

Abstract
Of the four major phosphodiesterase 4 (PDE4) subtypes, PDE4A, PDE4B and PDE4D are widely expressed in human inflammatory cells, including monocytes and T lymphocytes. We explored the functional role of these subtypes using ten subtype-selective PDE4 inhibitors, each belonging to one of two classes: (i) dual PDE4A/PDE4B inhibitors or (ii) PDE4D inhibitors.
These compounds were evaluated for their ability to inhibit antigen-stimulated T-cell proliferation and bacterial lipopolysaccharide (LPS)-stimulated tumour necrosis factor α (TNFα) release from peripheral blood monocytes.
All compounds inhibited T-cell proliferation in a concentration-dependent manner; with IC50 values distributed over an approximately 50 fold range. These compounds also inhibited TNFα release concentration-dependently, with a wider (∼1000 fold) range of IC50 values.
In both sets of experiments, mean IC50 values were significantly correlated with compound potency against the catalytic activity of recombinant human PDE4A or PDE4B when analysed by either linear regression of log IC50 values or by Spearman's rank-order correlation. The correlation between inhibition of inflammatory cell function and inhibition of recombinant PDE4D catalytic activity was not significant in either analysis.
These results suggest that PDE4A and/or PDE4B may play the major role in regulating these two inflammatory cell functions but do not rule out PDE4D as an important mediator of other activities in mononuclear leukocytes and other immune and inflammatory cells. Much more work is needed to establish the functional roles of the PDE4 subtypes across a broader range of cellular functions and cell types.
Keywords: Cyclic nucleotide phosphodiesterase, monocytes, PDE4, PDE4 subtypes, PDE4 inhibitors, T lymphocytes, T lymphocyte proliferation, tumour necrosis factor α
Introduction
The low Km cyclic AMP-specific phosphodiesterases, or PDE4s, catalyse the hydrolysis of the second messenger cyclic AMP to 5′-AMP, thus terminating its activity. They are distinguished from other cyclic AMP-hydrolysing enzymes not only by their low Km, but also by their insensitivity to Ca2+/calmodulin and cyclic GMP and their susceptibility to inhibition by rolipram (Conti et al., 1991). As the predominant class of PDE enzymes in immune and inflammatory cells (Thompson et al., 1976; Dent et al., 1991; Peachell et al., 1992), in which most of the effects of cyclic AMP are inhibitory in nature (Alvarez et al., 1995), PDE4s play a key role in the regulation of a number of active processes such as cell trafficking, release of inflammatory mediators, and immune cell proliferation. Conversely, PDE4 inhibition, while having little effect on basal cyclic AMP levels, increases both the magnitude and the duration of its elevation following a stimulus (Torphy et al., 1992b; Manning et al., 1996) and is associated with bronchodilation and with inhibition of chemotaxis, cytokine release and immune cell proliferation (Alvarez et al., 1995). This combination of smooth muscle relaxation and anti-inflammatory effects has made PDE4 inhibition an attractive goal of drug development for the treatment of asthma and other inflammatory diseases.
Highly potent and selective PDE4 inhibitors such as rolipram have been available for over a decade and have been shown to be effective in several animal models of pulmonary inflammation (Torphy, 1998). However, the therapeutic promise of these compounds has been tempered by their significant side-effects, particularly nausea and emesis. Thus the broad goal of drug development has been to improve the side-effect profile of PDE4 inhibitors while maintaining or improving efficacy. One strategy that has been pursued with some success is based upon the observation that PDE4 enzymes exist in both low- and high-affinity rolipram-binding conformations (Torphy et al., 1992a; Jacobitz et al., 1996). Although inhibition of the low-affinity rolipram-binding conformation correlates with inhibition of cyclic AMP hydrolysis and with inhibition of several inflammatory cell functions, inhibition of high-affinity rolipram binding appears to correlate with the production of certain side effects (Barnette et al., 1995a,1995b; 1996a,1996b; Souness et al., 1997). Compounds with relative selectivity for the low-affinity form of the enzyme would therefore be expected to display better therapeutic ratios than rolipram, which is selective for the high-affinity conformation (Barnette et al., 1996b; Christensen et al., 1998).
A second promising strategy, just beginning to be exploited, is the development of subtype-selective PDE4 inhibitors. Since the first rat homologues of the Drosophila ‘dunce' gene were cloned and characterized as low Km cyclic AMP-specific PDEs (Davis et al., 1989; Swinnen et al., 1989; Henkel-Tigges & Davis, 1989), the PDE4 family has grown to include four subtypes–PDE4A to PDE4D–each encoded by a distinct gene (Bolger et al., 1993; McLaughlin et al., 1993; Engels et al., 1995; Baecker et al., 1994) and each expressed as at least two N-terminal splice variants, which are believed to play a role in targeting PDE4 isoforms to specific intracellular sites (Shakur et al., 1993; Scotland & Houslay, 1995; Scotland et al., 1998). This multiplicity of differentially expressed and regulated PDE4 enzymes (Manning et al., 1996; Engels et al., 1994; Sette et al., 1994) suggests that compounds with improved side-effect profiles might be developed if the functionally relevant enzyme subtypes can be identified and selectively targeted. Because asthma is primarily an inflammatory disease, and because selective PDE4 inhibitors such as rolipram act mainly as anti-inflammatory agents and less significantly as bronchodilators, many studies of PDE4 subtype distribution have focused upon inflammatory cells (Engels et al., 1994; Essayan et al., 1994; Gantner et al., 1997; 1998; Jiang et al., 1998). These studies suggest that PDE4A, PDE4B or PDE4D, all of which are found to some extent in every inflammatory cell type studied, could be important regulators of inflammatory processes. Only PDE4C, which is present in the lung (Obernolte et al., 1997) but has only rarely and inconsistently been reported in any isolated inflammatory cell type, can be eliminated on the basis of its distribution. However, by screening a large number of PDE4 inhibitors against the recombinant human enzymes, we have been able to identify a few selective subtype inhibitors. These compounds, although they do not distinguish between PDE4A and PDE4B, do display a 4.5–17 fold selectivity for either PDE4A/PDE4B or PDE4D. Using these tools, we can begin to dissect the functional roles of the PDE4 subtypes in regulating diverse cellular processes in various cell types. Here we show, in two different functional cellular measures of inflammatory activity, that the anti-inflammatory potency of these PDE4 inhibitors is correlated with their potency as inhibitors of PDE4A/PDE4B.
Methods
Experimental compounds
Compounds tested were: A, trans-4-cyano-4-[3-(cyclopentyloxy)-4-methoxyphenyl]cyclohexyl-1-amine; B, cis-4-cyano-4-[3-(cyclopentyloxy) -4-methoxyphenyl] cyclohexane-1-carboxylic acid; C, trans-4-cyano-4-(3,4-bisdifluoromethoxyphenyl)cyclohexyl-1-amine; D, trans-4-cyano-4-[3-(cyclopentyloxy)-4-methoxyphenyl]cyclohexane-1-carboxylic acid; E, cis-4-cyano-4 - [3 - (cyclopropylmethoxy)-4 -methoxyphenyl]cyclohexane-1-carboxylic acid; F, cis-4-cyano-4-[(4-difluoromethoxy)-3-(cyclopropylmethoxy)] cyclohexane-1-carboxylic acid; G, trans-4-cyano - 4- [3-(cyclopropylmethoxy)-4-methoxyphenyl]-1-(methoxy)cyclohexane-1-carboxylic acid; H, trans-4-(2-aminopyrimidin-5-ylethynyl) -4- [3- (cyclopentyloxy) -4-methoxyphenyl]cyclohexyl-1-amine; I, trans-4-(3-carboxyphenylethynyl)-4-[3-(cyclopentyloxy)-4-methoxyphenyl]cyclohexyl-1-amine hydrochloride, J, trans - 4 - (4 - carboxyphenylethynyl) - 4 - [3 - (cyclopentyloxy)-4-methoxyphenyl]cyclohexyl-1-amine hydrochloride; and rolipram. All were synthesized in the Department of Medicinal Chemistry, SmithKline Beecham, Upper Merion, U.S.A.
Antigen-stimulated T-cell proliferation
Five PDE4A/B-selective and four PDE4D-selective compounds were tested for their ability to inhibit antigen-stimulated T-cell proliferation. Mixed peripheral blood mononuclear cells were isolated from the heparinized blood of house dust mite-allergic donors by centrifugation (350×g, 30 min) over Ficoll-Hypaque (Histopaque® 1077, Sigma, St. Louis, Missouri, U.S.A). Cells at the interface were harvested, washed twice in antibiotic-supplemented RPMI 1640, resuspended in RPMI 1640 supplemented with 5% human AB serum (Sigma), 2 mM L-glutamine, 5 μg ml−1 penicillin, 5 μg ml−1 streptomycin and 10 μg ml−1 neomycin (HAB medium), and plated at 2×105 cells well−1 into 96-well plates (Corning Costar, Acton, Massachusetts, U.S.A.). PDE4 inhibitors (final concentrations 10−9 to 10−5 M) were added to triplicate wells as 4× stocks in HAB medium/1% DMSO. After a 2-h incubation at 37°C in a humidified 5% CO2 incubator, dust mite extract (Dermatophagoides pteronyssinus, Greer Labs, Lenoir, North Carolina, U.S.A.) was added at a concentration of 1–25 μg ml−1. Plates were returned to the incubator and cells were pulsed with [3H]thymidine (0.5 μCi well−1; New England Nuclear, Boston, Massachusetts, U.S.A.) approximately 72 h later. Cells were harvested after 24 h onto Packard GF/C 96-well DNA-binding filter plates and counted on a TopCount scintillation counter (Packard, Meriden, Connecticut, U.S.A.). Data are expressed as per cent inhibition of [3H]thymidine incorporation as compared to antigen-stimulated vehicle controls.
Antigen-specific T-helper (Th) clones were established and assayed as previously described (Essayan et al., 1997), using 10 μg ml−1 ragweed antigen to stimulate proliferation. PDE4 inhibitors were tested from 0.1 to 100 μM.
Inhibition of LPS-stimulated TNFα release from human monocytes
Five PDE4A/B-selective and five PDE4D-selective compounds were evaluated for their ability to suppress bacterial lipopolysaccharide (LPS)-induced TNFα release from human monocytes. Peripheral blood monocytes from normal donors were enriched to a purity of 78.7±3.9% by density gradient centrifugation over isoosmotic Percoll (Sigma) as previously detailed (Manning et al., 1996), resuspended at a concentration of 106 cells ml−1 in RPMI 1640 supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 5 μg ml−1 penicillin, 5 μg ml−1 streptomycin and 10 μg ml−1 neomycin, and plated into 24-well plates. PDE4 inhibitors, prepared as 40× stocks in 20% DMSO, were added to triplicate wells and incubated for 45 min at 37°C in a humidified 5% CO2 atmosphere, after which 100 ng ml−1 E. coli 055 : B5 LPS (Sigma) was added to each well. Plates were returned to the incubator and monocyte supernatants were harvested 16–18 h later. After brief centrifugation at 1860×g to remove any cells, supernatants were transferred to clean tubes and stored at −30°C for later assay. TNFα was measured by enzyme-linked immunosorbent assay (Predicta human TNFα ELISA kit, Genzyme Corp., Cambridge, Massachusetts, U.S.A.). Assays were read on a Dynatech MR7000 plate reader and analysed using Δsoft software (Biometallics, Inc., Princeton, New Jersey, U.S.A.).
Inhibition of hrPDE4A, hrPDE4B and hrPDE4D
IC50 values of the compounds for inhibition of the catalytic activity of human recombinant (hr)PDE4A, PDE4B and PDE4D were determined by a modification of the method of Davis & Daly, (1979) as previously described (Torphy et al., 1992b), using 1 μM cyclic AMP as the substrate.
Recombinant enzymes
Human recombinant PDE4A (HSPDE4A4B) (Bolger et al., 1993), PDE4B (HSPDE4B2A) (McLaughlin et al., 1993) and PDE4D (HSPDE4D3A) (Baecker et al., 1994) were expressed in the PDE-deficient yeast Saccharomyces cerevisiae strain GL62.
Statistical analysis
Log IC50 values were calculated on the individual concentration-response curves of 2–7 experiments. Spearman's rank-order correlation and simple linear regressions on log IC50 values were performed using Prism v 3.00 (GraphPad™ Software Inc., San Diego, California, U.S.A.).
Results
Inhibition of house dust mite-stimulated T-cell proliferation
As shown in Table 1, the PDE4 inhibitors used in this study can be classified into two broad divisions based upon their subtype selectivity. Compounds A, C, H, I and J are classified as dual PDE4A/B inhibitors based on their similar IC50 values for inhibition of PDE4A and PDE4B catalytic activities and their 6–17 fold selectivity for PDE4A/B over PDE4D. The other five compounds are 5–10 fold selective for PDE4D relative to PDE4A.
Table 1.
−log IC50 values of experimental compounds
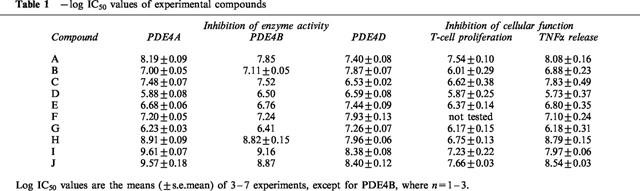
All compounds tested inhibited antigen-stimulated T-cell proliferation in a concentration-dependent manner (Figure 1a), with IC50 values ranging from 22 nM to 1.3 μM. When tested in Spearman's rank-order correlation, the rank order of potency against T-cell proliferation was found to correlate with the rank order of potency against hrPDE4A (ρ=0.867, P=0.005) and hrPDE4B (ρ=0.833, P=0.008) but not with the rank order of potency against hrPDE4D (ρ=0.567, P=0.121). More significantly, standard linear regressions performed on log IC50 values (Figure 2a) also showed positive correlation between inhibition of T-cell proliferation and inhibition of hrPDE4A (r=0.863, P=0.003) and hrPDE4B (r=0.798, P=0.01) catalytic activity but not of hrPDE4D (r=0.553, P=0.123).
Figure 1.
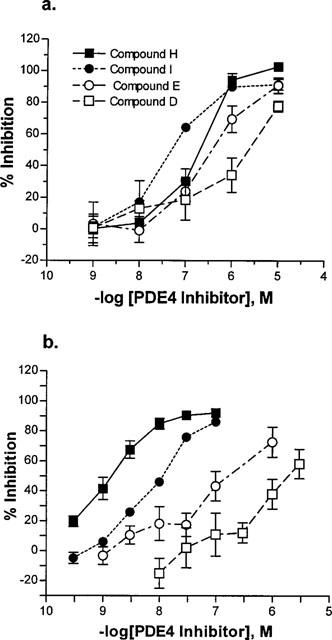
Functional inhibition by four representative selective PDE4 inhibitors. (a) Inhibition of antigen-stimulated T-cell proliferation. (b) Inhibition of LPS-stimulated TNFα release from human peripheral blood monocytes. Closed symbols are PDE4A/B-selective compounds; open symbols are PDE4D-selective compounds. IC50 values are in Table 1.
Figure 2.
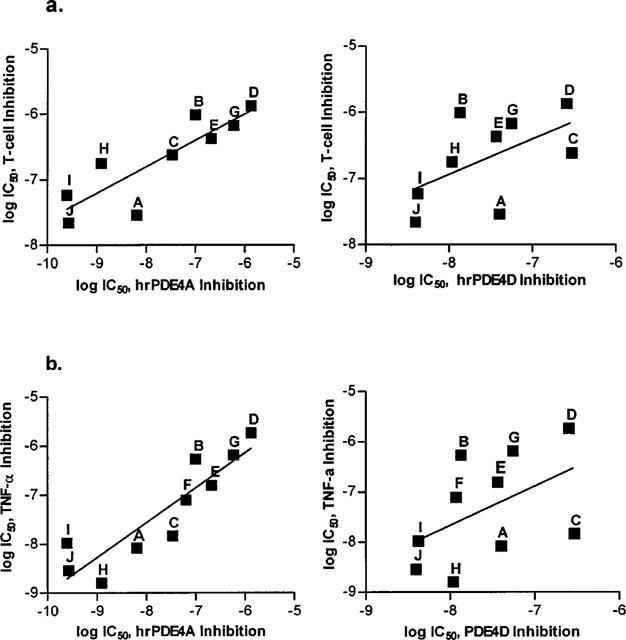
Linear regression analysis. (a) Log IC50 values for inhibition of antigen-stimulated T-cell proliferation vs values for inhibition of hrPDE4A (left, r=0.863, P=0.003) or hrPDE4D (right, r=0.552, P=0.123). (b) Log IC50 values for inhibition of LPS-stimulated TNFα release from human monocytes vs log IC50 values for inhibition of hrPDE4A (left, r=0.899, P=0.0004) or hrPDE4D (right, r=0.483, P=0.157).
Additional experiments were conducted to examine the ability of PDE4 inhibitors to suppress antigen-stimulated proliferation of clonal T helper 1 (Th1) lymphocytes, which do not express PDE4D, or Th2 lymphocytes, which do (Essayan et al., 1997). The compounds evaluated (Table 2) were rolipram, which is not PDE4 subtype-selective, dual PDE4A/B inhibitors A and C and PDE4D inhibitors B and G. Within this restricted selection of compounds, the potency of PDE4 inhibitors against proliferation of both cell types appeared to be correlated with their inhibition of PDE4A/B rather than PDE4D, although significance was not achieved in most analyses owing to the small number of compounds. The rank-order of potency for inhibition of both Th1 and Th2 lymphocyte proliferation was identical to that for inhibition of hrPDE4A (A>C>B>G, ρ=1.0, P=0.083 in each case); linear regressions of the log IC50 values for inhibition of each Th type vs inhibition of hrPDE4A yielded r values of 0.926 and 0.950 for Th1 and Th2, respectively, with P values of 0.074 and 0.050. On the other hand, the rank-order of potency against hrPDE4D (B>A>G>C) was not similar to that for inhibition of Th1 or Th2 proliferation and the linear regressions of log IC50 values produced poor correlation. Spearman's rank correlation yielded ρ=0.00 and P>0.9999 for both Th1 and Th2 lymphocytes. Values from the linear regressions were r=0.196, P=0.804 and r=0.444, P=0.556 for Th1 and Th2, respectively.
Table 2.
Suppression of T-helper subset proliferation by PDE4 inhibitors

Inhibition of TNFα release
All compounds also inhibited LPS-stimulated TNFα release from peripheral blood monocytes in a concentration-dependent manner, with IC50 values distributed from 1.6 nM to 2 μM (Table 1). Again, the rank-order of potency for functional inhibition was significantly correlated with the rank-order for inhibition of hrPDE4A (ρ=0.903, P=0.001) and hrPDE4B (ρ=0.891, P=0.001), while the correlation between the rank-order for inhibition of TNFα release and that for inhibition of hrPDE4D catalytic activity was not statistically significant (ρ=0.600, P=0.073). Consistently with this, there was a significant linear correlation between the IC50 values for inhibition of TNFα release and inhibition of hrPDE4A (r=0.899, P=0.0004) and hrPDE4B (r=0.889, P=0.0006), while there was no statistically significant relationship with inhibition of hrPDE4D (r=0.483, P=0.157).
Discussion
The bewildering size of the PDE4 family, with its four distinct genes and still burgeoning number of splice variants, provides in itself a persuasive argument for the desirability of subtype-selective inhibitors. The widespread distribution of several of the subtypes throughout the body indicates that they may be involved in the regulation of very basic cellular processes, while the simultaneous presence of several subtypes in a single cell, as seen in inflammatory cells, suggests that numerous cyclic AMP-dependent functions may be independently regulated by distinct PDE4 enzymes. Arguably, non-subtype selective PDE4 inhibitors such as rolipram are likely to regulate multiple cellular functions in many tissues, regardless of whether they are relevant to the therapeutic intent. Such considerations give rise to the hope that many of the side-effects of PDE4 inhibitors might be dissociated from their anti-inflammatory properties if the relevant PDE4 subtype(s) could be identified and selectively targeted. Cloning and recombinant expression of the human PDE4 subtypes, coupled with large-scale screening of PDE4 inhibitors, has permitted us to identify several compounds with selectivity for either PDE4A/B or PDE4D. Using these tools, we have shown, in two inflammatory cell types which are recruited into the lung in asthma, that at least two of the anti-inflammatory activities associated with PDE4 inhibition can be attributed to inhibition of PDE4A and/or PDE4B.
The most selective of our compounds exhibited only 17 fold selectivity between PDE4A/B and PDE4D. Thus, we found that each of the functions examined could be almost fully suppressed by any of the compounds tested, regardless of the selectivity of the particular compound. To establish correlation between selectivity and functional inhibition, we chose an experimental approach that has previously been utilized to associate specific functional effects with inhibition of either the low- or high-affinity rolipram-binding conformation of PDE4 (Barnette et al., 1995a,1995b; 1996a; Souness et al., 1997), and which depends only upon the availability of a range of selective compounds of varying potencies. IC50 values of our compounds for PDE4A/B ranged from 0.25 nM to 1.32 μM (<greater>5200 fold), while IC50 values for PDE4D were between 4.0 and 295 nM (74 fold). Not surprisingly, these ranges are compressed in the functional assays, to ∼1000 fold in the TNFα assay and ∼50 fold for inhibition of T-cell proliferation. It is all the more convincing that the linear regression shows a highly significant correlation between the IC50 values for inhibition of PDE4A/B and those for inhibition of T-cell proliferation when the range of values is compressed from 5200 fold to 50.
Our smaller study, comparing the effects of subtype-selective inhibitors on proliferation of Th1 and Th2 clones, shows that the rank order of potency for suppression of the proliferative response to antigen is identical to that for inhibition of PDE4A/B, regardless of the presence or absence of PDE4D in the cell. The results of this study must be interpreted cautiously, in light of its small scope. They do, however, tend to support the findings of the larger study, suggesting that PDE4A and/or PDE4B are the most important subtypes involved in the regulation of lymphocyte proliferation.
In summary, our data show that inhibition of both LPS-stimulated TNFα release from human monocytes and antigen-induced human T-cell proliferation are closely correlated with inhibition of PDE4A and/or PDE4B, suggesting that these particular inflammatory functions may be mediated by the activity of one or both of these PDE4 enzymes. These are, to our knowledge, the first data linking the anti-inflammatory properties of PDE4 inhibitors to inhibition of a subset of PDE4 enzymes. Thus, much work remains to be done to examine the functional roles of the subtypes in other cellular activities and in other inflammatory cell types. Though inhibition of both of the inflammatory activities examined in this study is correlated with inhibition of PDE4A/B and not apparently with inhibition of PDE4D, we cannot conclude that PDE4D is irrelevant to inflammatory activity, even in mononuclear cells. Association of specific cellular functions with single PDE4 subtypes will await the development of inhibitors which can differentiate PDE4A from PDE4B. Meanwhile, additional work can be done with the tools already available to analyse which subtypes play relevant roles in the regulation of other inflammatory activities, both in mononuclear cells and in other inflammatory cells.
Acknowledgments
We thank Joseph M. Karpinski, Aimee Guider, Cornelia J. Forster and Paul E. Bender for compound synthesis, and Christine M. Braun for technical assistance.
Abbreviations
- DMSO
dimethyl sulphoxidel
- ELISA
enzyme-linked immunosorbent assay
- LPS
lipopolysaccharide
- TNFα
tumor necrosis factor α
References
- ALVAREZ R., SETTE C., YANG D., EGLEN R.M., WILHELM R., SHELTON E.R., CONTI M. Activation and selective inhibition of a cyclic AMP-specific phosphodiesterase, PDE-4D3. Mol. Pharmacol. 1995;48:616–622. [PubMed] [Google Scholar]
- BAECKER P.A., OBERNOLTE R., BACH C., YEE C., SHELTON E.R. Isolation of a cDNA encoding a human rolipram-sensitive cyclic AMP phosphodiesterase (PDE IVD) Gene. 1994;138:253–256. doi: 10.1016/0378-1119(94)90818-4. [DOI] [PubMed] [Google Scholar]
- BARNETTE M.S., BARTUS J.O., BURMAN M., CHRISTENSEN S.B., CIESLINSKI L.B., ESSER K.M., PRABHAKAR U.S., RUSH J.A., TORPHY T.J. Association of the anti-inflammatory activity of phosphodiesterase 4 (PDE4) inhibitors with either inhibition of PDE4 catalytic activity or competition for [3H]rolipram binding. Biochem. Pharmacol. 1996a;51:949–956. doi: 10.1016/0006-2952(96)00053-6. [DOI] [PubMed] [Google Scholar]
- BARNETTE M.S., CHRISTENSEN S.B., UNDERWOOD D.C., TORPHY T.J. Phosphodiesterase 4: biological underpinnings for the design of improved inhibitors. Pharmacol. Rev. Comm. 1996b;8:65–73. [Google Scholar]
- BARNETTE M.S., GROUS M., CIESLINSKI L.B., BURMAN M., CHRISTENSEN S.B., TORPHY T.J. Inhibitors of phosphodiesterase IV (PDE IV) increase acid secretion in rabbit isolated gastric glands: correlation between function and interaction with a high-affinity rolipram binding site. J. Pharmacol. Exp. Ther. 1995a;273:1396–1402. [PubMed] [Google Scholar]
- BARNETTE M.S., MANNING C.D., CIESLINSKI L.B., BURMAN M., CHRISTENSEN S.B., TORPHY T.J. The ability of phosphodiesterase IV inhibitors to suppress superoxide production in guinea pig eosinophils is correlated with inhibition of phosphodiesterase IV catalytic activity. J. Pharmacol. Exp. Ther. 1995b;273:674–679. [PubMed] [Google Scholar]
- BOLGER G., MICHAELI T., MARTINS T., StJOHN T., STEINER B., RODGERS L., RIGGS M., WIGLER M., FERGUSON K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol. Cell. Biol. 1993;13:6558–6571. doi: 10.1128/mcb.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRISTENSEN S.B., GUIDER A.M., FORSTER C.J., GLEASON J.G., BENDER P.E., KARPINSKI J.M., DeWOLF W.E.J., BARNETTE M.S., UNDERWOOD D.C., GRISWOLD D.E., CIESLINSKI L.B., BURMAN M., BOCHNOWICZ S., OSBORN R.R., MANNING C.D., GROUS M., HILLEGAS L.M., BARTUS J.O., RYAN M.D., EGGLESTON D.S., HALTIWANGER R.C., TORPHY T.J. 1,4,-Cyclohexane carboxylates: potent and selective inhibitors of phosphodiesterase 4 for the treatment of asthma. J. Med. Chem. 1998;41:821–835. doi: 10.1021/jm970090r. [DOI] [PubMed] [Google Scholar]
- CONTI M., JIN S.L.-C., MONACO L., REPASKE D.R., SWINNEN J.V. Hormonal regulation of cyclic nucleotide phosphodiesterases. Endocr. Rev. 1991;12:218–234. doi: 10.1210/edrv-12-3-218. [DOI] [PubMed] [Google Scholar]
- DAVIS C.W., DALY J.W. A simple direct assay of 3′,5′-cyclic nucleotide phosphodiesterase activity based on the use of polyacrylamide-boronate affinity gel chromatography. J. Cyclic Nucleotide Res. 1979;5:65–74. [PubMed] [Google Scholar]
- DAVIS R.L., TAKAYASU H., EBERWINE M., MYRES J. Cloning and characterization of mammalian homologs of the Drosophila dunce+ gene. Proc. Natl. Acad. Sci. U.S.A. 1989;86:3604–3608. doi: 10.1073/pnas.86.10.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENT G., GIEMBYCZ M.A., RABE K.F., BARNES P.J. Inhibition of eosinophil cyclic nucleotide PDE activity and opsonised zymosan-stimulated respiratory burst by ‘type-IV'-selective PDE inhibitors. Br. J. Pharmacol. 1991;103:1339–1346. doi: 10.1111/j.1476-5381.1991.tb09790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGELS P., FICHTEL K., LUBBERT H. Expression and regulation of human and rat phosphodiesterase type IV isogenes. FEBS Lett. 1994;350:291–295. doi: 10.1016/0014-5793(94)00788-8. [DOI] [PubMed] [Google Scholar]
- ENGELS P., SULLIVAN M., MULLER T., LUBBERT H. Molecular cloning and functional expression in yeast of a human cAMP-specific phosphodiesterase subtype (PDE IV-C) FEBS Lett. 1995;358:305–310. doi: 10.1016/0014-5793(94)01460-i. [DOI] [PubMed] [Google Scholar]
- ESSAYAN D.M., HUANG S.-K., UNDEM B.J., KAGEY-SOBOTKA A., LICHTENSTEIN L.M. Modulation of antigen- and mitogen-induced proliferative responses of peripheral blood mononuclear cells by nonselective and isozyme selective cyclic nucleotide phosphodiesterase inhibitors. J. Immunol. 1994;153:3408–3416. [PubMed] [Google Scholar]
- ESSAYAN D.M., KAGEY-SOBOTKA S., LICHTENSTEIN L.M., HUANG S.-K. Differential regulation of human antigen-specific Th1 and Th2 lymphocyte responses by isozyme selective cyclic nucleotide phosphodiesterase inhibitors. J. Pharmacol. Exp. Ther. 1997;282:505–512. [PubMed] [Google Scholar]
- GANTNER F., GÖTZ C., GEKELER V., SCHUDT C., WENDEL A., HATZELMANN A. Phosphodiesterase profile of human B lymphocytes from normal and atopic donors and the effects of PDE inhibition on B cell proliferation. Br. J. Pharmacol. 1998;123:1031–1038. doi: 10.1038/sj.bjp.0701688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANTNER F., TENOR H., GEKELER V., SCHUDT C., WENDEL A., HATZELMANN A. Phosphodiesterase profiles of highly purified human peripheral blood leukocyte populations from normal and atopic individuals: a comparative study. J. Allergy Clin. Immunol. 1997;100:527–535. doi: 10.1016/s0091-6749(97)70146-5. [DOI] [PubMed] [Google Scholar]
- HENKEL-TIGGES J., DAVIS R.L. Rat homologs of the Drosophila dunce gene code for cyclic AMP phosphodiesterase sensitive to rolipram and Ro20-1724. Mol. Pharmacol. 1989;37:7–10. [PubMed] [Google Scholar]
- JACOBITZ S., MCLAUGHLIN M.M., LIVI G.P., BURMAN M., TORPHY T.J. Mapping the functional domains of human recombinant phosphodiesterase 4A: structural requirements for catalytic activity and rolipram binding. Mol. Pharmacol. 1996;50:891–899. [PubMed] [Google Scholar]
- JIANG X., PASKIND M., WELTZIEN R., EPSTEIN P.M. Expression and regulation of mRNA for distinct isoforms of cAMP-specific PDE-4 in mitogen-stimulated and leukemic human lymphocytes. Cell. Biochem. Biophys. 1998;28:135–160. doi: 10.1007/BF02737809. [DOI] [PubMed] [Google Scholar]
- MANNING C.D., MCLAUGHLIN M.M., LIVI G.P., CIESLINSKI L.B., TORPHY T.J., BARNETTE M.S. Prolonged beta adrenoceptor stimulation up-regulates cAMP phosphodiesterase activity in human monocytes by increasing mRNA and protein for phosphodiesterases 4A and 4B. J. Pharmacol. Exp. Ther. 1996;276:810–818. [PubMed] [Google Scholar]
- MCLAUGHLIN M.M., CIESLINSKI L.B., BURMAN M., TORPHY T.J., LIVI G.P. A low Km, rolipram-sensitive, cAMP-specific phosphodiesterase from human brain: cloning and expression of cDNA, biochemical characterization of recombinant protein, and tissue distribution of mRNA. J. Biol. Chem. 1993;268:6470–6476. [PubMed] [Google Scholar]
- OBERNOLTE R., RATZLIFF J., BAECKER P.A., DANIELS D.V., ZUPPAN P., JARNAGIN K., SHELTON E.R. Multiple splice variants of phosphodiesterase PDE4C cloned from human lung and testis. Biochim. Biophys. Acta. 1997;1353:287–297. doi: 10.1016/s0167-4781(97)00080-8. [DOI] [PubMed] [Google Scholar]
- PEACHELL P.T., UNDEM B.J., SCHLEIMER R.P., MACGLASHAN D.W., Jr, LICHTENSTEIN L.M., CIESLINSKI L.B., TORPHY T.J. Preliminary identification and role of phosphodiesterase isozymes in human basophils. J. Immunol. 1992;148:2503–2510. [PubMed] [Google Scholar]
- SCOTLAND G., BEARD M., ERDOGAN S., HUSTON E., MCCALLUM F., MACKENZIE S.J., PEDEN A.H., POOLEY L., RENA N.G., ROSS A.H., YARWOOD S.J., HOUSLAY M.D. Intracellular compartmentalization of PDE4 cyclic AMP-specific phosphodiesterases. Methods. 1998;14:65–79. doi: 10.1006/meth.1997.0566. [DOI] [PubMed] [Google Scholar]
- SCOTLAND G., HOUSLAY M.D. Chimeric constructs show that the unique N-terminal domain of the cyclic AMP phosphodiesterase RD1 (RNPDE4A1A; rPDE-IVA1) can confer membrane association upon the normally cytosolic protein chloramphenicol acetyltransferase. Biochem. J. 1995;308:673–681. doi: 10.1042/bj3080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETTE C., VICINI E., CONTI M. The rat PDE3/IVd phosphodiesterase gene codes for multiple proteins differentially activated by cAMP-dependent protein kinase. J. Biol. Chem. 1994;269:18271–18274. [PubMed] [Google Scholar]
- SHAKUR Y., PRYDE J.G., HOUSLAY M.D. Engineered deletion of the unique N-terminal domain of the cyclic AMP-specific phosphodiesterase RD1 prevents plasma membrane association and the attainment of enhanced thermostability without altering its sensitivity to inhibition by rolipram. Biochem. J. 1993;292:677–686. doi: 10.1042/bj2920677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOUNESS J.E., HOUGHTON C., SARDAR N., WITHNALL M.T. Evidence that cyclic AMP phosphodiesterase inhibitors suppress interleukin-2 release from murine splenocytes by interacting with a ‘low-affinity' phosphodiesterase 4 conformer. Br. J. Pharmacol. 1997;121:743–750. doi: 10.1038/sj.bjp.0701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SWINNEN J.V., JOSEPH D.R., CONTI M. Molecular cloning of rat homologues of the Drosophila melanogaster dunce cAMP phosphodiesterase: evidence for a family of genes. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5325–5329. doi: 10.1073/pnas.86.14.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON W.J., ROSS C.P., PLEDGER W.J., STRADA S., BANNER R.L., HERSH E.M. Cyclic adenosine 3′,5′-monophosphate phosphodiesterase: distinct forms in human lymphocytes and monocytes. J. Biol. Chem. 1976;251:4922–4929. [PubMed] [Google Scholar]
- TORPHY T.J. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am. J. Respir. Crit. Care Med. 1998;157:351–370. doi: 10.1164/ajrccm.157.2.9708012. [DOI] [PubMed] [Google Scholar]
- TORPHY T.J., STADEL J.M., BURMAN M., CIESLINSKI L.B., MCLAUGHLIN M.M., WHITE J.F., LIVI G.P. Coexpression of human cAMP-specific phosphodiesterase activity and high affinity rolipram binding in yeast. J. Biol. Chem. 1992a;267:1798–1804. [PubMed] [Google Scholar]
- TORPHY T.J., ZHOU H.-L., CIESLINSKI L.B. Stimulation of beta adrenoceptors in a human monocyte cell line (U937) up-regulates cyclic AMP-specific phosphodiesterase activity. J. Pharmacol. Exp. Ther. 1992b;263:1195–1205. [PubMed] [Google Scholar]