

Abstract
Lysophosphatidic acid (LPA) has been widely studied as a naturally occurring and multifunctional phospholipid messenger in diverse tissue and cell types and shown to inhibit adenylyl cyclase (AC) by a G protein-mediated mechanism.
In type II AC-expressing mouse RAW 264.7 macrophages, we showed that LPA at 3–50 μM increased cyclic AMP formation in a concentration-dependent manner, the effect being additive with that of forskolin or cholera toxin, and synergistic with that of prostaglandin E1 (PGE1) or isoproterenol.
The potentiation effect of LPA was unaffected by the removal of serum or pertussis toxin treatment.
Both colchicine and cytochalasin B potentiated the cyclic AMP response to PGE1, the effect being additive to that of LPA.
On studying the regulation of type II AC by protein kinase C (PKC), phorbol 12-myristate-13 acetate (PMA) potentiated the PGE1-elicited cyclic AMP response, this effect being non-additive to that of LPA, suggesting that PKC activation was the common mechanism involved in AC potentiation by LPA and PMA.
PKC inhibitor Ro 31-8220, but not Go 6976, significantly inhibited the LPA-induced cyclic AMP potentiation.
The potentiation effect of LPA was unaffected by long-term treatment with PMA, which resulted in the down-regulation of PKCα, βI, βII and PKCδ, but not PKCε, μ, λ and ζ.
By in situ kinase assay, we found a marked increase in atypical PKC activity after LPA treatment.
Taken together, we conclude that LPA can elicit a unique signalling cascade in RAW 264.7 macrophages and increase type II AC activity via the activation of atypical PKC.
Keywords: LPA, type II AC, atypical PKC, macrophage
Introduction
Lysophosphatidic acid (LPA) is a naturally occurring and multifunctional phospholipid messenger in several cell types (reviews see Moolenaar, 1995; Moolenaar et al., 1997) and can elicit a variety of cellular responses, including platelet aggregation, smooth muscle contraction, alterations in neuronal cell morphology, DNA synthesis and proliferation in different cell types. It is released from activated platelets or injured cells and is thought to be responsible for much of the cell growth and adhesion promoting activity in serum (Durieux & Lynch, 1993; Moolenaar, 1995). LPA elicits its biological responses through a receptor coupled to heterotrimeric G proteins. Radioligand binding studies have shown the presence of specific LPA binding sites to a membrane protein with apparent molecular mass of 38–40 kDa in various LPA-responsive cell types (van der Bend et al., 1992). Recent work has also identified three cDNA encoding functional LPA receptors that trigger different signalling pathways (Guo et al., 1996; Hecht et al., 1996; An et al., 1997).
At least four G protein-mediated signalling pathways have been identified in the action of LPA (review see Moolenaar, 1995; Goetzl & An, 1998). These are (i) stimulation of phospholipase C (PLC) via Gαq/11 and βγ dimers, (ii) inhibition of adenylyl cyclase (AC) via Gαi, (iii) activation of Ras and the downstream Raf/MAP kinase pathway via βγ dimers, and (iv) activation of Rho-dependent signalling elements via G12/13 and leading to the cytoskeleton-dependent functions as well as activation of phospholipase D (PLD) and phosphoinositide 3-kinase. Of these pathways, inhibition of AC and stimulation of Ras signalling are sensitive to pertussis toxin (PTX).
Cyclic AMP is well established as an important second messenger regulating a wide variety of cellular functions. Modulation of cyclic AMP levels is of particular significance in the regulation of macrophage functions, including cytokine synthesis, phagocytosis, adhesiveness, and NO generation (Ventura et al, 1990; Mullet et al., 1997). Besides the inhibition of AC observed in PC12 cells (Tigyi et al., 1996a) and fibroblasts (Carr et al., 1994), LPA was found to be responsible for the serum-induced sensitization of cyclic AMP accumulation (Kreps et al., 1993; Nogami et al., 1995). Divergent mechanisms appear to exist for the sensitization effect on AC activity. The results in airway smooth muscle cells suggested that the βγ subunits generated from LPA-activated receptors, which are coupled to both the PTX-sensitive Gi protein and PTX-insensitive Gq protein, are responsible for the enhancement. In addition, protein kinase C (PKC) activation resulting from Gq-triggered phosphoinositide (PI) turnover also leads to the potentiation of AC (Nogami et al., 1995). The conditionally regulatory role of βγ subunit on AC activity has also been demonstrated (Weng et al., 1996; Ahmed & Heppel, 1997). Apart from these studies, no detailed clarification on the signalling mechanism for LPA potentiation in the cyclic AMP system has been obtained.
As understanding of the cyclic AMP pathways is crucial to understanding the regulation of macrophage activation, we investigated here the intracellular signal transduction mechanism of LPA involved in the regulation of cyclic AMP synthesis in murine RAW 264.7 macrophages. The current investigation was undertaken to accomplish the following goals: (1) to evaluate the effect of LPA on cyclic AMP synthesis; (2) to examine the role of PKC on LPA modulation of cyclic AMP synthesis; (3) to identify the PKC isoforms involved in LPA-induced signalling; (4) to examine the changes in cytoskeleton arrangement during the LPA response. We report that LPA is able itself to activate AC, and, in combination with the Gs-coupled receptor agonists, prostaglandin E1 (PGE1) and isoproterenol, can markedly increase cyclic AMP synthesis. This potentiation effect of LPA is primarily due to the activation of atypical PKCs which leads to the stimulation of type II AC.
Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum, penicillin and streptomycin were obtained from Gibco BRL (Grand Island, NY, U.S.A.). [3H] cyclic AMP assay system, horseradish peroxidase-coupled anti-mouse and anti-rabbit antibodies, and the enhanced chemi-luminescence detection agent were purchased from Amersham International (Arlington Heights, II, U.S.A.). Ro 31-8220 and Go 6976 were purchased from Calbiochem (La Jolla, CA, U.S.A.). Mouse monoclonal antibodies specific for PKC α, δ, ε, λ, μ and ζ were purchased from Transduction Laboratories (Lexington, KY, U.S.A.), and rabbit polyclonal antibodies specific for PKC βI and βII were from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). ε-Peptide and ζ-PKC pseudosubstrate peptide were synthesized by the ABI Peptide Synthesizer. [γ-32P]-ATP was from NEN-Du Pont (Boston, MA, U.S.A.). All materials for SDS–PAGE and Bio-Rad protein assay were obtained from Bio-Rad Laboratories (Hercules, CA, U.S.A.). Other chemicals were obtained from Sigma (St. Louis, MO, U.S.A.).
Cell culture
Mouse RAW 264.7 macrophages, obtained from the American Type Culture Collection (Bethesda, MD, U.S.A.), were cultured in DMEM /10% foetal bovine serum /100 u ml−1 penicillin and 100 μg ml−1 streptomycin at 37°C in a humidified atmosphere of 95% air/5% CO2 as previously described (Lin & Chen, 1998).
Measurement of cyclic AMP levels
Intracellular cyclic AMP accumulation was assayed as previously reported (Lin & Chen, 1998). Briefly, confluent cells on 35-mm culture plates were washed with physiological saline solution (PSS, composition in mM: NaCl 118, KCl 4.7, CaCl2 1.8, MgCl2 1.2, KH2PO4 1.2, glucose 11 and HEPES 20, pH 7.4) and incubated in the presence of 500 μM 3-isobutyl-methylxanthine (IBMX) for 20 min at 37°C. The reaction was started by addition of the test reagents and continued for 10 min at 37°C; it was then terminated by aspirating the reaction mixture and immediately adding 0.1 N HCl. The cells were scraped off and the suspension centrifuged. The supernatant was neutralized and assayed of cyclic AMP levels with the [3H] cyclic AMP assay kit. Cell pellets were assayed for protein content using the Bradford (1976) method and bovine serum albumin as the standard and the cyclic AMP level expressed in pmol mg−1 protein.
Immunoblotting analysis
The cells were placed on ice, rinsed with PBS, resuspended in homogenization buffer (in mM): Tris-HCl 20, EGTA 0.5, EDTA 2, DTT 2, p-methylsulphonyl fluoride 0.5 and 10 μg ml−1 leupeptin, pH 7.5) and sonicated. The protein content of the sonicate was assayed by the Bradford method. Equal amounts of cell lysate protein (80 μg) were separated by 9% polyacrylamide gel electrophoresis in the presence of 0.1% sodium dodecyl sulphate (SDS–PAGE) and the proteins electrotransferred to nitrocellulose membranes (Amersham ECL grade). The blots were then blocked with TBST buffer (Tris-HCl 100 mM, NaCl 150 mM, Tween 20 0.1%, pH 7.5) containing 0.5% nonfat dry milk powder for 12 h at 4°C, washed with TBST buffer, and incubated with PKC isoform-specific primary antibody for 1.5 h. After buffer washing, the blots were then incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibody for 1 h. After an extensive wash, the blots were processed for visualization using the enhanced chemi-luminescence system according to the manufacturer's recommendations (Amersham), and then exposed to Kodak XAR-5 film to obtain the fluorographic images.
Assessment of atypical PKC activity
Activation of atypical PKC was assessed in situ as previously described (Wooten et al., 1996). Briefly, RAW 264.7 cells were harvested in PSS and 5×105 cells/each reaction treated with a given stimulus, followed by rapid centrifugation. The supernatant was rapidly removed by aspiration and cell pellets resuspended in 50 μl of permeabilization buffer (NaCl 137 mM, KCl 5.4 mM, sodium phosphate 0.3 mM, potassium phosphate 0.4 mM glucose 1 mg ml−1, HEPES 20 mM, pH 7.2, digitonin 10 μM, MgCl2 10 mM and β-glycerophosphate 25 mM) in the absence or presence of 150 μM ζ-pseudosubstrate peptide (PKC ζ sequence 113–129; SIYRRGARRWRKLYRAN). The reactions were initiated by addition of a mixture consisting of 150 μM ε-peptide (PKC ε sequence 149–164, pseudosubstrate site in PKCε with Ala 159 substituted by Ser, ERMRPRKRQGSVRRRV) and 100 μM [γ-32P]-ATP. After 10 min incubation at 37°C, the kinase reactions were terminated by addition of 30 μl 1% H3PO4 and the reaction mixture processed as described previously (Wooten et al., 1996). Changes in the activity of atypical PKC were calculated as the difference in phosphotransferase activity in the presence or absence of ζ-PKC pseudosubstrate peptide.
Measurement of [Ca2+]i
Cells grown on glass slides were loaded with 3 μM fura-II/AM and pluronic F-127 (0.02% v v−1) in DMEM at 37°C for 45 min. The fluorescence was monitored on a PTI M-series spectrofluorometer using dual excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm. The [Ca2+]i was calculated from the ratio of the fluorescence at the two excitation wavelengths, using a Kd value of 224 nM for the fura-II/Ca2+ equilibrium.
Statistical analysis
Each experiment was performed in duplicate, and the data represent the mean±s.e.mean of several independent experiments. P<0.05 was considered significant for evaluation of the data using Student's t-test. The n values represent the number of independent experiments. The error bar was omitted when it fell within the symbol representing the mean value.
Results
Increase in basal and stimuli-induced cyclic AMP accumulation by LPA
The intracellular cyclic AMP level accumulating within 10 min in the presence of the phosphodiesterase inhibitor, IBMX, was assayed. As shown in Figure 1, LPA concentration-dependently increased cyclic AMP formation within 3–50 μM. At the highest concentration we tested (50 μM), LPA increase in cyclic AMP did not reach the plateau effect. The increase in cyclic AMP is LPA-specific, as the related phospholipids, phosphatidic acid (30 μM) and lysophosphatidylcholine (3 μg ml−1) did not stimulate cyclic AMP formation, which were respectively of 16±4 (n=3) and 19±4 (n=3) pmol mg−1 protein as compared to basal level of 12±2 (n=3) pmol mg−1 protein.
Figure 1.
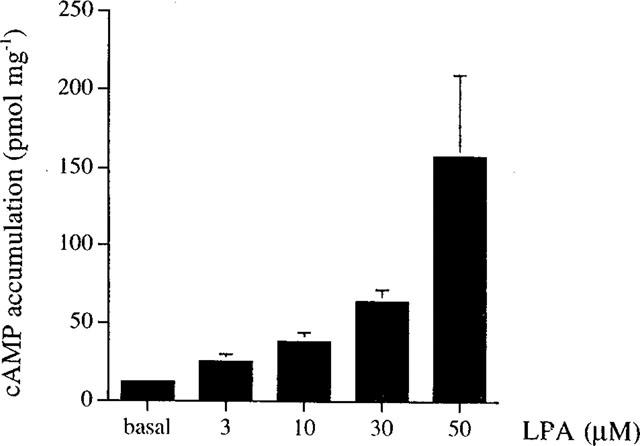
Concentration-dependent effect of LPA on cyclic AMP accumulation in RAW 264.7 macrophages. Cells incubated in PSS containing 500 μM IBMX were treated with vehicle (basal) or LPA at the indicated concentration for 10 min, then the intracellular cyclic AMP levels were measured. The values are the mean±s.e.mean of at least three independent experiments.
In combination with forskolin (10 μM), a direct activator of AC that increased the intracellular cyclic AMP levels from 10±3 to 52±6 (n=15) pmol mg−1 protein, the LPA-induced cyclic AMP response (3,10 and 30 μM) was essentially additive with that of forskolin (Figure 2a). Two Gs-coupled receptor agonists, PGE1 and isoproterenol, at 1 μM, respectively stimulated cyclic AMP formation to 292±40 (n=15) and 185±10 (n=12) pmol mg−1 protein; in the presence of LPA (1–30 μM), these agonist-induced cyclic AMP responses were potentiated (Figure 2b,c).
Figure 2.

Effects of LPA on forskolin, PGE1 and isoproterenol stimulation of cyclic AMP production in RAW 264.7 cells. Cells were incubated with various concentrations of LPA plus 10 μM forskolin (a), 1 μM PGE1 (b) or 1 μM isoproterenol (c) for 10 min, then assayed for cyclic AMP content and protein as described in Methods. The values are the mean±s.e.mean from at least three independent experiments. The values in the parentheses indicate the percentages of potentiation caused by LPA.
Interaction with G protein-acting toxins
Since the LPA receptors have been shown to be members of the G protein-coupling receptors (Moolenaar, 1995; Moolenaar et al., 1997), we explored the possible involvement of Gs and Gi/Go proteins in LPA-mediated cyclic AMP increase seen in macrophages. In cells pretreated for 24 h with 100 ng ml−1 PTX, which causes ADP-ribosylation and uncoupling of Gi/Go function, the potentiation effect of LPA on the PGE1 response was unchanged (data not shown).
Cholera toxin treatment itself (500 ng ml−1 for 150 min) increased the basal cyclic AMP to 320±60 pmol mg−1 protein (n=4) and also potentiated the production of cyclic AMP induced by PGE1 (Figure 3); the cyclic AMP increase on co-addition of cholera toxin and LPA (30 μM) showed an additive effect. When cells were treated with all three stimuli, cholera toxin, PGE1 and LPA, a marked potentiation of cyclic AMP formation, about 4 fold greater than the co-stimulated response by PGE1 and LPA, was seen (Figure 3).
Figure 3.

Interactions between PGE1-, LPA- and cholera toxin-induced cyclic AMP accumulation. Cells were treated with PGE1 (1 μM, 10 min) and/or LPA (30 μM, 10 min) following pretreatment with vehicle or cholera toxin (CTX, 500 ng ml−1. 150 min) as indicated. Note the additive effect of the LPA and CTX responses, and the potentiation effect seen with PGE1 and CTX in the presence or absence of LPA. The values are the mean±s.e.mean from at least three independent experiments.
Cytoskeleton-unrelated mechanism of LPA response
Previous studies have demonstrated the Rho-dependent effect of LPA on stress fibre formation in fibroblasts (Ridley & Hall, 1992; Malcolm et al., 1996). In order to study the possible involvement of cytoskeletal rearrangement in the LPA-induced AC potentiation, we tested the effect of LPA on stress fibre formation and also explored the effect of two cytoskeletal inhibitors, colchicine (a microtubule disrupter) and cytochalasin B (an actin filament disrupter), which have also been shown to increase AC activity in macrophages (Grunspan-Swirsky & Pick, 1978).
Using FITC-phalloidin to detect actin, actin filaments were seen in the filopodium and the lamellipodium of the ventral and dorsal aspects of serum-cultured RAW 264.7 macrophages (Figure 4a,b); however, no stress fibres were seen in the cytoplasm. In serum-deprived RAW 264.7 cells, the actin filaments were present in the filopodium and lamellipodium regardless of the presence or absence of LPA (50 μM) (Figure 4c–f) and LPA did not induce stress fibre formation in these cells. These results indicated that, in contrast to its effect in fibroblasts (Ridley & Hall, 1992) and C6 glioma cells (our unpublished data), LPA did not elicit stress fibre formation. In parallel, the effect of serum deprivation on LPA-induced cyclic AMP potentiation was tested, since LPA has been suggested to be responsible for the serum-induced AC activation (Kreps et al., 1993; Nogami et al., 1995); neither the cyclic AMP response of PGE1 (1 μM) nor the potentiation effect of LPA (1–30 μM) was affected by serum-deprivation for 4 h (data not shown).
Figure 4.

Effect of LPA on actin cytoskeleton in RAW 264.7 macrophages. (a,b) Control cells grown in DMEM containing 10% serum. (c,d) Cells incubated in serum-free DMEM for 4 h (e,f) Serum-deprived cells treated with 50 μM LPA for 10 min. Cells were then washed with PBS, fixed in 10% formalin, and permeabilized with 0.2% Triton X-100. Actin was stained using FITC-phalloidin in PBS for 30 min at room temperature. (a,d,e) ventral aspect. (b,d,f) dorsal aspect. Scale bar=10 μm.
Figure 5 shows that colchicine (10 μM) and cytochalasin B (10 μg ml−1) respectively potentiate PGE1-induced cyclic AMP formation by 4.2 or 2.5 fold within 1 h treatment, although neither drug itself increased the basal cyclic AMP level in the absence of Gs-mediated AC activation (data not shown). The potentiation effects of colchicine and cytochalasin B were additive with that of LPA (10 μM). In addition, the effect of colchicine, but not those of cytochalasin B nor LPA, was sensitive to pretreatment with taxol (20 μM, a microtubule stabilizer) (Figure 5), confirming that the action of colchicine is dependent on microtubule disruption.
Figure 5.

Effects of colchicine, cytochalasin B or taxol on LPA-induced cyclic AMP potentiation. Cells were pre-treated with vehicle or 20 μM taxol for 20 min, then vehicle, colchicine (10 μM) or cytochalasin B (10 μg ml−1) was added for 50 min before the addition of vehicle or 1 μM PGE1 plus LPA (10 μM). The values are the mean±s.e.mean from three independent experiments.
Ca2+-independent effect of LPA
To explore the possible involvement of an intracellular Ca2+ increase in LPA-induced AC activation, we tested (i) whether LPA can increase [Ca2+]i via a PI breakdown pathway; (ii) whether the LPA potentiation occurs in the absence of extracellular Ca2+. In normal PSS containing 1.8 mM CaCl2, LPA (1–30 μM) produces a concentration-dependent increase in [Ca2+]i. At 1 or 10 μM, the [Ca2+]i gradually increased, showing an increase of 409±35 nM (n=3) and 523±52 nM (n=4), respectively, at 5 min. At 30 μM, the [Ca2+]i rapidly increased by 636±45 nM (n=4) and remained at this level for at least 10 min (Figure 6). When cells were incubated in the Ca2+-free PSS with 1 mM EGTA, the [Ca2+]i response to LPA disappeared (data not shown). Under these conditions, LPA (50 μM) not only produced a greater increase in the basal cyclic AMP level (200±13 pmol mg−1 (n=3) compared with 81±8 pmol mg−1 (n=3) in normal PSS), but also potentiated the PGE1 response to a similar extent (Figure 7).
Figure 6.
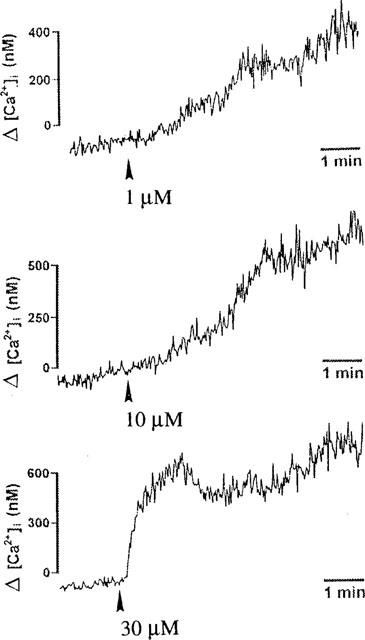
Concentration-dependent effect of LPA on [Ca2+]i. Cells labelling with 3 μM fura-2/AM were treated with LPA (1, 10 or 30 μM), and the [Ca2+]i was determined as described in Methods.
Figure 7.

Effect of extracellular Ca2+ on LPA-induced cyclic AMP accumulation. RAW 264.7 cells, incubated in either normal PSS (control) or Ca2+-free PSS (containing 1 mM EGTA and in the absence of CaCl2) were treated with 50 μM LPA (a) or LPA (50 μM) plus PGE1 (1 μM) (b). The values are the mean±s.e.mean of at least three independent experiments. *P<0.05 compared with the LPA response in normal PSS.
To assess the action on PI turnover, we measured [3H]-inositol phosphate (IP) accumulation in [3H]-myoinositol pre-labelled cells, as previously described (Lin & Chen, 1997). When assayed in normal PSS containing 10 mM LiCl to selectively inhibit inositol monophosphatase, no significant increase in [3H]-IP accumulation was seen following treatment with concentrations of LPA up to 50 μM (data not shown).
Involvement of PKC in LPA-induced cyclic AMP synthesis
To explore the involvement of PKC on LPA-induced AC activation, the effects of PKC inhibitors and a PKC activator were studied. As shown in Figure 8a, the PKC inhibitors, staurosporine and Ro 31-8220, but not Go 6976, inhibited the LPA (50 μM)-induced cyclic AMP increase. The LPA response was decreased to 38±6% (n=3) and 17±3% (n=4) by 1 μM staurosporine and 10 μM Ro 31-8220, respectively.
Figure 8.

Effects of PKC inhibitors on LPA-induced cyclic AMP accumulation. Cells were pre-treated with staurosporine (1 μM), Ro 31–8220 (10 μM) or Go6976 (1 μM) for 20 min before stimulation with LPA (30 μM) alone (a) or LPA plus PGE1 (1 μM) (b). The values are the mean±s.e.mean from at least three independent experiments. *P<0.05 as compared with the control LPA (a) or PGE1 (b) response. **P<0.05 as compared with the cyclic AMP response to LPA+PGE1 without PKC inhibitor treatment.
The effects of PKC inhibitors on LPA-induced cyclic AMP potentiation of PGE1 stimulation are shown in Figure 8b. Ro 31-8220 at 10 μM effectively reduced the LPA-induced potentiation of the PGE1 response from 460±50% (n=3) of control to 128±13% (n=3), and itself moderately attenuated the PGE1 response alone by 39±7% (n=3). On the other hand, Go 6976 at 1 μM slightly increased PGE1 response by 45±12% (n=3), but failed to significantly attenuate the LPA-induced cyclic AMP potentiation.
To further link the mechanism responsible for the effects of LPA to PKC-mediated signalling, we examined the effects of PKC activation and down-regulation on agonist-induced induced AC activation. Phorbol 12-myristate-13 acetate (PMA), an activator of classical and novel types of PKC isoforms, can mimic the potentiation effect of LPA. As shown in Figure 9a, pretreatment with 1 μM PMA for 20 min did not affect the basal cyclic AMP level, but increased the PGE1 response to 211±21% (n=4). The potentiating effects of PMA and LPA, at their respective concentrations for maximal effect, were non-additive. These results, together with those from the PKC inhibitor studies, indicate that PKC activation plays a major role in the action of LPA.
Figure 9.

Effect of PMA treatment on PGE1- and LPA-induced cyclic AMP accumulation. Cells were treated with 1 μM PMA for 20 min or 24 h prior to the addition of PGE1 (1 μM) and/or LPA (30 μM) for 10 min, as indicated. The values are the mean±s.e.mean from five or six independent experiments. *P<0.05 as compared with the control PGE1 response without PMA pretreatment. **Non-additivity between LPA- and PMA-induced cyclic AMP accumulation.
Further studies in cells pre-treated with 1 μM PMA for 24 h showed that the LPA-induced increase in basal cyclic AMP levels (Figure 9b) and the potentiation of PGE1 (Figure 9a) response were not altered compared with controls, suggesting that the PKC isoform(s) involved in LPA signalling belongs to the PMA-non-down-regulated types. Our previous report has already shown that 24 h pretreatment of RAW 264.7 cells with PMA can inhibit the subsequent effect of PMA in potentiating cyclic AMP response (Lin & Chen, 1998). To investigate the possible PKC isoform(s) involved in LPA-induced AC activation, down-regulation was performed. Eight PKC isoforms (α, βI, βII, δ, ε, μ, λ and ζ) were identified in RAW 264.7 macrophages by immunoblot analysis; after treatment with 1 μM PMA for 24 h, PKCα, βI, βII and δ were down-regulated by 95±3%, 85±7%, 96±3% and 80±4%, respectively (n=3), while PKCε, μ, λ and ζ were not significantly affected (Figure 10).
Figure 10.

PMA-induced PKC down-regulation in RAW 264.7 macrophages. Cells were pre-treated with vehicle (C) or 1 μM PMA for 24 h, then the amount of each PKC isoform was determined by immunoblotting with isoform-specific antibodies and ECL detection. The results are representative of two or three experiments.
LPA induced atypical PKC activation
Previous studies have established an in situ kinase assay system for the specific detection of the kinase activity of atypical PKC isoforms using PKCε peptide (sequence 149–164) as a substrate and PKCζ pseudosubstrate peptide (sequence 113–129) as an inhibitor (Wooten et al., 1996). As shown in Figure 11a, LPA (50 μM) markedly increased atypical PKC activity at 4 and 7 min after treatment; its concentration-dependent effect is shown in Figure 11b. At concentrations higher than 10 μM, LPA can caused a significant increase in atypical PKC activity, the effect being reached about 5.5 fold at 30 μM.
Figure 11.

Effect of LPA on atypical PKC activity. (a) Time-dependent increase in atypical PKC activity following treatment with LPA. RAW 264.7 cells were treated with 50 μM LPA for 0–7 min as indicated. At the end of the stimulation period, the cells were assayed in situ for atypical PKC activity. (b) Concentration-dependent stimulation of atypical PKC activity by LPA. Cells were treated as indicated with 1–50 μM of LPA for 7 min followed by the in situ assay of atypical PKC. The results are expressed as the mean±s.e.mean from three independent experiments. *P<0.05 as compared with the control activity.
Discussion
Previous studies have demonstrated several underlying mechanisms involved in the LPA effect on the regulation of cyclic AMP formation. Coupling to Gi-mediated AC inhibiton is one of the signalling pathways for LPA receptors, as seen in Swiss 3T3 mouse fibroblasts (van Corven et al., 1989) and PC12 pheochromocytoma cells (Tigyi et al., 1996a). In human airway smooth muscle cells, although LPA itself does not affect basal cyclic AMP levels, LPA receptors, coupling to Gi proteins, as well as to Gq proteins, can modulate AC activity by three different mechanisms (Nogami et al., 1995). Firstly, activation of Giα results in the inhibition of forskolin-induced cyclic AMP formation. Secondly, the Gq-triggered PI turnover and PKC activation lead to the potentiation of isoproterenol-stimulated type II AC activity. Thirdly, released βγ subunits, derived from either Gi or Gq protein, also participate in the potentiation of AC II activity. In human foreskin fibroblasts, LPA elicits opposite effects on cyclic AMP regulation, depending on the stimulation agent used. In contrast to the PTX-sensitive inhibition of AC stimulated by isoproterenol, PGE2, cholera toxin and forskolin, LPA enhances ATP- or adenosine-induced cyclic AMP formation; this effect is dependent on the released βγ subunits (Ahmed & Heppel, 1997). Hoffman et al. (1996) reported that in glial cells, the sensitizing effect of LPA on β-adrenergic agonist-induced cyclic AMP formation is completely inhibited by PTX, suggesting the involvement of PTX-sensitive G proteins in the LPA effects. All this information implies the multiplicity and cell-type dependency of the LPA-mediated regulation of AC activity. Because of this complexity, the aim of the present study was to explore the underlying mechanism responsible for LPA modulation of the cyclic AMP system in macrophages. We have previously shown that RAW 264.7 macrophages express type II AC, which is a PKC-sensitive AC isoform (Lin & Chen, 1998). Interestingly, here we demonstrated that LPA can not only activate AC in RAW 264.7 macrophages by itself, but also potentiate agonist-stimulated AC activity via a novel pathway, which requires atypical PKC activation and is independent of PTX-sensitive pathways, PI turnover or a [Ca2+]i increase. The PTX-insensitivity further rules out the involvement of Gi/Go coupling to LPA potentiation of cyclic AMP formation in RAW 264.7 cells.
The modulation of cyclic AMP responses by LPA appeared to occur at the level of AC, and not elsewhere in the cyclic AMP cascade. Exposure of mouse RAW 264.7 macrophages to LPA synergistically increased the stimulation of cyclic AMP levels by Gs-coupled receptor agonists, PGE1 and isoproterenol. Moreover, LPA treatment also additively increased the cyclic AMP stimulation by distinct mechanisms, such as those involving cholera toxin (an activator acting on Gsα subunit), forskolin (a direct activator of AC), colchicine and cytochalasin B (disrupters of cytoskeleton). Thus, the effect of LPA on AC system might act on the postreceptor level and involve the cyclase itself. As mentioned above, although most, but not all, of the effects of LPA in other cells are sensitive to PTX inhibition, the ineffectiveness of PTX on LPA-induced cyclic AMP potentiation rules out the possibility that the effects of LPA in RAW 264.7 cells are mediated via βγ subunits released from either Gi or Go proteins. Moreover, the failure of LPA to trigger PI turnover also excludes the participation of Gq-derived βγ subunits in this potentiation event.
Several pieces of evidence led to the conclusion that the AC stimulation and potentiation effects of LPA are dependent on atypical PKC activation. Firstly, the effect of LPA was blocked by non-selective inhibitors of PKC Ro 31-8220 and staurosporine, while Go 6976, a selective inhibitor of classic PKCα, β and γ (Martiny-Baron et al., 1993), failed to affect it. Secondly, the LPA effect was mimicked by the PKC activator, PMA, and their effects were non-additive. Thirdly, long-term pretreatment with PMA failed to affect the LPA response, suggesting that PKC isoforms resistant to PMA down-regulation were involved. Fourthly, immunoblot analysis of RAW 264.7 cells using antibodies specific for different PKC isozymes revealed the presence of PKC α, βI, βII, δ, ε, μ, λ and ζ in these cells, of these only PKC α, βI, βII and δ, were down-regulated by 24 h PMA treatment. Fifthly, direct measurement of atypical PKC activity revealed a marked increase on treatment with LPA. Taken together, these data suggest that different PKC isoforms are responsible for PMA- or LPA-mediated AC activation in RAW 264.7 macrophages. The activation of atypical PKC isoforms is involved in the stimulation of AC by LPA, whereas, as we recently reported in the same cell system, PMA utilizes PKC ε and/or μ to up-regulate AC activity (Lin & Chen, 1998). Indeed, the type II isozyme of AC able to be stimulated and/or sensitized by PKC has been extensively characterized (Jacobowitz & Iyengar, 1994; Zimmermann & Taussig, 1996). With regard to the mechanism involved in the activation of atypical PKC by LPA, we ruled out the involvement of diacylglycerol based on the evidence that atypical PKCs are not activated by diacylglycerol (Nishizuka, 1995). We now plan to carry out further experiments in order to better understand the link between LPA receptors, if it exists in macrophages, and atypical PKC activation. Naturally, a possible direct interaction between LPA and atypical PKC must be considered, since previous in vitro studies have shown direct activation of PKCα and PKCζ by LPA at quite high concentrations, e.g. 214 μM (Limatola et al., 1994; Sando & Chertihin, 1996).
The results from the present study suggest that a cytosketetal change is not likely to be the mechanism involved in LPA-induced cyclic AMP potentiation, although LPA does have cytosketetal effects in certain cell types. For example, it can induce the rapid formation of actin stress fibres and the assembly of focal adhesion in serum-starved Swiss 3T3 cells (Ridley & Hall, 1992). These cytoskeletal effects might also result in the dramatic shape changes seen in PC12 cells (Tigyi et al., 1996b) and neuroblastoma cells (Jalink et al., 1993). Cytoskeletal structures, such as microtubules and microfilaments, have been implicated in the regulation of AC activity in response to hormones in a variety of cell systems (Jasper et al., 1995), and this observation prompted us to investigate the effects of colchicine and cytochalasin B in LPA-mediated cyclic AMP potentiation. These agents are known to disrupt microtubules and microfilaments respectively, and can enhance agonist-stimulated cyclic AMP accumulation. In the present study, we ruled out any involvement of cytoskeletal changes in the LPA-induced cyclic AMP effect because of the following reasons. Firstly, in serum-deprived RAW 264.7 cells LPA does not induce stress fibre formation or a change in actin staining in the filopodium and lamellipodium, but still induces cyclic AMP potentiation. Secondly, treatment of RAW 264.7 cells with cytochalasin B, which is an actin filament disassembling agent that blocks LPA-induced shape changes in other cell types (Jalink et al., 1993; Inoue et al., 1995), had no significant effect on LPA-stimulated cyclic AMP accumulation; colchicine also had no effect on the potentiation effect of LPA. In fact, in RAW 264.7 macrophages, the disassembly of both microtubules and microfilaments facilitated agonist-induced cyclic AMP formation, while taxol, a microtubule stabilizer, selectively blocked the action of colchicine. Thus we conclude that the LPA-induced stimulation of AC II in RAW 264.7 macrophages does not involve cytoskeletal changes.
In RAW 264.7 macrophages, as in many other cell types, such as platelets, PC12 cells, and Xenopus oocytes (Watson et al., 1985; Durieux et al., 1992; Dyer et al., 1992), LPA induced an increase in [Ca2+]i. Since the basal, PGE1- and isoproterenol-mediated cyclic AMP levels in RAW 264.7 macrophages are unaffected by Ca2+ elevating agents (thapsigargin and ionomycin) (data not shown), and since a PKC activator did not increase intracellular free Ca2+ levels, the LPA-induced increase in cyclic AMP could not be due to the increased intracellular Ca2+. Further confirmation came from the fact that the LPA-increased cyclic AMP was still seen, and actually was more pronounced, in the absence of extracellular Ca2+, which completely dimishes the [Ca2+]i increasing effect of LPA itself. Our results also indicate that the rise in [Ca2+]i results from the influx of extracellular Ca2+, and is not relevant to the inositol trisphosphate-mediated [Ca2+]i transient seen with LPA treatment in fibroblasts (van Corven et al., 1989; Jalink et al., 1990), PC12 cells (Tigyi et al., 1996b), mesangial cells (Inoue et al., 1995) and hepatocytes (Im et al., 1997). In RAW 264.7 cells, LPA did not induce PI hydrolysis at concentrations up to 50 μM. It has been shown that, in addition to its ability to mobilize intracellular Ca2+ by activation of the PI cascade, LPA can also promote Ca2+ entry across the plasma membrane in rat glomerular mesangial cells (Inoue et al., 1995). Further studies will be required to elucidate the precise mechanisms whereby Ca2+ entry is stimulated by LPA and thus diminishes LPA-increased cyclic AMP formation in RAW 264.7 cells. In this context, by using RT–PCR, we have ruled out the presence of Ca2+-inhibited ACs (types V and VI) in RAW 264.7 cells (data not shown).
As already mentioned, since LPA elicits a wide range of biological responses via its cellular G protein coupled receptors, several lines of evidence suggest that the diverse effects of LPA are mediated by multiple subtypes of LPA receptors with distinct signalling properties and tissue distribution (Durieux & Lynch, 1993; Moolenaar et al., 1997). Thus, one possible explanation for the opposite effects of LPA on AC activity (for example, inhibition via Gi protein or stimulation via PKC activation) might be due to the cell-type specific expression of LPA receptor subtypes that activate different signalling mechanisms. Up to now at least three mammalian LPA receptors have been cloned (Goetzl & An, 1998).
In summary, LPA activates atypical PKC in murine RAW 264.7 macrophages, which leads to the stimulation of type II AC and an increase in cyclic AMP formation due to a variety of stimuli. These results demonstrate a novel mechanism of LPA action on the AC system and also show the complexity of LPA-mediated signalling.
Acknowledgments
This research was supported by a research grant from the National Science Council of Taiwan (NSC89-2320-B002-022).
Abbreviations
- AC
adenylyl cyclase
- DMEM
Dulbecco's modified Eagle's medium
- IBMX
3-isobutyl-methylxanthine
- IP
inositol phosphate
- LPA
lysophosphatidic acid
- PGE1
prostaglandin E1
- PI
phosphoinositide
- PKC
protein kinase C
- PLC
phospholipase C
- PLD
phospholipase D
- PMA
phorbol 12-myristate-13 acetate
- PSS
physiological saline solution
- PTX
pertussis toxin
References
- AHMED A.H., HEPPEL L.A. Evidence for a role of G protein βγ subunits in the enhancement of cAMP accumulation and DNA synthesis by adenosine in human cells. J. Cell. Physiol. 1997;170:263–271. doi: 10.1002/(SICI)1097-4652(199703)170:3<263::AID-JCP7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- AN S., DICKENS M.A., BLEU T., HALLMARK O.G., GOETZL E.J. Molecular cloning of the human Edg2 protein and its identification as a functional cellular receptor for lysophosphatidic acid. Biochem. Biophys. Res. Commun. 1997;231:619–622. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- CARR C., GRASSIE M., MILLIGAN G. Stimulation of high affinity GTPase activity and cholera toxin-catalysed [32P]ADP-ribosylation of Gi by lysophosphatidic acid (LPA) in wild-type and α2C10 adrenoceptor-transfected Rat 1 fibroblasts. Biochem. J. 1994;298:493–497. doi: 10.1042/bj2980493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DURIEUX M.E., LYNCH K.K. Signalling properties of lysophosphatidic acid. Trends Pharmacol. Sci. 1993;14:249–254. doi: 10.1016/0165-6147(93)90021-b. [DOI] [PubMed] [Google Scholar]
- DURIEUX M.E., SALAFRANCA M.N., LYNCH K.R., MOORMAN J.R. Lysophosphatidic acid induces a pertussis toxin-sensitive Ca2+-activated Cl− current in Xenopus laevis oocytes. Am. J. Physiol. 1992;263:C896–C900. doi: 10.1152/ajpcell.1992.263.4.C896. [DOI] [PubMed] [Google Scholar]
- DYER D., TIGYI G., MILEDI R. The effect of serum albumin on PC12 cells. II Intracellular calcium transients and their role in neurite retraction. Mol. Brain Res. 1992;14:302–309. doi: 10.1016/0169-328x(92)90097-u. [DOI] [PubMed] [Google Scholar]
- GOETZL E.J., AN S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- GRUNSPAN-SWIRSKY A., PICK E. Enhancement of macrophage adenylate cyclase by microtubule disruption drugs. Immunopharmacol. 1978;1:71–82. doi: 10.1016/0162-3109(78)90010-3. [DOI] [PubMed] [Google Scholar]
- GUO Z., LILIOM K., FISCHER D.J., BATHURST I.C., TOMEI L.D., KIEFER M.C., TIGYI G. Molecular cloning of a high-affinity receptor for the growth factor-like lipid mediator lysophosphatidic acid from Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 1996;93:14367–14372. doi: 10.1073/pnas.93.25.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HECHT J.H., WEINER J.A., POST S.R., CHUN J. Ventricular Zone Gene-1 (Vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell. Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMAN J., WHITTLE S., TOEWS M. Modulation of cyclic AMP accumulation in glial cells by exogenous phospholipase C. J. Lipid Med. Cell. Signal. 1996;13:51–62. doi: 10.1016/0929-7855(95)00044-5. [DOI] [PubMed] [Google Scholar]
- IM D.S., FUJIOKA T., KATADA T., KONDO Y., UI M., OKAJIMA F. Characterization of sphingosine 1-phosphate-induced actions and its signaling pathways in rat hepatocytes. Am. J. Physiol. 1997;272:G1091–G1099. doi: 10.1152/ajpgi.1997.272.5.G1091. [DOI] [PubMed] [Google Scholar]
- INOUE C.N., FORSTER H.G., EPSTEIN M. Effects of lysophosphatidic acid, a novel lipid mediator, on cytosolic Ca2+ and contractility in cultured rat mesangial cells. Circ. Res. 1995;77:888–896. doi: 10.1161/01.res.77.5.888. [DOI] [PubMed] [Google Scholar]
- JACOBOWITZ O., IYENGAR R. Phorbol ester-induced stimulation and phosphorylation of adenylyl cyclase 2. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10630–10634. doi: 10.1073/pnas.91.22.10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JALINK K., EICHHOTZ T., POSTMA F.R., VAN CORVEN E.J., MOOLENAAR W.H. Lysophosphatidic acid induces neuronal shape changes via a novel, receptor-mediated signaling pathway: similarity to thrombin action. Cell Growth Diff. 1993;4:206–215. [PubMed] [Google Scholar]
- JALINK K., VAN CORVEN E.J., MOOLENAAR W.H. Lysophosphatidic acid, but not phosphatidic acid, is a potent Ca2+-mobilizing stimulus for fibroblasts. J. Biol. Chem. 1990;265:12232–12239. [PubMed] [Google Scholar]
- JASPER J.R., POST S.R., DESAI K.H., INSEL P.A., BERNSTEIN D. Colchicine and cytochalasin B enhances cyclic AMP accumulation via postreceptor actions. J. Pharmacol. Exp. Ther. 1995;274:937–942. [PubMed] [Google Scholar]
- KREPS D.M., WHITTLE S., HOFFMAN J.M., TOEWS M.L. Lysophosphatidic acid mimics serum-induced sensitization of cyclic AMP accumulation. FASEB J. 1993;7:1376–1380. doi: 10.1096/fasebj.7.14.8224610. [DOI] [PubMed] [Google Scholar]
- LIMATOLA C., SCHAAP D., MOOLENAAR W.H., VAN BLITTERSWIJK W.J. Phosphatidic acid activation of protein kinase C-ζ overexpressed in COS cells: comparison with other protein kinase C isotypes and other acidic lipids. Biochem. J. 1994;304:1001–1008. doi: 10.1042/bj3041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN W.W., CHEN B.C. Involvement of protein kinase C in the UTP-mediated potentiation of cyclic AMP accumulation in mouse J774 macrophages. Br. J. Pharmacol. 1997;121:1749–1757. doi: 10.1038/sj.bjp.0701300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN W.W., CHEN B.C. Distinct PKC isoforms mediate the activation of cPLA2 and adenylyl cyclase by phorbol ester in RAW 264.7 macrophages. Br. J. Pharmacol. 1998;125:1601–1609. doi: 10.1038/sj.bjp.0702219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALCOLM K.C., ELLIOTT C.M., EXTON J.H. Evidence for Rho-mediated agonist stimulation of phospholipase D in rat fibroblasts. J. Biol. Chem. 1996;271:13135–13139. doi: 10.1074/jbc.271.22.13135. [DOI] [PubMed] [Google Scholar]
- MARTINY-BARON G., KAZANIETZ M.G., MISCHAK H., BLUMBERG P.M., KOCHS G., HUG H., MARME D., SCHACHTELE C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J. Biol. Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- MOOLENAAR W.H. Lysophosphatidic acid, a multifunctional phospholipid messenger. J. Biol. Chem. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- MOOLENAAR W.H., KRANENBURG O., POSTMA F.R., ZONDAG G.C. Lysophosphatidic acid: G-protein signalling and cellular responses. Curr. Opinion Cell Biol. 1997;9:168–173. doi: 10.1016/s0955-0674(97)80059-2. [DOI] [PubMed] [Google Scholar]
- MULLET D., FERTEL R.H., KNISS D., COX G.W. An increase in intracellular cyclic AMP modulates nitric oxide production in IFN-γ-treated macrophages. J. Immunol. 1997;158:897–904. [PubMed] [Google Scholar]
- NISHIZUKA Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- NOGAMI M., WHITTLE S.M., ROMBERGER D.J., RENNARD S.I., TOEWS M.L. Lysophosphatidic acid regulation of cyclic AMP accumulation in cultured human airway smooth muscle cells. Mol. Pharmacol. 1995;48:766–793. [PubMed] [Google Scholar]
- RIDLEY A., HALL A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- SANDO J.J., CHERTIHIN O.I. Activation of protein kinase C by lysophosphatidic acid: dependence on composition of phospholipid vesicles. Biochem. J. 1996;317:583–588. doi: 10.1042/bj3170583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIGYI G., FISCHER D.J., SEBOK A., MARSHALL F., DYER D.L., MILEDI R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite-protective effects of cyclic AMP signaling. J. Neurochem. 1996a;66:549–558. doi: 10.1046/j.1471-4159.1996.66020549.x. [DOI] [PubMed] [Google Scholar]
- TIGYI G., FISCHER D.J., SEBOK A., YANG C., DYER D.L., MILEDI R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: control by phosphoinositide-Ca2+ signaling and Rho. J. Neurochem. 1996b;66:537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- VAN CORVEN E.J., GROENINK A., JALINK K., EICHOLTZ T., MOOLENAAR W.H. Lysophosphatidate-induced cell proliferation. Identification and dissection of signaling pathways mediated by G proteins. Cell. 1989;59:45–54. doi: 10.1016/0092-8674(89)90868-4. [DOI] [PubMed] [Google Scholar]
- VAN DER BEND R.L., BRUNNER J., JALINK K., VAN CORVEN E.J., MOOLENAAR W.H., VAN BLITTERSWIJK W.J. Identification of a putative membrane receptor for the bioactive phospholipid, lysophosphatidic acid. EMBO J. 1992;11:2495–2501. doi: 10.1002/j.1460-2075.1992.tb05314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENTURA M.A., RIBIER A., DELBOURG I., CHAMBAUT-GUERIN A.M., THOMOPOULOS P. The adhesiveness of monocytic U937 cells is stimulated by pro-inflammatory agents and inhibited by adenosine 3′,5′-cyclic monophosphate. Biochem. Pharmacol. 1990;39:677–683. doi: 10.1016/0006-2952(90)90145-b. [DOI] [PubMed] [Google Scholar]
- WATSON S.P., MCCONNELL R.T., LAPETINA E.G. Decannoyl lysophosphatidic acid induces platelet aggregation through an extracellular action. Evidence against a second messenger role for lysophosphatidic acid. Biochem. J. 1985;232:61–66. doi: 10.1042/bj2320061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WENG G., Li J., DINGUS J., HILDEBRANDT J.D., WEINSTEIN H., IYENGAR R. Gβ subunit interacts with a peptide encoding region 956-982 of adenylyl cyclase 2. Cross-linking of the peptide to free Gβγ but not the heterotrimer. J. Biol. Chem. 1996;271:26445–26448. doi: 10.1074/jbc.271.43.26445. [DOI] [PubMed] [Google Scholar]
- WOOTEN M.W., SEIBENHENER M.L., MATTHEWS L.H., ZHOU G., COLEMAN E.S. Modulation of ζ-protein kinase C by cyclic AMP in PC12 cells occurs through phosphorylation by protein kinase A. J. Neurochem. 1996;67:1023–1031. doi: 10.1046/j.1471-4159.1996.67031023.x. [DOI] [PubMed] [Google Scholar]
- ZIMMERMANN G., TAUSSIG R. Protein kinase C alters the responsiveness of adenylyl cyclases to G protein α and βγ subunits. J. Biol. Chem. 1996;271:27161–27166. doi: 10.1074/jbc.271.43.27161. [DOI] [PubMed] [Google Scholar]