin has been widely used as a model of the LQT-3 syndrome, an heart disease with elementary biophysical features similar to those observed in sodium channel-derived myotonias (Priori et al., 1996; Sicouri et al., 1997; Shimizu & Antzelevitch, 1997). When applied to skeletal muscle fibres, ATX slows the sodium channel entry into the inactivated state, causing at the same time slower decay and incomplete inactivation of the macroscopic–current-like sodium current, as it is generally observed in patients suffering from myotonia (Cannon, 1997). Recently, we have shown that mexiletine, which is today the drug of choice for treating myotonias (Ptáčček, 1998), blocks the peak sodium current more than the ATX-induced steady-state sodium current (Desaphy et al., 1999). A similar conclusion was raised by other investigators which studied the effects of mexiletine on mutant channels (Sah et al., 1998; Fleischhauer et al., 1998), suggesting that the therapeutic effect of mexiletine may be rather unspecific, reducing sarcolemma hyperexcitability by blocking wild-type channels. Although Me5 is still unable to fully restore sodium current kinetics towards those of wild-type channels, mexiletine structural modification improves drug action on inactivation-deficient Na+ channels since Me5 reduces peak and steady-state currents to the same extent. This improvement obtained with Me5 is likely to result from a reduced unbinding rate of this compound compared to mexiletine, as suggested above. In this situation, the greater stabilization of the channel in an inactivated state expected from Me5 may limit the number of channel reopenings, as it has been proposed to explain the beneficial effect of mexiletine or lidocaine on the cardiac Na+ channels carrying mutations of the Long–QT3 syndrome (Dumaine & Kirsch, 1998). If such is the case, two ways could be followed to find more specific antimyotonic agents: searching for a drug with either enhanced open-channel blocking action or reduced unbinding rate.
Acknowledgments
This work was supported by grants from Telethon-Italy to D. Conte Camerino (#901 and 1208) and a postdoctoral fellowship from Telethon-Italy to J.-F. Desaphy.
Abbreviations
- ATX
sea anemone toxin II
- INa
sodium current
- LAs
local anaesthetics
- Me5
1-(2,6-dimethylphenoxy)-2-isopropyl-ethanamine
- Mex
mexiletine
References
- CANNON S.C. From mutation to myotonia in sodium channel disorders. Neuromuscul. Disord. 1997;7:241–249. doi: 10.1016/s0960-8966(97)00430-6. [DOI] [PubMed] [Google Scholar]
- CANNON S.C., BROWN R.H., COREY D.P. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys. J. 1993;65:270–284. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANNON S.C., COREY D.P. Loss of Na+ channel inactivation by anemone toxin (ATX II) mimics the myotonic state in hyperkalaemic periodic paralysis. J. Physiol. 1993;466:501–520. [PMC free article] [PubMed] [Google Scholar]
- CATTERALL W.A. Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol. Sci. 1987;8:57–65. [Google Scholar]
- COLQUHOUN D., SIGWORTH F.J.Fitting and statistical analysis of single-channel records Single-channel recording 1983Plenum Press: New York; 191–263.In: Sakmann, B. & Neher, E. (eds) [Google Scholar]
- COURTNEY K.R. Comparative actions of mexiletine on sodium channels in nerve, skeletal and cardiac muscle. Eur. J. Pharmacol. 1981;74:9–18. doi: 10.1016/0014-2999(81)90317-4. [DOI] [PubMed] [Google Scholar]
- COURTNEY K.R. Why do some drugs preferentially block open sodium channels. J. Mol. Cell. Cardiol. 1988;20:461–464. doi: 10.1016/s0022-2828(88)80073-7. [DOI] [PubMed] [Google Scholar]
- DE LUCA A., NATUZZI F., FALCONE G., DURANTI A., LENTINI G., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D. Inhibition of frog skeletal muscle sodium channels by newly synthesized chiral derivatives of mexiletine and tocainide. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997a;356:777–787. doi: 10.1007/pl00005118. [DOI] [PubMed] [Google Scholar]
- DE LUCA A., PIERNO S., NATUZZI F., FRANCHINI C., DURANTI A., LENTINI G., TORTORELLA V., JOCKUSCH H., CONTE CAMERINO D. Evaluation of the antimyotonic activity of mexiletine and some new analogs on sodium currents of single muscle fibers and on the abnormal excitability of the myotonic ADR mouse. J. Pharmacol. Exp. Ther. 1997b;282:93–100. [PubMed] [Google Scholar]
- DESAPHY J.-F., CONTE CAMERINO D., TORTORELLA V., DE LUCA A. Effect of mexiletine on sea anemone toxin-induced noninactivating sodium channels of rat skeletal muscle, a model of sodium channel myotonia. Neuromuscul. Disord. 1999;9:182–189. doi: 10.1016/s0960-8966(98)00115-1. [DOI] [PubMed] [Google Scholar]
- DESAPHY J.-F., DE LUCA A., CONTE CAMERINO D. Blockade by cAMP of native sodium channels of adult rat skeletal muscle fibers. Am. J. Physiol. 1998a;275:C1465–C1472. doi: 10.1152/ajpcell.1998.275.6.C1465. [DOI] [PubMed] [Google Scholar]
- DESAPHY J.-F., DE LUCA A., PIERNO S., IMBRICI P., CONTE CAMERINO D. Partial recovery of skeletal muscle sodium channel properties in aged rats chronically treated with growth hormone or the GH-secretagogue hexarelin. J. Pharmacol. Exp. Ther. 1998b;286:903–912. [PubMed] [Google Scholar]
- DUMAINE R., KIRSCH G.E. Mechanism of lidocaine block of late current in long Q-T mutant Na+ channels. Am. J. Physiol. 1998;274:H477–H487. doi: 10.1152/ajpheart.1998.274.2.H477. [DOI] [PubMed] [Google Scholar]
- FLEISCHHAUER R., MITROVIC N., DEYMEER F., LEHMANN-HORN F., LERCHE H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch. 1998;436:757–765. doi: 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- FRANCHINI C., CELLUCCI C., CORBO F., LENTINI G., SCILIMATI A., TORTORELLA V., STASI F. Stereospecific synthesis and absolute configuration of mexiletine. Chirality. 1994;6:590–595. [Google Scholar]
- FRAZIER D.T., NARAHASHI T., YAMADA M. The site of action and active form of local anesthetics. J. Pharmacol. Exp. Ther. 1970;171:45–51. [PubMed] [Google Scholar]
- GRANT A.O., WENDT D.J. Block and modulation of cardiac Na+ channels by antiarrhythmic drugs, neurotransmitters and hormones. Trends Pharmacol. Sci. 1992;13:352–358. doi: 10.1016/0165-6147(92)90108-i. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HILLE B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDEGHEM L.M., KATZUNG B.G. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- POSTMA S.W., CATTERALL W.A. Inhibition of binding of [3H] batrachotoxin A 20-alpha-benzoate to sodium channels by local anesthetics. Mol. Pharmacol. 1984;25:219–225. [PubMed] [Google Scholar]
- PRIORI S.G., NAPOLITANO C., CANTÙ F., BROWN A.M., SCHWARZ P.J. Differential response to Na+ channel blockade, β-adrenergic stimulation, and rapid pacing in a cellular model mimicking the SCN5A and HERG defects present in the long-QT syndrome. Circ. Res. 1996;78:1009–1015. doi: 10.1161/01.res.78.6.1009. [DOI] [PubMed] [Google Scholar]
- PTÁčEK L. The familial periodic paralyses and nondystrophic myotonias. Am. J. Med. 1998;104:58–70. doi: 10.1016/s0002-9343(98)00123-5. [DOI] [PubMed] [Google Scholar]
- QU Y., ROGERS J., TANADA T., SCHEUER T., CATTERALL W.A. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11839–11843. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROGERS J.C., QU Y., TANADA T.N., SCHEUER T., CATTERALL W.A. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- SAH R.L., TSUSHIMA R.G., BACKX P.H. Effects of local anesthetics on Na+ channels containing the equine hyperkalemic periodic paralysis mutation. Am. J. Physiol. 1998;275:C389–C400. doi: 10.1152/ajpcell.1998.275.2.C389. [DOI] [PubMed] [Google Scholar]
- SHELDON R.S., HILL R.J., TAOUIS M., WILSON L. Aminoalkyl structural requirements for interaction of lidocaine with the class I antiarrhythmic receptor on rat cardiac myocytes. Mol. Pharmacol. 1991;39:609–614. [PubMed] [Google Scholar]
- SHIMIZU W., ANTZELEVITCH C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- SICOURI S., ANTZELEVITCH D., HEILMANN C., ANTZELEVITCH C. Effects of sodium channel block with mexiletine to reverse action potential prolongation in in vitro models of the long-QT syndrome. J. Cardiovasc. Electrophysiol. 1997;8:1280–1290. doi: 10.1111/j.1540-8167.1997.tb01019.x. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., BENNETT P.B., HONDEGHEM L.M.Mechanisms of drug-channel interaction The heart and cardiovascular system, second edition 1992Raven Press: New York; 2165–2193.In: Fozzard, H.A.(ed) [Google Scholar]
- STARMER C.F., GRANT A.O., STRAUSS H.C. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNAMI A., DUDLEY S.C., FOZZARD H.A. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNAMI A., FAN Z., SAWANOBORI T., HIRAOKA M. Use-dependent block of Na+ currents by mexiletine at the single channel level in guinea-pig ventricular myocytes. Br. J. Pharmacol. 1991;110:183–192. doi: 10.1111/j.1476-5381.1993.tb13790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMSEN W.J., CATTERALL W.A. Localization of the receptor site for α-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc. Natl. Acad. Sci. U.S.A. 1989;86:10161–10165. doi: 10.1073/pnas.86.24.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRICARICO D., FAKLER B., SPITTELMEISTER W., RUPPERSBERG J.P., STUTZEL R., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D., RüDEL R. Stereoselective of interaction of tocainide and its chiral analogs with the sodium channels in human myoballs. Pflugers Arch. 1991;415:234–237. doi: 10.1007/BF00370521. [DOI] [PubMed] [Google Scholar]
- WANG G.K., QUAN C., WANG S.-Y. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated μ1 Na+ channels. Pflugers Arch. 1998;435:293–302. doi: 10.1007/s004240050515. [DOI] [PubMed] [Google Scholar]
- WRIGHT S.N., WANG S.-Y., WANG G.K. Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthetics. Mol. Pharmacol. 1998;54:733–739. [PubMed] [Google Scholar]
- ZAMPONI G.W., FRENCH R.J. Dissecting lidocaine action: Diethylamide and phenol mimic separate modes of lidocaine block of sodium channels from heart and skeletal muscle. Biophys. J. 1993;65:2335–2347. doi: 10.1016/S0006-3495(93)81292-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMPONI G.W., FRENCH R.J. Amine blockers of the cytoplasmic mouth of sodium channels: a small structural change can abolish voltage dependence. Biophys. J. 1994;67:1015–1027. doi: 10.1016/S0006-3495(94)80567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]