

Abstract
The subtypes of α1-adrenoceptor mediating contractions of rat vas deferens to endogenous and exogenous noradrenaline and to the exogenous agonists methoxamine, phenylephrine and A61603 have been examined.
The effects of antagonists on the shape of concentration-response curves, both tonic and phasic, to the four agonists were analysed. Prazosin produced parallel shifts in all cases. Particularly for RS 17053 against noradrenaline, there was some evidence for a resistant component of the agonist response. High concentrations of RS 17053 (1–10 μM) virtually abolished tonic contractions but phasic contractions were resistant.
A series of nine antagonists (the above and WB4101, benoxathian, phentolamine, BMY 7378, HV 723, spiperone) were investigated against contractions to noradrenaline. The correlation with the potency of the series of α1-adrenoceptor antagonists against contractions to noradrenaline was significant only for the α1A-adrenoceptor ligand binding site (r=0.88, n=9, P<0.01).
In epididymal portions (nifedipine 10 μM), the isometric contraction to a single electrical pulse is α1-adrenoceptor mediated. The correlation with ligand binding sites for 11 antagonists (the above plus ARC 239 and (+)-niguldipine) was significant only for the α1D-adrenoceptor subtype (r=0.65, n=11, P<0.05).
In conclusion, tonic contractions of rat vas deferens produced by exogenous agonists are mediated predominantly by α1A-adrenoceptors, although a second subtype of receptor may additionally be involved in phasic contractions. Nerve-stimulation evoked α1-adrenoceptor mediated contractions seem to predominantly involve non-α1A-adrenoceptors, and the receptor involved resembles the α1D-receptor.
Keywords: Rat vas deferens, α1A-adrenoceptors, α1B-adrenoceptors, α1D-adrenoceptors, prazosin, RS17053, 5-methyl-urapidil, A61603
Introduction
α1-Adrenoceptors were initially subdivided into α1A- and α1B-subtypes in ligand binding studies, based on the affinities of a series of ligands, especially WB 4101 and prazosin (Morrow & Creese, 1986), and based on the ability of the alkylating agent chloroethylclonidine (CEC) to inactive the α1B- but not the α1A-subtype (Han et al., 1987). Molecular cloning techniques revealed initially four subtypes of α1-adrenoceptor: α1a, α1b, α1c, α1d (Cotecchia et al., 1988; Schwinn et al., 1990; Lomasney et al., 1991; Perez et al., 1991; see Bylund et al., 1994). However, the α1a and α1d clones showed 99.8% homogeneity and appeared to represent the same subtype. It is now clear that the α1a/α1d clone represents a novel subtype of α1-adrenoceptor (α1D), whereas the α1c is now identified with the α1A-ligand binding site. These clones have now been renamed to match the functional receptors: α1A (formely α1c), α1B (formerly α1b), and α1D (formerly α1a/α1d).
One of the earliest functional subclassifications of α1-adrenoceptors was into α1H and α1L, with high and low affinity for prazosin (see Flavahan & Vanhoutte, 1986 but see Docherty, 1987; Muramatsu et al., 1990). α1-Adrenoceptors in blood vessels were further subdivided into three subtypes, α1H, α1N and α1L, based on their affinities for prazosin, WB 4101 and HV 723 (Muramatsu et al., 1990). Under this classification, α1H-receptors have high affinity for prazosin, and appear to match the α1A, α1B, α1D classes (Muramatsu et al., 1995), whereas α1L and α1N have low affinity for prazosin and do not seem to match current molecular cloning based classifications.
New α1-adrenoceptor antagonists have been developed for effects in the lower urinary tract to treat benign prostatic hypertrophy, and the receptors involved were identified as α1A-adrenoceptors. However, some antagonists which were selective for α1A-adrenoceptors in ligand binding studies had low potencies in functional studies of the lower urinary tract (RS 17053: Ford et al., 1996; Kenny et al., 1996; SB 216469: Chess-Williams et al., 1996).
It has been variously suggested that contractions of rat vas deferens to exogenous noradrenaline or adrenaline are mediated predominantly by α1A-adrenoceptors (Han et al., 1987; Hanft & Gross, 1989; Aboud et al., 1993), or α1L- in addition to α1A-adrenoceptors (Ohmura et al., 1992). The objects of this study were to re-examine the α1-adrenoceptor subtypes present in rat vas deferens in the light of current knowledge of α1-adrenoceptor subtypes and using some of the newer subtype selective compounds. Some of these results have been published in shortened form (Docherty, 1998).
Methods
Male Wistar rats (200–300 g) were obtained from Trinity College Dublin, and vas deferens was employed as outlined below.
Rat vas deferens
Whole vas deferens, or prostatic or epididymal portions were obtained. Tissues were attached to myograph transducers under 1 g tension in organ baths at 37°C in Krebs–Henseleit solution of the following composition: (mM): NaCl 119; NaHCO3 25; D-glucose 11.1; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.0; EDTA 0.03, ascorbic acid 0.28. Additionally, cocaine (3 μM), propranolol (3 μM) and indomethacin (10 μM) were present except where otherwise stated.
In investigations of contractions produced by exogenous agonists, tissues were equilibrated for 30 min and then contracted with noradrenaline (10 μM). Bathing fluid was then changed every 15 min for the next hour. Following 60 min exposure to antagonist or vehicle, a single agonist concentration response-curve was obtained per tissue. Tissues were contracted with noradrenaline, methoxamine, phenylephrine or A61603 administered cumulative in 0.5 log unit increments beginning with 1 nM. Antagonist potency was expressed as the dissociation constant KB from the equation KB=[B]/(DR-1), where [B] is the concentration of antagonist and DR is the agonist dose-ratio produced by the antagonist as compared to the vehicle experiment, or as a pA2 Value. Antagonist pA2 values were obtained from the x-intercept of the plot of log (agonist DR-1) against log antagonist concentration, where the slope was not significantly different from negative unity (Arunlakshana & Schild, 1959). Slopes of Schild plots not significantly different from −1.0 were constrained to −1.0, and a pKB was obtained. In cases when slopes of Schild plots were significantly different from negative unity, or when a single concentration of antagonist was employed, antagonist potency was expressed as a pKB from the effects of a single effective concentration of antagonist.
In experiments investigating the ability of competitive antagonists to inhibit the isometric twitch in epididymal portions of vas deferens (in the presence or absence of cocaine), tissues were placed between platinum electrodes and stimulated every 5 min with a single stimulus (0.5 ms pulses, supramaximal pulses) to produce isometric contractions, and nifedipine (10 μM) was present to block the non-noradrenergic component of the twitch. Antagonists or vehicle were added cumulative in 1 log unit increments at 5 min intervals. An isometric twitch was obtained following 5 min exposure to each antagonist concentration, or following exposure to the vehicle.
Drugs
A61603 (N - [5 - (4,5 - dihydro - 1H - imidazol - 2yl) - 2 -hydroxy-5,6,7,8 - tetrahydro - naphthalen- 1 - yl]- methanesulfonamide; a gift from: Abbott Laboratories, U.S.A.); ARC239 (2-(2,4-(o-methoxyphenyl ) - piperazin - 1 - yl ) - ethyl - 4 , 4 dimethyl - 1 , 3-(2H,4H)-isoquinolindine chloride; a gift from: Karl Thomae, Germany); benoxathian hydrochloride (Research Biochemicals, U.S.A.); BMY 7378 (8-[2-(4-(2-methoxyphenyl) piperazin-1-yl)ethyl]-8-azaspiro[ 4 , 5 ]decane - 7 , 9 - dione; Research Biochemicals, U.S.A.); cocaine hydrochloride (Sigma, U.K.); corticosterone (Sigma, U.K.); HV723 (α-ethyl-3,4,5-trimethoxy -□thinsp;a - (3-((2-(2-methoxyphenoxy)ethyl)-amino)-propyl)-benzene acetonitrile fumarate; a gift from: Hokurika, Japan), methoxamine hydrochloride (Research Biochemicals, U.S.A.); 5-methyl-urapidil (a gift from: Byk, Germany); nifedipine (Sigma, U.K.); (+)-niguldipine (Sigma, U.K.); noradrenaline bitartrate (Sigma, U.K.); phentolamine hydrochloride (Research Biochemicals, U.S.A.); phenylephrine hydrochloride (Sigma, U.K.); prazosin hydrochloride (a gift from: Pfizer, Sandwich, U.K.); propranolol hydrochloride (Sigma, U.K.); RS17053 (N-[2(2-cyclopropylmethoxy)ethyl]-5-chloro-α, α-dimethyl-1H-indole-3-ethylamine hydrochloride; (a gift from: Roche Bioscience, U.S.A.); spiperone (Research Biochemicals, U.S.A.); WB 4101 (2-(2′,6′-dimethoxyphenoxyethyl) aminomethyl-1,4-benzodioxan; Research Biochemicals, U.S.A.).
Drugs were dissolved in distilled water, except for corticosterone and spiperone (100% ethanol), (+)-niguldipine (100% methanol) and RS 17053 (DMSO).
Statistics
Values are mean±s.e.mean from n experiments. Agonist pD2 (−log EC50) values before and after vehicle or test antagonist were compared, and effects of antagonist on nerve stimulation-evoked responses were compared with the effects of vehicle, by Student's t-test for paired or unpaired data where appropriate, and by Analysis of Variance. Statistical and graphical analysis was carried out using Instat for Macintosh and GraphPad Prism for PC. Correlations were made between functional antagonist potency and pooled binding affinity data from a number of studies: there are limitations in using pooled data statistically as the aggregate mean may not have the required normal distribution.
Results
Whole vas deferens: responses to exogenous agonists
In whole vas deferens, agonists produced contractions consisting of intermittent spikes superimposed on a tonic contraction: phasic contractions were measured as the total increase above baseline and so includes the spike and tonic components (see Figure 1). The agonists noradrenaline, methoxamine, phenylephrine and A61603 produced tonic contractions with pD2 values of 5.84±0.12, 5.53±0.07, 5.52±0.06 and 7.98±0.16 g and maximum contractions of 1.34±0.09, 1.05±0.08, 1.40±0.07 and 1.33±0.09 g, respectively (n=21 in all cases). The maximum tonic contraction to methoxamine was significantly less than the maximum to the other agonists (Analysis of Variance, P<0.05). The agonists noradrenaline, methoxamine, phenylephrine and A61603 produced phasic contractions with pD2 values of 5.59±0.11, 5.28±0.10, 5.25±0.08 and 7.74±0.15 g and maximum contractions (measured as the maximum height of tonic contractions combined with intermittent spikes) of 2.11±0.11, 2.04±0.10, 2.26±0.15 and 2.16±0.11 g, respectively (n=21 in all cases).
Figure 1.
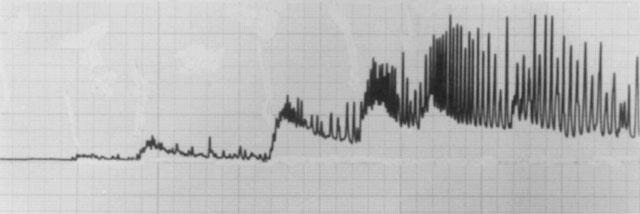
Tracing of a typical experiment illustrating the tonic and phasic nature of contractions to agonists in rat vas deferens. This experiment shows the effects of increasing concentrations of noradrenaline (0.1–30 μM). Tonic contractions were taken as the peak sustained contraction, and phasic contractions as the maximum intermittent tension (including sustained) obtained. Time scale: 20 min of recording shown.
The effects of prazosin (10–30 nM), RS 17053 (100 nM) and 5-methyl-urapidil (30 nM) on tonic contractions to noradrenaline and phasic contractions to the four agonists are shown in Figures 2 and 3. Phasic contractions are omitted for the other three antagonists, since tonic and phasic contractions were affected similarly by antagonists. Prazosin produced parallel shifts in all cases, and all antagonists produced parallel shifts in the potency of A61603. RS 17053 and 5-methyl-urapidil tended to produce apparently non-parallel shifts in the potency of noradrenaline and methoxamine, and to a lesser extent phenylephrine, but the resistant component was small (Figures 2 and 3).
Figure 2.
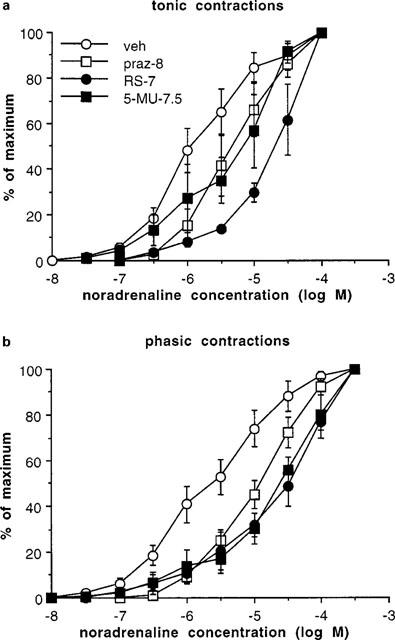
Tonic (a) and phasic (b) contractions obtained to noradrenaline in whole vas deferens, following exposure to vehicle (veh), prazosin (10 nM) (praz-8), RS 17053 (100 nM) (RS-7) or 5-methyl-urapidil (30 nM) (5-MU-7.5). Values are expressed as percentage of the maximum for each experiment. Vertical bars represent s.e.mean from at least three experiment. For clarity, vehicle experiments are combined but, since pKB values were obtained in experiments with paired vehicles, the shift in agonist potency produced by antagonists is not readily obtained from these curves.
Figure 3.
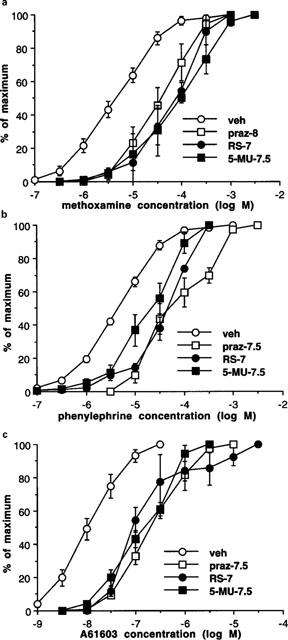
Phasic contractions obtained to methoxamine (a), phenylephrine (b) and A 6163 (c) in whole vas deferens, following exposure to vehicle (veh), prazosin (10 or 30 nM) (praz-8 or −7.5), RS 17053 (100 nM) (RS-7) or 5-methyl-urapidil (30 nM) (5-MU-7.5). Values are expressed as percentage of the maximum for each experiment. Vertical bars represent s.e.mean from at least three experiments. For clarity, vehicle experiments are combined but, since pKB values were obtained in experiments with paired vehicles, the shift in agonist potency produced by antagonists is not readily obtained from these curves.
However, a component particularly of the phasic contractions were resistant to high concentrations of antagonists; this was particularly evident for RS 17053 against noradrenaline. The effects of high concentrations of RS 17053 were investigated against tonic and phasic contractions to noradrenaline in terms of absolute tension developed (Figure 4). RS 17053 (1 μM) significantly reduced the maximum tonic contraction to 0.57±0.10 g (n=4), without affecting maximum phasic contractions. RS 17053 (10 μM) significantly reduced tonic contractions to 0.18±0.05 g (n=9) and significantly reduced phasic contractions to 1.14±0.22 g (n=9). In the figure, noradrenaline pD2 for phasic contractions in vehicle experiments was 5.59±0.21, and the pD2 was shifted approximately 10 fold by RS 17053 (0.1 μM) (pD2) of 4.50±0.14) and by RS 17053 (1 μM) (pD2 of 4.59±0.26 g) (Figure 4). A noradrenaline pD2 following RS 17053 (10 μM) could not be obtained due to a significant reduction in the maximum response (Figure 4). It is clear from the figure that, although low concentrations of RS 17053 (0.1 μM) significantly shifted the potency of noradrenaline, there was a phasic component of the response resistant even to RS 17053 (10 μM) (Figure 4).
Figure 4.
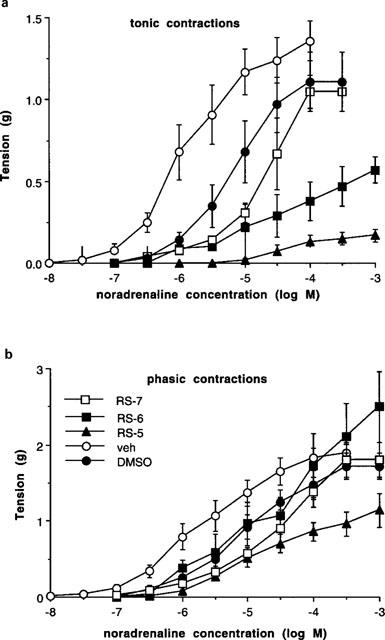
Tonic (a) and phasic (b) contractions obtained to noradrenaline in whole vas deferens expressed as absolute tension (g), following exposure to vehicle (veh), RS 17053 (100 nM) (RS-7), RS 17053 (1 μM) (RS-6), DMSO or RS 17053 (10 μM) (RS-5). Vertical bars represent s.e.mean from at least four experiments. For clarity, vehicle experiments for the two lower concentrations of RS 17053 are combined but, since DMSO, the vehicle for the highest concentration of RS 17053 (10 μM), also produced significant effects, this is included separately.
Prazosin produced concentration-dependent shifts in the potency of all four agonists, and behaved competitively in that it did not decrease the maximum contraction, and slope of the Schild plot were not significantly different from negative unity, so that pA2 values could be calculated (Table 1). However, in terms of slope of Schild plots, 5-methylurapidil behaved non-competitively against phenylephrine and RS17053 behaved competitively against methoxamine (see Table 1). However, pKB values from the effects of the lowest effective concentration were calculated whenever pA2 values could not be calculated (Table 1).
Table 1.
Potencies of antagonists against contractions to noradrenaline, methoxamine, phenylephrine and A61603 in rat vas deferens

In addition to prazosin, 5-methyl-urapidil and RS 17053, WB4101, benoxathian, phentolamine, BMY 7378, HV 723 and spiperone were investigated against contractions to noradrenaline. pA2 values were converted to pKB values by constraining the slope of Schild plots to −1. The pKB values obtained are shown in Table 2. For some of the antagonists, pKB was calculated from the effects of a single concentration of antagonist producing an approximate 1 log unit (10 fold) shift in agonist potency: (μM): RS 17053 0.1, 1.16±0.06 g; phentolamine 1, 1.28±0.25 g; BMY 7378 1, 0.48±0.14 g; BMY 10, 1.42±0.30 g; HV 723 0.1, 1.24±0.10 g; spiperone 1, 1.16±0.18 g. These potencies were correlated with affinities for α1A, α1B and α1D-adrenoceptor ligand binding sites obtained by taking the mean of published values (see Tables 2, 3 and Figure 5). The correlation with the potency of the series of α1-adrenoceptor antagonists against contractions to noradrenaline was significant for the α1A-adrenoceptor ligand binding site (r=0.88, n=9, P<0.01) (Figure 5). One of the published studies examined seven of the antagonists used in this study (all except benoxathian and spiperone; Ford et al., 1996). Correlation at antagonist potency with the ligand binding data of Ford et al. (1996) was again significant only for the α1A-adrenoceptor (α1A r=90, P<0.01; α1B r=0.66, n.s.; α1D r=0.62, n=9, n.s.).
Table 2.
Potencies of antagonists against contractions to noradrenaline in rat vas deferens
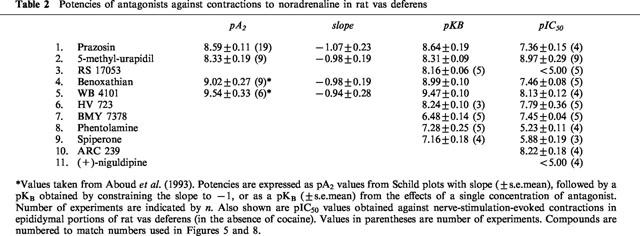
Table 3.
Published antagonists affinities for α1A-, α1B- and α1D-ligand binding sites
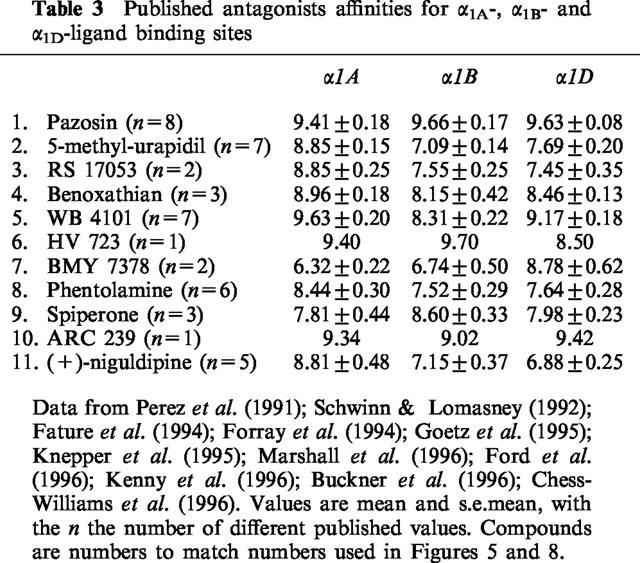
Figure 5.
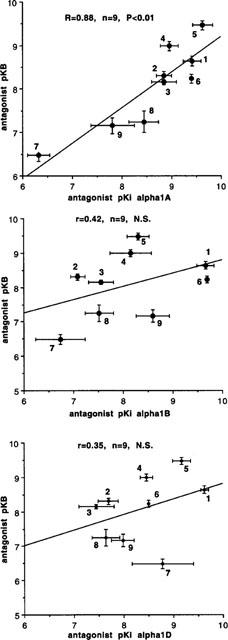
Correlation between published antagonist affinity for α1A-, α1B- and α1D-ligand binding sites (data from Perez et al., 1991; Schwinn & Lomasney, 1992; Faure et al., 1994; Forray et al., 1994; Goetz et al., 1995; Knepper et al., 1995; Marshall et al., 1996; Ford et al., 1996; Kenny et al., 1996) and antagonist postjunctional potency (pKB) against contractions to noradrenaline in whole vas deferens. Numbers indicate compounds as listed in Tables 2 and 3. Vertical and horizontal bars indicate s.e.mean. There was a significant correlation between antagonist postjunctional potency and affinity for α1A-adrenoceptor ligand binding site.
Epididymal portions of rat vas deferens: nerve-mediated responses
In epididymal portions of rat vas deferens, the initial biphasic twitch to a single electrical stimulus was reduced to a monophasic response by nifedipine (10 μM), which eliminates the first (non-noradrenergic) phase, leaving the second (noradrenergic) phase. Under these conditions, the twitch to a single stimulus was 0.80±0.05 g (n=68), and antagonists reduced the size of the twitch in a concentration-dependent manner (Figure 6). Antagonist potency was slightly reduced in the presence of cocaine, but the correlation between antagonist potency with and without cocaine was very close (r=0.98) so that cocaine did not change relative potencies (Figure 7). A total of 11 antagonists were examined (those listed above plus ARC239 and (+)-niguldipine) and potencies (without cocaine) are shown in Table 2. These potencies were correlated with affinities for α1A, α1B and α1D-adrenoceptor ligand binding sites obtained by taking the mean of published values (see Tables 2, 3 and Figure 8). The correlation with ligand binding sites was significant only for the α1D-adrenoceptor subtype (r=0.65, n=11, P<0.05) (Figure 8). One of the published studies examined eight of the antagonists used in this study (all except ARC 239, benoxathian and spiperone; Ford et al., 1996). Correlation of antagonist potency with the ligand binding data of Ford et al. (1996) was not significant for any α1-adrenoceptor subtype (α1A r=0.01, n.s.; α1B r=0.16, n.s.; α1D r=0.53, n.s.).
Figure 6.
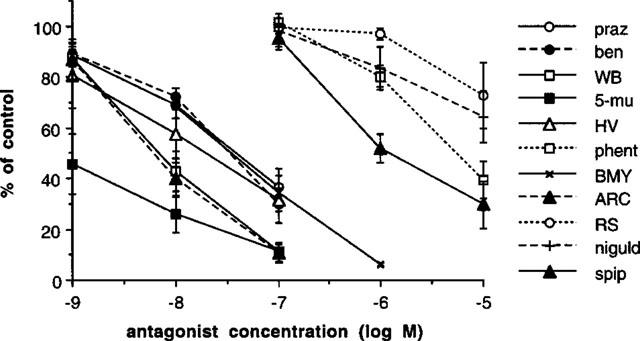
Effects of antagonists on the contractile response of epididymal portions of rat vas deferens in the presence of nifedipine (10 μM) to eliminate non-adrenergic contractions: prazosin (praz), benoxathian (ben), WB 4101 (WB), 5-methyl-urapidil (5-mu), HV 723 (HV), phentolamine (phen), BMY 7378 (BM). ARC 239 (ARC), RS 17053 (RS), (+)-niguldipine (niguld), spiperone (spip). Vertical bars represent s.e.mean from at least three experiments.
Figure 7.
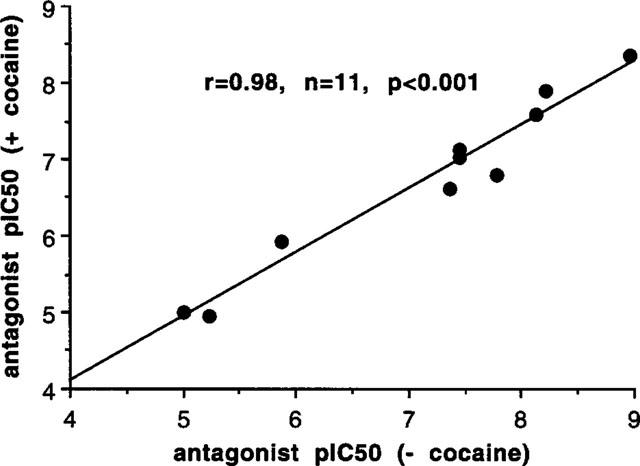
Correlation between antagonist postjunctional potency (pIC50) against stimulation-evoked contractions to a single stimulus in epididymal portions of rat vas deferens in the presence of nifedipine (10 μM) with and without cocaine. There was a very significant correlation between potencies with and without cocaine.
Figure 8.
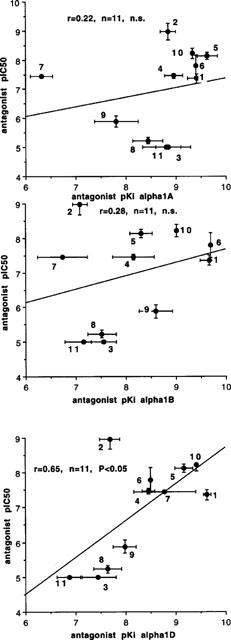
Correlation between published antagonist affinity for α1A-, α1B- and α1D-ligand binding sites (data from Perez et al., 1991; Schwinn & Lomasney, 1992; Faure et al., 1994; Forray et al., 1994; Goetz et al., 1995; Knepper et al., 1995; Marshall et al., 1996; Ford et al., 1996; Kenny et al., 1996) and antagonist postjunctional potency (pIC50) against stimulation-evoked contractions to a single stimulus in epididymal portions of rat vas deferens in the presence of nifedipine (10 μM). Numbers indicate compounds as listed in Tables 2 and 3. Vertical and horizontal bars indicate s.e.mean. There was a significant correlation between antagonist potency and affinity for the α1D-adrenoceptor ligand binding site
Discussion
In this study, we have looked at subtypes of α1-adrenoceptor mediating contractions of the rat vas deferens to exogenous agonists (noradrenaline, methoxamine phenylephrine or A61603) and contractions to endogenous noradrenaline. These will be discussed separately, beginning with contractions to exogenous agonists.
Responses to exogenous agonists
In whole vas deferens, the agonists noradrenaline, methoxamine, phenylephrine and A61603 produced concentration-dependent contractions which consisted of intermittent spikes superimposed on a smaller tonic contraction: all agonists behaved apparently similarly in terms of producing tonic and phasic contractions. Phasic contractions were measured as the total contraction, i.e. the tonic contraction plus the intermittant spike. Agonists tended to be slightly more potent at producing tonic than phasic contractions, but this was simply because the tonic contraction dominated at low agonist concentrations (see Figure 1) and the maximum phasic contraction was significantly greater than the maximum tonic contraction. The potencies of noradrenaline, phenylephrine and A61603 obtained in this study are similar to potencies obtained to these agonists in rat vas deferens by Knepper et al. (1995). In the absence of antagonists, both tonic and phasic contractions to all agonists showed monophasic concentration-response curves.
The antagonist prazosin produced approximately parallel shifts in the potency of all four agonists, and RS 17053 and 5-methyl-urapidil produced parallel shifts in the potency of A61603. However, for the agonists noradrenaline and methoxamine (and to a lesser extent phenylephrine), both RS 17053 and 5-methyl-urapidil tended to produce non-parallel shifts such that the potency of the agonist was shifted more at higher than lower concentrations. However, these differences were relatively small and difficult to quantify.
We chose to compare antagonist potencies against noradrenaline with published ligand binding data for the three α1-adrenoceptor sites; we followed the method of Marshall et al. (1996) in taking the mean of several published ligand binding affinities for each antagonist at each site (see Table 3). We chose nine antagonists; RS 17053, BMY 7378, phentolamine, HV723 and spiperone in addition to the above: BMY 7378 is especially interesting as an α1D-selective antagonist (Goetz et al., 1995) which has higher potency as an antagonist in rat aorta, but lower potency in vas deferens (Deng et al., 1996). Given the evidence for two components to the contractile response, it is perhaps not surprising that some Schild plots to RS 17053 and 5-methyl-urapidil demonstrated non-competitive interactions. The correlation between published data for ligand binding sites and the antagonist potencies obtained in our results would confirm the identification of the predominant receptor mediating contractions of rat vas deferens to noradrenaline as an α1A-adrenoceptor. This agrees with other studies of agonist contractions in rat vas deferens (Teng et al., 1994; Burt et al., 1995). However, it is clear from the above that a second receptor is involved in contractions to noradrenaline, but it appears that none of the antagonists employed is selective for that receptor, although 5-methyl-urapidil and RS 17053 showed low potency at this second site. A component of the response to noradrenaline in rat vas deferens resistant to 5-methyl-urapidil has previously been reported in studies in which the antagonist was given subsequent to the agonist (Bultmann et al., 1994).
High concentration of RS 17053 virtually abolished tonic contractions to noradrenaline, presumably by competitive antagonism, but were less effective against phasic contractions. However, the problem in comparing phasic concentrations with and without high concentrations of antagonist is the phasic contractions have a large tonic component in the absence of antagonist which may obscure the spikes. Both tonic and phasic contractions were abolished by pretreatment with phenoxybenzamine (10 μM), and the alkylating agent CEC (100 μM) did not significantly affect tonic or phasic contractions to noradrenaline, but directly produced phasic contractions (spikes) (Aboud et al., 1993, and unpublished). In the presence of the calcium entry blocker nifedipine, the intermittent spikes produced by noradrenaline were abolished, and a small tonic contraction remained (Aboud et al., 1993). Since the phasic contraction to noradrenaline could not be examined in isolation, the α1-adrenoceptor mediated response to nerve stimulation was examined in more detail.
Responses to endogenous noradrenaline
In rat whole vas deferens, the electrical stimulation-evoked contraction to a single stimulus consists of a biphasic response, the first phase of which is non-adrenoceptor mediated and predominates in prostatic portions, and the second phase of which is α1-adrenoceptor mediated, and predominates in the epididymal portion (Brown et al., 1983). The second α-noradrenergic phase can be examined in isolation in the epididymal portion in the presence of nifedipine which eliminates the second non-noradrenergic response. Antagonist potencies at inhibiting this stimulation-evoked α1-adrenoceptor mediated contraction were obtained both in the absence and presence of cocaine. Cocaine blocks the uptake of noradrenaline back into the nerve terminal, and so prolongs and potentiates contractions: antagonists tended to be less potent at inhibiting contractions in the presence of cocaine, but antagonist potencies with or without cocaine correlated well with each other. Antagonist potencies at inhibiting nerve-evoked contractions in epididymal portions did not correlate with α1A-adrenoceptors, but significantly correlated only with the α1D-adrenoceptor ligand binding site, even though the correlation was relatively low (r=0.65), due largely to one outlier, 5-methyl-urapidil. Interestingly, when 5-methyl-urapidil is eliminated from the correlation, there was a much improved correlation with α1D-adrenoceptors (r=0.90, P<0.001). In the present study, the relatively high potency of BMY 7378 against nerve stimulation-evoked contractions (pIC50 of 7.45) may also suggest an α1D-adrenoceptor. Since the α1D-receptor is reported to be expressed in rat vas deferens (Perez et al., 1991; Rokosh et al., 1994), the above receptor may represent a function α1D-adrenoceptor. Bultmann et al. (1994) investigated nerve mediated responses in rat vas deferens and found that 5-methyl-urapidil was approximately ten times less potent than WB 4101, a potency which would also aid identification with α1D-adrenoceptors. The high potency of 5-methyl-urapidil in our study is difficult to explain, but may suggest other actions of this compound. Because of this, we re-examined the potency of 5-methyl-urapidil: in our experiments the pIC50 varied between 10.0 and 8.00, with a mean of 8.97. This range of values is much greater than for other antagonists (e.g. benoxathian: 7.22–7.70). Although 5-methyl-urapidil is reported to bind with high affinity to 5-HT-1A receptors (Zilles et al., 1991), its inhibitory action in rat vas deferens is not antagonized by the 5-HT-1 receptor antagonist cyanopindolol (authors, unpublished). An α1D-adrenoceptor should show some sensitivity to block by CEC, and the nerve-evoked adrenergic contraction of rat vas deferens is indeed blocked by CEC (Mallard et al., 1992; Aboud et al., 1993), but this action of CEC is at least partly prejunctional (Bultmann & Starke, 1993). An α1D-adrenoceptor has also been reported to mediated nerve-evoked pressor responses in the pithed rat preparation (Castillo et al., 1998).
Finally, how do we explain the contradictory findings in the literature concerning the α1-adrenoceptor subtype mediating contractions of rat vas deferens? Clearly with two subtypes present, different experimental conditions may favour the dominance of one or other of the subtypes so that both α1A- and α1L-adrenoceptors have been reported. Genetic polymorphism of α1A-adrenoceptors does not explain α1L-adrenoceptors, since all variants have been found to have similar pharmacological characteristics (Shibata et al., 1996). The α1A-adrenoceptor, expressed in the CHO-K1 cell line, displayed binding properties of the α1A-adrenoceptor but functionally, in terms of inositol phosphate accumulation, the receptor displayed properties of the α1L-adrenoceptor (Ford et al., 1996). Hence, presumably due to factors determined by the methodology employed, and perhaps due to environmental differences around the receptor, the same receptor can show characteristics of both α1A- and α1L-adrenoceptors: hence α1A- and α1L may be different affinity/conformational states of the α1A-adrenoceptor. The additional presence of an α1D-adrenoceptor complicates matters further.
In conclusion, we have studied contractile responses of rat vas deferens to exogenous agonists and to electrical stimulation. Tonic and phasic contractions of rat vas deferens produced by exogenous agonists are mediated predominantly by α1A-adrenoceptors, although particularly phasic contractions may have a second component. Nerve-evoked contractions seem to be mediated predominated by non-α1A-adrenoceptors, and the receptor involved resembles the α1D-receptor.
Acknowledgments
Supported by The Health Research Board (Ireland), the Irish Heart Foundation and Royal College of Surgeons in Ireland.
Abbreviations
- CEC
chloroethylclonidine
References
- ABOUD R.W., SHAFII M., DOCHERTY J.R. Investigation of the subtypes of α1-adrenoceptor mediating contraction of rat aorta, vas deferens and spleen. Br. J. Pharmacol. 1993;109:80–87. doi: 10.1111/j.1476-5381.1993.tb13534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 1959;14:48–52. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN D.A., DOCHERTY J.R., FRENCH A.M., MACDONALD A., MCGRATH J.C., SCOTT N.C. Separation of noradrenergic and non-adrenergic contractions to field stimulation in the rat vas deferens. Br. J. Pharmacol. 1983;79:379–393. doi: 10.1111/j.1476-5381.1983.tb11010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKNER S.A., OHEIM K.W., MORSE P.A., KNEPPER S.M., HANCOCK A.A. Alpha 1-adrenoceptor-induced contractility in rat aorta is mediated by the alpha 1D subtype. Eur. J. Pharmacol. 1996;297:241–248. doi: 10.1016/0014-2999(95)00755-5. [DOI] [PubMed] [Google Scholar]
- BULTMANN R., KURZ A.K., STARKE K. α1-Adrenoceptors and calcium sources in adrenergic neurogenic contractions of rat vas deferens. Br. J. Pharmacol. 1994;111:151–158. doi: 10.1111/j.1476-5381.1994.tb14037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BULTMANN R., STARKE K. Chloroethylclonidine: An irreversible agonist at prejunctional α2-adrenoceptors in rat vas deferens. Br. J. Pharmacol. 1993;108:336–341. doi: 10.1111/j.1476-5381.1993.tb12806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURT R.P., CHAPPLE C.R., MARSHALL I. Evidence for a functional α1A- (α1C-) adrenoceptor mediating contraction of the rat epididymal vas deferens and an α1B-adrenoceptor mediating contraction of the rat spleen. Br. J. Pharmacol. 1995;115:467–475. doi: 10.1111/j.1476-5381.1995.tb16356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BYLUND D.B., EILENBERG D.C., HEIBLE J.P., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., MOLINOFF P.B., RUFFOLO R.R., TRENDELENBURG U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- CASTILLO E.F., LOPEZ R.M., RODRIGUEZ-SILVERIO J., BOBADILLA R.A., CASTILLO C. α1D-Adrenoceptors contribute to the neurogenic vasopressor response in pithed rats. Fund. Clin. Pharmacol. 1998;12:584–589. doi: 10.1111/j.1472-8206.1998.tb00990.x. [DOI] [PubMed] [Google Scholar]
- CHESS-WILLIAMS R., CHAPPLE C.R., VERFURTH F., NOBLE A.J., COULDWELL C.J., MICHEL M.C. The effects of SB 216469, an antagonist which discriminates between the α1A-adrenoceptor and the human prostate α1-adrenoceptor. Br. J. Pharmacol. 1996;119:1093–1100. doi: 10.1111/j.1476-5381.1996.tb16009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COTECCHIA S., SCHWINN D.A., RANDALL R.R., LEFKOWITZ R.J., CARON M.G., KOBILKA B.K. Molecular cloning and expression of the cDNA for the hamster α1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 1988;85:7159–7163. doi: 10.1073/pnas.85.19.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENG X.F., CHEMTOB S., VARMA D.R. Characterization of α1D-adrenoceptor subtype in rat myocardium, aorta and other tissues. Br. J. Pharmacol. 1996;119:269–276. doi: 10.1111/j.1476-5381.1996.tb15981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOCHERTY J.R. Alpha-1 adrenoceptor subclassification in vascular smooth muscle: a premature proposal. Trends Pharmacol. Sci. 1987;8:123–124. [Google Scholar]
- DOCHERTY J.R. The pharmacology of α1- and α2-adrenoceptors: Evidence for and against a further subdivision. Pharmacol. Ther. 1989;44:241–284. doi: 10.1016/0163-7258(89)90067-3. [DOI] [PubMed] [Google Scholar]
- DOCHERTY J.R. Subtypes of functional α1- and α2-adrenoceptor adrenoceptors. Eur. J. Pharmacol. 1998;361:1–15. doi: 10.1016/s0014-2999(98)00682-7. [DOI] [PubMed] [Google Scholar]
- FAURE C., PIMOULE C., ARBILLA S., LANGER S., GRAHAM D. Expression of α1-adrenoceptor subtypes in rat tissues: implications for α1-adrenoceptor classification. Eur. J. Pharmacol. 1994;268:141–149. doi: 10.1016/0922-4106(94)90183-x. [DOI] [PubMed] [Google Scholar]
- FLAVAHAN N.A., VANHOUTTE P.M. Alpha-1 adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol. Sci. 1986;7:347–349. [Google Scholar]
- FORD A.P.D.W., ARREDONDO N.F., BLUE D.R., BONHAUS D.W., JASPER J., KAVA M.S., LESNICK J., PISTER J.R., SHIEH I.M., VIMONT R.L., WILLIAMS T.J., MCNEA J.E., STAMEY T.A., CLARKE D.E. RS17053, a selective α1-Adrenoceptor antagonist, displays low affinity for functional α1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol. Pharmacol. 1996;49:209–215. [PubMed] [Google Scholar]
- FORRAY C., BARD J.A., LAZ T.M., WETZEL J.M., CHIU G., SHAPIRO E., TANG R., LEPOR H., HARTIG P.R., WEINSHANK R.L., BRANCHEK T.A., GLUCHOWSKI C. The α1-adrenergic receptor that mediates smooth muscle contraction in human prostate has the pharmacological properties of the cloned human α1C subtype. Mol. Pharmacol. 1994;45:703–708. [PubMed] [Google Scholar]
- GOETZ A.S., KING H.K., WARD S.D.C., TRUE T.A., RIMELE T.J., SAUSSY D.L. BMY 7378 is a selective antagonist of the D subtype of α1-adrenoceptors. Eur. J. Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- HAN C., ABEL P.W., MINNEMAN K.P. α1-Adrenoceptor subtypes linked to different mechanisms for increasing intracellular Ca2+ in smooth muscle. Nature. 1987;329:333–335. doi: 10.1038/329333a0. [DOI] [PubMed] [Google Scholar]
- HANFT G., GROSS G. Subclassification of α1-adrenoceptor recognition sites by urapidil derivatives and other selective antagonists. Br. J. Pharmacol. 1989;97:691–700. doi: 10.1111/j.1476-5381.1989.tb12005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNY B.A., MILLER A.M., WILLIAMSON I.J.R., O'CONNELL J., CHALMERS D.H., NAYLOR A.M. Evaluation of the pharmacological selectivity profile of α1-adrenoceptor antagonists at prostatic α-adrenoceptors: binding, functional and in vivo studies. Br. J. Pharmacol. 1996;118:871–878. doi: 10.1111/j.1476-5381.1996.tb15480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNEPPER S.M., BUCKNER S.A., BRUNE M.E., DEBERNARDIS J.F., MEYER M.D., HANCOCK A.A. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J. Pharmacol. Exp. Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- LOMASNEY J.W., COTECCHIA S., LORENZ W., LEUNG W.-Y., SCHWINN D.A., YANG-FENG T.L., BROWNSTEIN M., LEFKOWITZ R.J., CARON M.G. Molecular cloning and expression of the cDNA for the α1A-adrenergic receptor. J. Biol. Chem. 1991;266:6365–6369. [PubMed] [Google Scholar]
- MALLARD N.J., MARSHALL R.W., SITHERS A.J., SPRIGGS T.L.B. Separation of putative α1A- and α1B-adrenoceptor mediated components in the tension response of the rat vas deferens to electrical field stimulation. Br. J. Pharmacol. 1992;105:727–731. doi: 10.1111/j.1476-5381.1992.tb09046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSHALL I., BURT R.P., GREEN G.M., HUSSAIN M.B., CHAPPLE C.R. Different subtypes of α1A-adrenoceptor mediating contraction of rat epididymal vas deferens, rat hepatic portal vein and human prostate distinguished by the antagonist RS 17053. Br. J. Pharmacol. 1996;119:407–415. doi: 10.1111/j.1476-5381.1996.tb16001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORROW A.L., CREESE I. Characterization of α1-adrenergic subtypes in rat brain: a reevaluation of [3H] WB 4101 and [3H] prazosin binding. Mol. Pharmacol. 1986;29:321–330. [PubMed] [Google Scholar]
- MURAMATSU I., OHMURA T., KIGOSHI S., HASHIMOTO S., OSHITA M. Pharmacological subclassification of α1-adrenoceptors in vascular smooth muscle. Br. J. Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAMATSU I., OSHITA M., OHMURA T., KIGOSHI S., AKINO H., GOBARA M., OKADA K. Pharmacological characterisation of α1-adrenoceptor subtypes in the human prostate: functional and binding studies. Br. J. Urol. 1995;74:572–578. doi: 10.1111/j.1464-410x.1994.tb09186.x. [DOI] [PubMed] [Google Scholar]
- OHMURA T., OSHITA M., KIGOSHI S., MURAMATSU I. Identification of α1-adrenoceptor subtypes in the rat vas deferens: binding and functional studies. Br. J. Pharmacol. 1992;107:697–704. doi: 10.1111/j.1476-5381.1992.tb14509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREZ D.M., PIASCIK M.T., GRAHAM R.M. Solution-phase library screening for the identification of rare clones: Isolation of an α1D-adrenergic receptor cDNA. Mol. Pharmacol. 1991;40:876–883. [PubMed] [Google Scholar]
- ROKOSH D.G., BAILEY B.A., STEWART A.F.R., KARNS L.R., LONG C.S., SIMPSON P.C. Distribution of α1C-adrenergic receptor mRNA in adult rat tissues by RNase protection and comparison with α1B and α1D. Biochem. Biophys. Res. Comm. 1994;200:1177–1184. doi: 10.1006/bbrc.1994.1575. [DOI] [PubMed] [Google Scholar]
- SCHWINN D.A., LOMASNEY J.W. Pharmacologic characterization of cloned α1-adrenoceptor subtypes: selective antagonists suggest the existence of a fourth subtype. Eur. J. Pharmacol. 1992;227:433–436. doi: 10.1016/0922-4106(92)90162-o. [DOI] [PubMed] [Google Scholar]
- SCHWINN D.A., LOMASNEY J.W., LORENZ W., SZKLUT P.J., FREMEAU R.T., YANG-FENG T.L., CARON M.G., LEFKOWITZ R.J., COTECCHIA S. Molecular cloning and expression of the cDNA for a novel α1-adrenergic receptor subtype. J. Biol. Chem. 1990;265:8183–8189. [PubMed] [Google Scholar]
- SHIBATA K., HIRASAWA A., MORIYAMA N., KAWABE K., OGAWA S., TSUJIMOTO G. α1a-Adrenoceptor polymorphism: pharmacological characterization and association with benign prostatic hypertrophy. Br. J. Pharmacol. 1996;118:1403–1408. doi: 10.1111/j.1476-5381.1996.tb15552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TENG C.-M., GUH J.-H., KO F.-N. Functional identification of α1-adrenoceptor subtypes in human prostate: comparison with those in rat vas deferens and spleen. Eur. J. Pharmacol. 1994;265:61–66. doi: 10.1016/0014-2999(94)90223-2. [DOI] [PubMed] [Google Scholar]
- ZILLES K., GROSS G., SCHEICHER A., SCHILDGEN S., BAUER A., BAHRO M., SCHWENDEMANN G., ZECH K., KOLASSA N. Regional and laminar distributions of α1-adrenoceptors and their subtypes in human and rat hippocampus. Neuroscience. 1991;40:307–320. doi: 10.1016/0306-4522(91)90122-5. [DOI] [PubMed] [Google Scholar]