

Abstract
It was the aim of our study to investigate the effects of the sulphonylurea glibenclamide on voltage dependent potassium currents in human atrial myocytes.
The drug blocked a fraction of the quasi steady state current (ramp response) which was activated positive to −20 mV, was sensitive to 4-aminopyridine (500 μM) and was different from the ATP dependent potassium current IK(ATP).
Glibenclamide dose dependently inhibited both, the peak as well as the late current elicited by step depolarization positive to −20 mV. The IC50 for reduction in charge area of total outward current was 76 μM.
The double-exponential inactivation time-course of the total outward current was accelerated in the presence of glibenclamide with a τfast of 12.7±1.5 ms and a τslow of 213±25 ms in control and 5.8±1.9 ms (P<0.001) and 101±20 ms (P<0.05) under glibenclamide (100 μM).
Our data suggest, that both repolarizing currents in human atrial myocytes, the transient outward current (Ito1) and the ultrarapid delayed rectifier current (IKur) were inhibited by glibenclamide.
In human ventricular myocytes glibenclamide inhibited Ito1 without affecting the late current.
Our data suggest that glibenclamide inhibits human voltage dependent cardiac potassium currents at concentrations above 10 μM.
Keywords: Sulphonylurea, glibenclamide, human, cardiac, potassium current, transient outward current
Introduction
The sulphonylurea glibenclamide (glyburide) is commonly used for treatment of non-insulin-dependent diabetes mellitus (NIDDM). The beneficial effect of glibenclamide involves the inhibition of ATP sensitive potassium channels (IK(ATP)-channels) in pancreatic β-cells leading to the release of insulin (Ashford, 1993). IK(ATP)-channels are also present in other tissues like the myocardium (Noma, 1983) where glibenclamide exerts a potent inhibitory effect (Findlay, 1992).
The role of glibenclamide in the incidence of arrhythmias and mortality, especially in myocardial ischemia is a matter of debate (for review see Schotborgh & Wilde, 1997). Beside the use in NIDDM patients glibenclamide is a frequently used tool in cardiovascular research whereby concentrations of up to 100 μM are often used for studying IK(ATP) (for review see Wilde & Janse, 1994, Schotborgh & Wilde, 1997). However, at these concentrations glibenclamide may also affect other membrane currents than IK(ATP) and may also affect the metabolism of the heart (Schotborgh & Wilde, 1997). Suggested as a selective IK(ATP)-blocker glibenclamide is also frequently used in models of ischaemic preconditioning to ‘prove' a contribution of IK(ATP) in the preconditioning effect (Tomai et al., 1994; Speechly-Dick et al., 1995; Cleveland et al., 1997).
We investigated the interaction of glibenclamide with voltage dependent outward currents in isolated human atrial and ventricular myocytes by using the patch clamp technique in the whole cell recording mode at a bath temperature of 36–37°C. We present evidence that glibenclamide inhibits both repolarizing currents, the transient outward current (Ito1) and the ultrarapid delayed rectifier current (IKur). To our knowledge this is the first report describing glibenclamide effects on human voltage dependent cardiac K+ currents.
Methods
Myocyte isolation
Human atrial myocytes were isolated from right atrial appendages which were obtained during open heart surgery. Human ventricular myocytes were isolated from the subepicardium of the left ventricle of explanted hearts. The use of human tissue was approved by the ethical committee of the University of Graz and the study conforms to the standards set by the declaration of Helsinki. Myocyte isolation was performed by the combined use of enzymes (trypsin, collagenase) in a tissue dissociation vessel as described previously (Pelzmann et al., 1995).
The isolated myocytes were stored in a cell culture medium (M-199, Sigma), supplemented with 50 μg ml−1 penicillin and 50 IU ml−1 streptomycin and were kept in an incubator at 37°C. Experiments were performed 3–8 h after cell isolation.
Electrophysiological recordings
The isolated myocytes were placed in an experimental chamber mounted on the stage of an inverted microscope (Axiovert, Zeiss, Oberkochen, Germany) and were superfused with extracellular saline (composition in mM: NaCl 137, KCl 5.4, CaCl2 0.1, MgCl2 1.1, CdCl2 0.1, NaHCO3 0.4, HEPES/Na+ 10, D(+)-glucose 5.6, adjusted to a pH of 7.4 with NaOH) at a temperature of 36–37°C and a flow rate of ∼3 ml min−1. A low external calcium concentration in combination with CdCl2 (Schaffer et al., 1998) was used to suppress calcium currents and calcium activated transient outward currents. Membrane currents were recorded as described previously (Schaffer et al., 1998) with the patch clamp technique in the whole cell mode by the use of a L/M EPC-7 amplifier (List, Darmstadt, Germany), and a Digidata 1200 interface (Axon Instruments, Foster City, U.S.A.). A personal computer equipped with pClamp 5.7.1 software (Axon) was used for generation of voltage clamp protocols, data storage and evaluation. When filled with standard internal solution (composition in mM: KCl 110, ATP/K+ 4.3, MgCl2 2, CaCl2 1, EGTA 11, HEPES/K+ 10 adjusted with KOH to a pH of 7.4 (estimated free [Ca2+]<10−8 M) the patch-electrodes had a tip-resistance of 2–3 MΩ. Only rod-shaped myocytes with clear cross-striation and without blebs were used for experiments. Cell membrane capacitance was evaluated by integrating the area under the capacitive transient elicited by a 10 mV hyperpolarizing voltage clamp step from a holding potential of −50 mV. Cell-capacitance (up to 100 pF) and series resistance were compensated (usually >50%). Access resistance (Rs) was calculated (prior to compensation) by dividing the time constant of the capacitive transient by the cell membrane capacitance. Rs prior to compensation was 5.82±0.38 MΩ (n=20 cells) in atrial and 4.45±0.58 MΩ (n=7 cells) in ventricular myocytes.
Transmembrane currents were recorded either at a holding potential of −40 mV, in response to voltage clamp ramps from −100 to +60 mV (0.08 V s−1) or by depolarizing clamp steps (−40 to +60 mV in steps of 10 mV, 300 ms) from a holding potential of −80 mV which were preceded by a 50 ms prepulse to −40 mV in order to inactivate INa. In atrial myocytes the inactivation time course of the total outward current could be described by two exponential functions:
where Itotal was the total outward current, Afast and τfast were the amplitude and time constant of the fast inactivation phase, Aslow and τslow were the amplitude and time constant of the slow inactivation phase and Ass was the amplitude of the steady state current. The amplitude of the peak outward current and the late current were determined relative to the zero current level.
To study the concentration dependent inhibition of total outward current the reduction in charge movement was calculated. The integral (0–300 ms) of the inactivation function of the total outward current (determined by the amplitudes and time constants derived from the fitting procedure) was used as a measure of charge movement.
In human ventricular myocytes Ito1 was measured as the difference between the peak of the outward current and the current at the end of the clamp pulse.
Statistics
All data are presented as means±s.e.mean, statistical significance was tested by Student's t-test for paired data. Values of P<0.05 were regarded as significant.
Drugs
Glibenclamide and cromakalim stock solutions were prepared in DMSO and further diluted to the desired concentration. The DMSO concentration in the extracellular saline did not exceed 0.1%. 4-aminopyridine (4-AP) was added directly to the extracellular solution. All chemicals were purchased from Sigma.
Results
To test for a contribution of the ATP-dependent potassium current IK(ATP) to basal electrical activity, isolated human atrial myocytes were voltage clamped to −40 mV and superfused with the IK(ATP)-blocker glibenclamide (50 μM). A typical experiment is shown in Figure 1. It can be seen, that the holding current (Ihold−40 mV) was not affected by glibenclamide suggesting that IK(ATP) was not active under these conditions. However, when the quasi steady state current was elicited with a voltage ramp (−100 to +60 mV, 0.08 V s−1), a clear outward current inhibition by glibenclamide was noted (see Figure 2a). The glibenclamide sensitive fraction of the ramp response (obtained by digital subtraction) is shown in Figure 2b. The glibenclamide sensitive current activated positive to −20 mV and was outward directed. This current resembled the 4-aminopyridine (4-AP; 500 μM) sensitive current shown in Figure 2c. In the presence of 4-AP (500 μM) glibenclamide failed to inhibit a fraction of the quasi steady state current (Figure 2d) suggesting that glibenclamide and 4-AP affect the same current components. For comparison, IK(ATP) is shown in Figure 2e (current activated by 100 μM cromakalim, obtained by digital subtraction). IK(ATP) reversed at about −85 mV and differed completely from the glibenclamide sensitive current. These experiments led to the conclusion that (1) IK(ATP) does not contribute to basal electrical activity in human atrial myocytes using a pipette ATP concentration of 4.3 mM and (2) glibenclamide blocked an outward current component which was sensitive to 4-AP and was different from IK(ATP).
Figure 1.
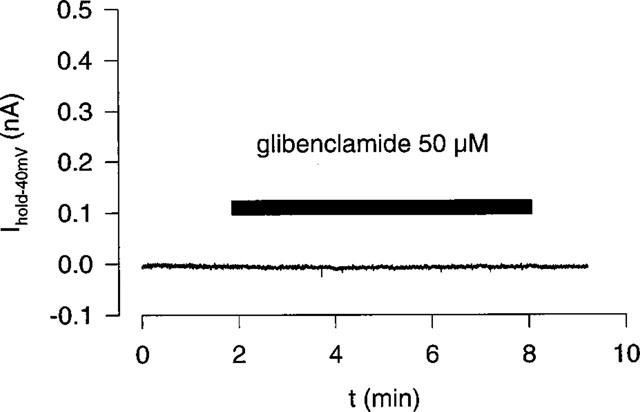
Effect of glibenclamide (50 μM) on the holding current (−40 mV) of an isolated human atrial myocyte. The pipette ATP concentration was 4.3 mM.
Figure 2.
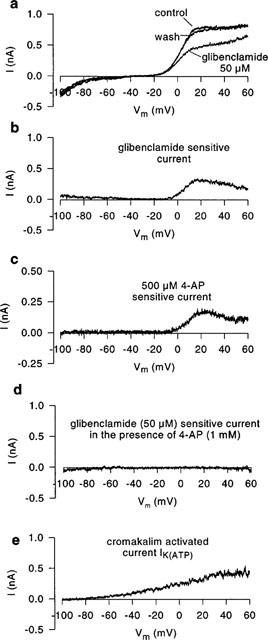
Effects of glibenclamide on the quasi steady state current of human atrial myocytes elicited by a voltage ramp from −100 to +60 mV (0.08 V s−1). (a) Ramp response under control conditions, under the influence of glibenclamide (50 μM) and after washout. (b) Glibenclamide sensitive current obtained by digital subtraction (data a). The glibenclamide sensitive current is similar to the 4-AP (500 μM) sensitive current shown in (c) and differs completely from IK(ATP) which is shown in (e) as the current activated by cromakalim (100 μM). (d) Glibenclamide sensitive current in the presence of 4-AP (1 mM). Drug sensitive currents were obtained by digital subtraction.
To further investigate the glibenclamide sensitive outward current, myocytes were depolarized to potentials between −40 and +60 mV (duration 300 ms; at a frequency of 0.5 Hz) from a holding potential of −80 mV. The depolarizing pulses were preceded by a 50 ms step to −40 mV to voltage-inactivate INa. Figure 3a shows typical outward current traces under control conditions and under the influence of 30 and 100 μM glibenclamide. Both, the peak outward current and the late current (current at the end of the clamp pulse) were concentration dependently reduced by glibenclamide. Furthermore glibenclamide caused an apparent acceleration of the time-course of inactivation of the total outward current. The time-course of inactivation of the total outward current could be described as a double-exponential function (see Methods) with τfast of 12.7±1.5 ms and a τslow of 213±25 ms in control and 5.8±1.9 ms (P<0.001) and 101±20 ms (P<0.05) under 100 μM glibenclamide (at +40 mV; n=5). The effect of glibenclamide on the current voltage relationship of peak total outward current is shown in Figure 3b. Since time course of inactivation was apparently accelerated in the presence of glibenclamide we used charge area reduction of total outward current (at +40 mV) as a measure of the concentration dependent inhibition as shown in Figure 3c. These data were fitted with a Hill function giving an IC50 of 76.4±6.3 μM and a slope of −1.21±0.13.
Figure 3.
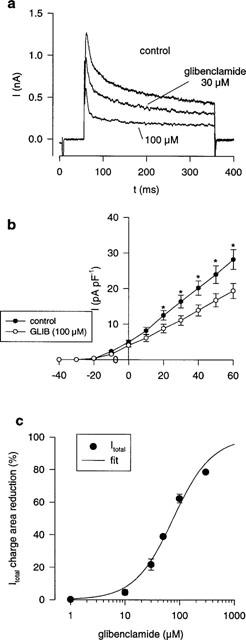
Glibenclamide induced block of outward currents in human atrial myocytes. (a) Total outward current in response to a depolarization to +40 mV under control conditions and under the influence of glibenclamide (30, 100 μM). Glibenclamide affected both, the peak current and the late current at the end of the clamp pulse. The time-course of inactivation of the total outward current was accelerated in the presence of glibenclamide. (b) Current voltage relationship of the peak total outward current under control conditions and under 100 μM glibenclamide (n=5). (c) Mean concentration-effect data of the effect of glibenclamide on charge area beneath total outward current. The solid line represents the results of fitting a Hill function to experimental data giving an IC50 76.4±6.3 μM and a slope of −1.21±0.13. (*P<0.05).
In human left ventricular subepicardial myocytes glibenclamide induced a concentration dependent reduction of Ito 1 whereas the non-inactivating fraction of the outward current was not affected (Figure 4a,b,c). At a concentration of 100 μM glibenclamide Ito1 was inhibited by 78.2±2.66% (at +40 mV; n=4).
Figure 4.
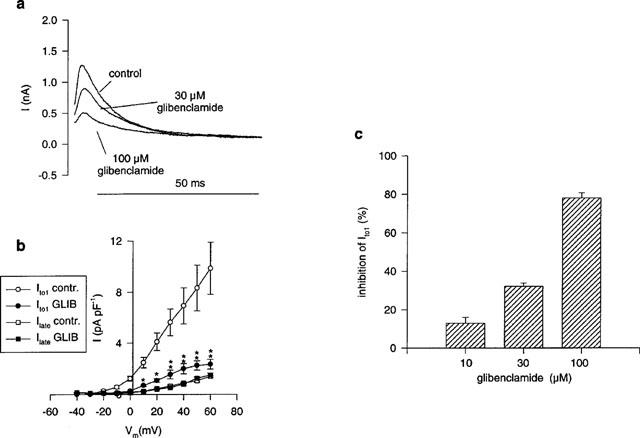
Glibenclamide induced block of Ito1 in human ventricular myocytes. (a) Outward current recordings (+40 mV) in control and after superfusion with glibenclamide (30, 100 μM). The late current (Ilate) was not affected by glibenclamide. (b) Current voltage relationship of Ito1 and Ilate under control conditions and under influence of glibenclamide (100 μM; closed symbols, n=4). (c) Concentration dependence of Ito1 inhibition by glibenclamide. (**P<0.01; *P<0.05).
Discussion
To test whether or not the ATP sensitive potassium current (IK(ATP)) contributes to the basal electrical activity, isolated human atrial myocytes were superfused with glibenclamide, which is thought to act as a selective IK(ATP)-blocker. Using a pipette ATP concentration of 4.3 mM no IK(ATP)-like current was inhibited, suggesting that IK(ATP) was not active under these conditions. This finding confirms observations by Heidbüchel et al. (1990) who have described the lack of spontaneous openings of IK(ATP)-channels in cell-attached patches of human atrial myocytes.
However, recordings of the quasi steady state current (ramp-response) showed that glibenclamide exerts an inhibitory effect on an outward current component (different from IK(ATP)) which activated positive to −20 mV and was sensitive to low concentrations of 4-AP. Two distinct 4-AP-sensitive outward currents which activate positive to −20 mV have been described in human atrial myocytes recently. A transient outward current (Ito1) which is sensitive to 4-AP in the millimolar range and an ultrarapid delayed rectifier current (IKur, Iso) which is sensitive to submillimolar concentrations of 4-AP are present in the human atrium (Wang et al., 1993; 1995; Amos et al., 1996; Schaffer et al., 1998).
In human atrial myocytes glibenclamide reduced both, the peak of the total outward current as well as the amplitude of the current at the end of the clamp pulse. Half-maximal inhibition of the total outward current (charge area reduction) was achieved with 76 μM glibenclamide which is one order of magnitude higher than for IK(ATP)-block (5–10 μM; see: Schotborgh & Wilde, 1997). The effect of glibenclamide on individual currents is difficult to estimate since both, Ito1 and IKur show time dependent inactivation (Feng et al., 1998; Schaffer et al., 1998). Thus, Ito1 can not be determined as the difference between the peak and the late current neither can the amplitude of the late current be regarded as a reliable measure of IKur.
For this reason we have studied the effects of glibenclamide on the total outward current and find indirect evidence for inhibition of both, Ito1 and IKur. Reduction of the late current under glibenclamide suggests block of IKur. This assumption is further supported by the glibenclamide induced apparent acceleration of the slow inactivation phase which represents inactivation of IKur (Schaffer et al., 1998). The apparent acceleration of the fast inactivation phase suggests block of Ito1. In ventricular myocytes where Ito1 can be studied without an overlaying IKur (see: Amos et al., 1996) a clear inhibitory action of glibenclamide on Ito1 was seen. This supports our assumption of Ito1 block in the atrium. However, further studies will be necessary to investigate the effects of glibenclamide on individual currents in human atrial myocytes (Ito1, IKur).
In human ventricular myocytes the late current, which is thought to be a non-selective cation current (Amos et al., 1996), was unaffected by glibenclamide.
Besides affecting IK(ATP), glibenclamide has been shown to inhibit the cyclic AMP activated chloride current (Tominaga et al., 1995; Faivre et al., 1998), the cystic fibrosis chloride channel (Sheppard & Welsh, 1992) as well as volume-sensitive chloride channels (Sakaguchi et al., 1997; Liu et al., 1998). In neuroblastoma cells glibenclamide inhibited a voltage gated potassium current which was independent of intracellular ATP and Ca2+ (Reeve et al., 1992).
At present we are not aware of any report of glibenclamide effects on voltage dependent potassium currents in human cardiomyocytes. However, recently it was shown that glibenclamide inhibits neural and cardiac HERG-channels (Rosati, et al., 1998) with a similar IC50 (74.8 μM) as reported in the present study for block of human atrial outward current (76.4 μM). Rosati et al. (1998) suggested that glibenclamide block of HERG channels may indicate the linkage of sulphonylurea receptors to HERG channels. Our data do not support this assumption since the apparent acceleration of inactivation may be interpreted as open channel block and suggest a direct action of glibenclamide on Ito1 and IKur.
Whether or not inhibition of Ito1 and IKur play a role in patients on glibenclamide can not be answered yet. However, Ito1 (atrium, ventricle) and IKur (atrium) play a central role for repolarization in the human heart. Thus glibenclamide may cause an action potential lengthening (class III antiarrhythmic effect) in both the atrium and the ventricle. At 10 μM glibenclamide we found 15–20% inhibition of Ito1 in human ventricular myocytes. At this concentration of glibenclamide effects on heart rate and QT interval which were not related to inhibition of IK(ATP) have been described in the rat heart (Rees & Curtis, 1995). Since tedisamil, a potent Ito1 blocking drug (Wettwer et al., 1998), elicited similar effects (Tsuchihashi & Curtis, 1991), it was speculated that glibenclamide (at 10 μM) may block Ito1 (Rees & Curtis, 1995).
In clinical use, glibenclamide reaches a blood concentration up to 1.5 μM (Schotborgh & Wilde, 1997), a concentration where the effects on Ito1 and IKur are marginal. Whether glibenclamide may unmask primary or iatrogenic long-QT syndromes as discussed by Rosati et al. (1998) remains speculative although long QT-syndrome in response to antidiabetic therapy with glibenclamide has been reported recently (Ikeda, 1994).
The IK(ATP) channel is thought to be the end effector of preconditioning (increased tolerance to myocardial ischaemia and reperfusion by preceding transient ischaemic period; Sumeray & Yellon, 1997). Knowledge of the role of IK(ATP) in experimental preconditioning is mainly based on the fact that glibenclamide abolishes such a preconditioning effect (Tomai et al., 1994; Speechly-Dick et al., 1995; Cleveland et al., 1997) assuming that glibenclamide is a pure IK(ATP)-blocker. However, our data show that at least in human heart, glibenclamide exerts additional K+ channel blocking effects. Thus, a concentration of glibenclamide in/or below the μM range are necessary to induce ‘selective' IK(ATP) inhibition.
It is further notable that structurally diverse substances inhibit human cardiac Ito1 and/or IKur. Inhibition was reported for classical class III antiarrhythmic agents like ambasilide (Koidl et al., 1996) and tedisamil (Wettwer et al., 1998), the class Ia agent quinidine (Wang et al., 1995; Nenov et al., 1998), Ic agents like propafenone (Schaffer et al., 1995; Gross & Castle, 1998) and flecainide (Wang et al., 1995) and other substances like bertosamil (Tessier et al., 1997), the antihistamine loratadine (Crumb, 1999), 4-aminopyridine (Gross et al., 1995; Wang et al., 1995; Schaffer et al., 1998) and the sulphonylurea glibenclamide as described in this study.
In summary, the present study describes the inhibitory action of glibenclamide on voltage dependent potassium currents (Ito1 and IKur) in human atrial and ventricular myocytes. Our data indicate that glibenclamide concentrations in or even below the μM range are required for a selective inhibition of IK(ATP) in the human heart. Since inhibition of Ito1 and IKur (as well as IKr; Rosati et al., 1998) occurred at concentrations above the glibenclamide plasma levels, these effects are rather unlikely to play a significant role in NIDDM patients. However, it remains to be determined whether glibenclamide may induce iatrogenic long-QT syndrome under certain conditions.
Acknowledgments
This work was supported by the Austrian Science Fund, P13111-Med, SFB F707 and grants of the Austrian National Bank (Project 7377) and Austrian Heart Foundation. We are grateful to the team of Cardiovascular Surgery, Universitätsklinik Graz for collaboration.
Abbreviations
- (Ito1)
transient outward potassium current
- (IKur)
ultrarapid delayed rectifier potassium current
References
- AMOS G.J., WETTWER E., METZGER F., LI Q., HIMMEL H.M., RAVENS U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J. Physiol. 1996;491:31–50. doi: 10.1113/jphysiol.1996.sp021194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHFORD M.L.J.Sulphonylureas: a receptor and a potassium channel K+ channels in cardiovascular medicine 1993Springer Verlag:Paris; 161–174.In: Escande, D. & Standen, N. (eds) [Google Scholar]
- CLEVELAND J.C., MELDRUM D.R., ROWLAND R.T., BANERJEE A., HARKEN A.H. Adenosine preconditioning of human myocardium is dependent upon the ATP-sensitive K+ channel. J. Mol. Cell. Cardiol. 1997;29:175–182. doi: 10.1006/jmcc.1996.0262. [DOI] [PubMed] [Google Scholar]
- CRUMB W.C. Rate-dependent blockade of a potassium current in human atrium by the antihistamine loratadine. Br. J. Pharmacol. 1999;126:575–580. doi: 10.1038/sj.bjp.0702273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAIVRE J.-F., ROUANET S., BRIL A. Comparative effects of glibenclamide, tedisamil, dofetilide, E-4031, and BRL-32872 on protein kinase A-activated chloride current in guinea pig ventricular myocytes. J. Cardiovasc. Pharmacol. 1998;31:551–557. doi: 10.1097/00005344-199804000-00013. [DOI] [PubMed] [Google Scholar]
- FENG J., XU D., WANG Z., NATTEL S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: properties and consequences. Am. J. Physiol. 1998;275:H1717–H1725. doi: 10.1152/ajpheart.1998.275.5.H1717. [DOI] [PubMed] [Google Scholar]
- FINDLAY I. Inhibition of ATP-sensitive K+ channels in cardiac muscle by the sulphonylurea drug glibenclamide. J. Pharmacol Exp. Ther. 1992;261:540–545. [PubMed] [Google Scholar]
- GROSS G.J., BURKE R.P., CASTLE N.A. Characterization of transient outward current in young human atrial myocytes. Cardiovasc. Res. 1995;29:112–117. [PubMed] [Google Scholar]
- GROSS G.J., CASTLE N.A. Propafenone inhibition of human atrial myocyte repolarizing currents. J. Mol. Cell. Cardiol. 1998;30:783–793. doi: 10.1006/jmcc.1998.0643. [DOI] [PubMed] [Google Scholar]
- HEIDBÜCHEL H., VEREECKE J., CARMELIET E. Three different potassium channels in human atrium. Circ. Res. 1990;66:1277–1286. doi: 10.1161/01.res.66.5.1277. [DOI] [PubMed] [Google Scholar]
- IKEDA T. QT prolongation in type 2 diabetes mellitus treated with glibenclamide. Diabetes Metab. 1994;20:565–567. [PubMed] [Google Scholar]
- KOIDL B., FLASCHBERGER P., SCHAFFER P., PELZMANN B., BERNHART E., MÄCHLER H., RIGLER B. Effects of the class III anitarrhythmic drug ambasilide on outward currents in human atrial myocytes. Naunyn-Schmiedberg's Arch. Pharmacol. 1996;353:226–232. doi: 10.1007/BF00168761. [DOI] [PubMed] [Google Scholar]
- LIU Y., OIKI S., TSUMURA T., SHIMIZU T., OKADA Y. Glibenclamide blocks volume-sensitive Cl- channels by dual mechanisms. Am. J. Physiol. 1998;275:C343–C351. doi: 10.1152/ajpcell.1998.275.2.C343. [DOI] [PubMed] [Google Scholar]
- NENOV N.I., CRUMB W.J., JR, PIGOTT J.D., HARRISON L.H., JR, CLARKSON C.W. Quinidine interactions with human atrial potassium channels. Circ. Res. 1998;83:1224–1231. doi: 10.1161/01.res.83.12.1224. [DOI] [PubMed] [Google Scholar]
- NOMA A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- PELZMANN B., SCHAFFER P., MÄCHLER H., RIGLER B., KOIDL B. Adenosine inhibits the L-type calcium current in human atrial myocytes. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;351:293–297. doi: 10.1007/BF00233249. [DOI] [PubMed] [Google Scholar]
- REES S.A., CURTIS M.J. Pharmacological analysis in rat of the role of the ATP-sensitive potassium channel as a potential target for antifibrillatory intervention in acute myocardial ischemia. J. Cardiovasc. Pharmacol. 1995;26:280–288. doi: 10.1097/00005344-199508000-00014. [DOI] [PubMed] [Google Scholar]
- REEVE H.L., VAUGHAN P.F., PEERS C. Glibenclamide inhibits a voltage-gated K+ current in the human neuroblastoma cell line SH-SY5Y. Neurosci. Lett. 1992;135:37–40. doi: 10.1016/0304-3940(92)90130-y. [DOI] [PubMed] [Google Scholar]
- ROSATI B., ROCCHETTI M., ZAZA A., WANKE E. Sulfonylureas blockade of neural and cardiac HERG channels. FEBS Lett. 1998;440:125–130. doi: 10.1016/s0014-5793(98)01444-6. [DOI] [PubMed] [Google Scholar]
- SAKAGUCHI M., MATSUURA H., EHARA T. Swelling-induced Cl- current in guinea-pig atrial myocytes: inhibition by glibenclamide. J. Physiol. 1997;505:41–52. doi: 10.1111/j.1469-7793.1997.041bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFFER P., PELZMANN B., BERNHART E., LANG P., LØKEBØ J.-E., MÄCHLER H., RIGLER B., KOIDL B. Estimation of outward currents in isolated human atrial myocytes using inactivation time course analysis. Pflügers Arch. 1998;436:457–468. doi: 10.1007/s004240050657. [DOI] [PubMed] [Google Scholar]
- SCHAFFER P., PELZMANN B., BERNHART E., LANG P., MÄCHLER H., RIGLER B., KOIDL B.Effects of propafenone on outward currents in human atrial myocytes Potassium channels in normal and pathological conditions 1995Leuven University Press: Leuven; 396–399.In: Vereecke, J., Van Bogaert, P.P. & Verdonck, F. (eds) [Google Scholar]
- SCHOTBORGH C.E., WILDE A.A.M. Sulfonylurea derivatives in cardiovascular research and in cardiovascular patients. Cardiovasc. Res. 1997;34:73–80. doi: 10.1016/s0008-6363(97)00036-9. [DOI] [PubMed] [Google Scholar]
- SHEPPARD D.N., WELSH M.J. Effects of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPEECHLY-DICK M.E., GROVER G.J., YELLON D.M. Does ischemic preconditioning in the human involve protein kinase C and the ATP-dependent K+ channel. Circ. Res. 1995;77:1030–1035. doi: 10.1161/01.res.77.5.1030. [DOI] [PubMed] [Google Scholar]
- SUMERAY M.S., YELLON D.M. Myocardial preconditioning. What have we learned. Eur. Heart J. 1997;18 Suppl:A8–A14. doi: 10.1093/eurheartj/18.suppl_a.8. [DOI] [PubMed] [Google Scholar]
- TESSIER S., RÜCKER-MARTIN C., MACÉ L., CORABOEUF E., MERCADIER J.-J., HATEM S. The antiarrhythmic agent bertosamil induces inactivation of the sustained outward K+ current in human atrial myocytes. Brit. J. Pharmacol. 1997;122:291–301. doi: 10.1038/sj.bjp.0701369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMAI F., CREA F., GASPARDONE A., VERSACI F., DE PAULIS R., DE PEPPO A.P., CHIARIELLO L., GIOFFRÉ P.A. Ischemic preconditioning during coronary angioplasty is prevented by glibenclamide, a selective ATP-sensitive K+ channel blocker. Circulation. 1994;90:700–705. doi: 10.1161/01.cir.90.2.700. [DOI] [PubMed] [Google Scholar]
- TOMINAGA M., HORIE M., SASAYAMA S., OKADA Y. Glibenclamide, an ATP-sensitive K+ channel blocker, inhibits cAMP-activated Cl- conductance. Circ. Res. 1995;77:417–423. doi: 10.1161/01.res.77.2.417. [DOI] [PubMed] [Google Scholar]
- TSUCHIHASHI K., CURTIS M.J. Influence of tedisamil in initiation and maintenance of ventricular fibrillation: chemical defibrillation by Ito blockade. J. Cardiovasc. Pharmacol. 1991;18:445–456. doi: 10.1097/00005344-199109000-00018. [DOI] [PubMed] [Google Scholar]
- WANG Z., FERMINI B., NATTEL S. Sustained depolarization-induced outward current in human atrial myocytes. Circ. Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- WANG Z., FERMINI B., NATTEL S. Effects of flecainide, quinidine, and 4-aminopyridine on transient outward and ultrarapid delayed rectifier currents in human atrial myocytes. J. Pharmacol. Exp. Ther. 1995;272:184–196. [PubMed] [Google Scholar]
- WETTWER E., HIMMEL H.M., AMOS G.J., LI Q., METZGER F., RAVENS U. Mechanisms of block by tedisamil of transient outward current in human ventricular subepicardial myocytes. Brit. J. Pharmacol. 1998;125:659–666. doi: 10.1038/sj.bjp.0702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILDE A.A.M., JANSE M.J. Electrophysiological effects of ATP-sensitive potassium channel modulation: implication for arrhythmogenesis. Cardiovasc. Res. 1994;28:16–12. doi: 10.1093/cvr/28.1.16. [DOI] [PubMed] [Google Scholar]