

Abstract
The present study was designed to investigate the mechanism of the antiplatelet action of the anaesthetic propofol in vitro.
Human whole blood was incubated with different concentrations of propofol and its solvent Intralipid®. We determined, platelet aggregometry in whole blood, platelet-enriched plasma (PRP), PRP plus red blood cells (RBC), and PRP plus leucocytes (LC); platelet production of thromboxane B2 (TxB2), ATP release by platelet dense granules, adenosine uptake by RBC, intraplatelet levels of cyclic adenosine monophosphate (cyclic AMP) and cyclic guanosine monophosphate (cyclic GMP), and LC production of nitric oxide (NO).
Propofol-induced inhibition of platelet aggregation was greater in whole blood (IC50 80–136 μM) than in PRP (IC50>600 μM), except when aggregation was induced by arachidonic acid, in which case the antiaggregatory effect of the anaesthetic was similar in both media (IC50 72–85 μM). Inhibition of platelet aggregation correlated significantly with inhibition of TxB2 synthesis (r2=0.83). Propofol also inhibited granular ATP release; this effect was greatest in whole blood.
The presence of RBC or LC increased the antiaggregatory effect of propofol, mainly when collagen was used as aggregating agent. Intralipid inhibited the uptake of adenosine by RBC, however this effect probably does not contribute significantly to its antiaggregatory effect.
The anaesthetic potentiated the NO-cyclic GMP pathway, mainly by increasing the synthesis of NO by LC. Intralipid had no effect on the NO-cyclic GMP pathway in the LC-platelet interaction.
Propofol inhibited platelet aggregation in human whole blood, possibly through the sum of the effects of Intralipid on the platelet-RBC interaction and the increased synthesis of NO by LC in the platelet-LC interaction.
Keywords: Nitric oxide, leucocytes, platelets, propofol, thromboxane
Introduction
Propofol (2,6-diisopropylphenol) is an intravenous anaesthetic that is widely used in daily clinical practice in surgical patients. Structurally, it is similar to α-tocopherol and acetylsalicylic acid. Its antioxidant effect has been ascribed to its chemical similarity with α-tocopherol (Musacchio et al., 1991; De La Cruz et al., 1998).
In studies motivated by the chemical similarity of propofol to acetylsalicylic acid, we recently showed that this anaesthetic inhibits platelet aggregation in human whole blood in vitro (De La Cruz et al., 1997) in a range of concentrations similar to those found in human plasma after intravenous administration (Gepts et al., 1987). Moreover, this effect has been demonstrated after intravenous infusion of propofol to surgical patients (Aoki et al., 1998; Dogan et al., 1999). An important finding in this work was that propofol showed a weak antiaggregatory effect in platelet-rich plasma (PRP), which was not seen in whole blood except when aggregation was induced with arachidonic acid; in this case the antiaggregatory effect was similar in PRP and whole blood.
Petros et al. (1993) showed that propofol increased the concentration of cyclic guanosine monophosphate (cyclic GMP) in cultured endothelial cells through a nitric oxide-dependent mechanism. Nitric oxide is known to inhibit platelet function (Radomski et al., 1987), and acetylsalicylic acid increases the antiaggregatory effect of nitric oxide produced by neutrophils (Lopez-Farre et al., 1995). On the basis of these two findings, we designed the present in vitro study to analyse the mechanism underlying the antiaggregatory effect of propofol. We compared the effect in whole blood and PRP, and also investigated the influence of propofol on the leucocyte nitric oxide-platelet cyclic GMP axis.
Methods
Whole blood for this in vitro study was obtained from healthy men (mean age 35.3±1.6 years, range 18–42 years) who had not taken any medication for up to at least 15 days previously. Each subject gave his informed consent to participate in the study. Each blood sample was separated into the several fractions (in addition to whole blood). Platelet-rich plasma was obtained by centrifugation of whole blood at 180×g for 10 min at 20°C. Red blood cells (RBC) from the pellet produced by centrifugation for PRP were washed in 0.1 M phosphate-buffered saline at pH 7.4 and centrifuged at 1000×g for 10 min at 20°C. Leucocytes were obtained by centrifugation of whole blood on a Ficoll gradient (Boyum, 1968) and washing in phosphate-buffer saline (pH 7.4), followed by centrifugation at 1000×g for 15 min at 20°C.
Propofol (Diprivan®, Zeneca Pharma S.A., Madrid, Spain) was incubated at different concentrations depending on the experiment, using the same solvent as is used for clinical anaesthesia (10% Intralipid®, Kabi-Pharmacia, Barcelona, Spain). This solvent consists of purified soy oil (50 g l−1), egg phospholipids (6 g l−1), glycerol anhydride (11 g l−1) and water. In each experiment we analysed the possible effect of the solvent on the effect under study.
Eight to ten samples were run in each of the experiments detailed below.
Platelet aggregometry
Platelet aggregation was measured with the electric impedance method described by Cardinal & Flower (1980) with a Chrono-Log 540 aggregometer (Chrono-Log Corp., Haverton, PA, U.S.A.), using ADP (2.5 μM), collagen (1 μg ml−1) and arachidonic acid (0.4 mM) (Menarini Diagnóstico, Barcelona, Spain) to induce aggregation. Propofol and Intralipid were incubated at 37°C for 10 min before the aggregation inducer was added, and aggregation was recorded for 10 min. Maximum intensity of aggregation was quantified as the maximum change in electron impedance in samples without the drug and in samples incubated with Intralipid or a given concentration of propofol.
The aggregating agent concentrations were chosen according to previous experiments in which EC50 values were as follows: 2.1×0.37 μM for ADP (n=10), 1.1±0.14 μg ml−1 for collagen (n=10) and 0.36±0.05 mM for arachidonic acid (n=10).
Aggregation was tested in whole blood, PRP, and PRP with RBC or leucocytes at a proportion similar to that found in cell counts of whole blood (4.6±0.3×1012 cells l−1 for RBC, 6.8±0.3×109 cells l−1 for leucocytes): 4.5×1012 RBC l−1 and 7×109 leucocytes l−1.
In separate experiments we studied the influence of the adenosine-cyclic AMP and nitric oxide (NO)-cyclic GMP pathways on the antiaggregatory effect of propofol. Aggregation was measured in the presence of biochemical modifiers of these two pathways. The adenosine-cyclic AMP pathway was stimulated with adenosine and with isobutylmethylxanthine (IBMX) (an inhibitor of type III phosphodiesterase); as inhibitors we used adenosine deaminase (ADA) (an enzyme that breaks down adenosine) and dideoxyadenosine (DDA) (an adenylcyclase inhibitor). The NO-cyclic GMP pathway was stimulated with L-arginine (an NO precursor) and zaprinast (an inhibitor of type V phosphodiesterase); as inhibitors we used L-NG nitro arginine methyl ester (L-NAME) (an inhibitor of NO synthase), haemoglobin (a NO scavenger) and methylene blue (a guanyl cyclase inhibitor).
Granular reactivity
We measured granular reactivity as ATP release by platelet dense granules according to the method of Feinman et al. (1985), using the luciferin-luciferase reaction (Izasa Diagnóstica, Barcelona, Spain). Luminescence was detected in a Chrono-Log lumi-aggregometer. Granular release was stimulated with 2 μg ml−1 collagen, and ATP release was recorded and compared with the luminescence emitted by a commercial ATP standard (Chrono-Log Corp.).
Uptake of [14C]adenosine by red blood cells
The percent uptake of 1 μmol l−1 [14C]-adenosine by a concentrate of 2×1011 RBC l−1 was determined after incubation for different periods at 37°C, according to the method described by Roos and Pleger (1972). Briefly, after incubation of the labelled RBC with propofol, the sample was centrifuged at 10,000×g for 3 min, and radioactivity was determined in the supernatant and the pellet. These values were used to calculate the percentage of adenosine in the RBC.
Platelet production of thromboxane B2
Samples of PRP were stimulated with 100 μmol l−1 arachidonic acid for 5 min at 37°C, than 100 μM indomethacin was added to stop the reaction. The sample was centrifuged at 10,000×g and the amount of thromboxane B2 (TxB2) in the supernatant was determined with an enzymeimmunoassay (Biotrak® RPN 220 Amersham International plc, Little Chalfont, Buckinghamshire, U.K.). The sensitivity of this method is 3.6 pg ml−1, the within-assay variability for duplicate determinations was 2.5% and the between-assay variability was 9.9%. The cross reactivity of this method is as follows: 100% thromboxane B2, 60.5% 2,3-dinor-thromboxane B2, <0.4% 2,3-dinor-6-keto-PGF1α, <0.01% 6,15-diketo-13,14-dihydro-PGF1α, 0.1% 11-dehydro-thromboxane B2, <0.01% 6-keto- PGE2, <0.011% PGD2, 0.18% PGD2, <0.01% PGE2, 1.6% PGF1α, 0.06% PGF2α, <0.01% arachidonic acid.
Intraplatelet level of cyclic AMP
After PRP was obtained, the platelets were washed in a medium consisting of NaCl (0.113 M), NaH2PO4 (0.024 M), KH2PO4 (0.004 M), glucose (0.005 M), apyrase (0.05 g l−1) and prostaglandin E1 (5×10−9 M). The sample was centrifuged at 1000×g for 15 min at 4°C, and the resulting pellet was resuspended in a solution consisting of NaCl (0.134 M), NaH2PO4 (0.036 M), NaHCO3 (0.012 M), CaCl2 (0.0029 M), MgCl2 (0.001 M), HEPES (0.005 M), glucose (0.005 M), apyrase (0.05 g l−1) and bovine albumin (0.5 g l−1). The number of platelets was adjusted to 2.5×1011 cells l−1, and the sample was divided into aliquots to which 10 μM IBMX, 1 μM prostaglandin E1 and the drug or solvent were added. PGE1 was incubated in order to increase basal production of cyclic AMP from washed platelets, because platelet samples showed low cyclic AMP levels. After incubation for 5 min at 37°C the reaction was stopped by adding 1 N HCl; the sample was then centrifuged at 10,000×g for 3 min and the supernatant was neutralized with NaOH. The amount of cyclic AMP was quantified with a commercial enzyme immunoassay (Amersham International plc). The sensitivity of this method is 38.4 pg ml−1, the within-assay variability for duplicate determinations was 7.9% and the between-assay variability was 11.0%. The cross reactivity of this method is as follows: 100% cyclic AMP, 0.014% cyclic IMP, 0.0007% cyclic GMP, 0.034% cyclic CMP, 0.0036% cyclic TMP, 0.006% AMP, 0.0002% ADP, 0.00015% ATP, <0.00015% EDTA, 0.00015% theophylline, 0.0071% Iso-butyl-methyl-xanthine.
Intraplatelet level of cyclic GMP
To measure intraplatelet levels of cyclic GMP the samples were prepared using a method similar to that described above for cyclic AMP, except that 100 μM zaprinast was used in place of IBMX, and 10 μM sodium nitroprusside was used instead of prostaglandin E1. Sodium nitroprosside was incubated in order to increase the basal production of cyclic GMP from washed platelet since most platelet samples showed low cyclic GMP levels. The amount of cyclic GMP was quantified with a commercial enzymeimmunoassay (Amersham International plc). The sensitivity of this method is 161 pg ml−1, the within-assay variability for duplicate determinations was 7.5% and the between-assay variability was 12.2%. The cross reactivity of this method is as follows: 100% cyclic GMP, <0.0005% cyclic AMP, <10−6% AMP, <10−6% ADP, <10−6% ATP, <0.0005% GMP, <0.00025% GDP, <0.00025% GTP.
Leucocyte production of nitric oxide
Nitric oxide production was induced with the calcium A 23187 ionophore (1 μM) and quantified in concentrates of polymorphonuclear leucocytes obtained by centrifugation of whole blood on a Ficoll gradient. Two analytical methods were used. In one, NO production was determined electrochemically (Shibuki, 1992) with an NO detection electrode coupled to an ISO–NO detector (World Precision Instruments, Aston, Stevenage, Hertsfordshire, U.K.). In the other method, nitrites were quantified in the same leucocyte samples as in the first set of analyses, using the Griess reagent reaction (1% sulfanilamide and 0.1% naphthylethylenediamine in 4% phosphoric acid). Nitrite levels were determined spectrophotometrically at a wavelength of 546 nm. Mean leucocyte number in whole blood were 6.8×0.3×109 cells l−1, and polymorphonuclear leucocytes in samples for NO detection were 3.5±0.2×109 cells l−1 (3.8±0.4×109 cells l−1 in whole blood). We found a significant direct linear correlation between NO production (measured electrochemically) and nitrite levels in the supernatant (r2=0.988). In five control samples, we determined total amount of nitrite (NO−2) and nitrates (NO−3), using the same experimental protocol in presence of 10 UI ml−1 Aspergillus nitrate reductase; the ratio NO−2/NO−2+NO−3 was 1/46 in these five experiments, and also maintain the linear correlation with the electrochemically detection of NO.
Statistical analysis
All data in the text, tables and figures are the mean±s.e.mean of all values for each experiment. The results were tested with one-way analysis of variance (ANOVA) followed by Bonferroni correction. All analyses were carried out with version 8.0 of the SPSS program for Windows '95. The minimum value used to establish statistical significance was P<0.05.
Results
Effect of propofol on platelet aggregation
In whole blood, propofol inhibited platelet aggregation induced with ADP, collagen or arachidonic acid in a dose-dependent manner. The effect was significantly greater in whole blood than in PRP, except when aggregation was induced with arachidonic acid; in this case the antiaggregatory effect was similar in both media. Figure 1 shows the aggregation inhibition curves in whole blood and PRP. Intralipid (10 μl from a concentration of 100 mg ml−1) did not modify maximum aggregation in whole blood or PRP, and maximum inhibition in samples of whole blood induced with arachidonic acid was 4.5±0.8% (n=6).
Figure 1.
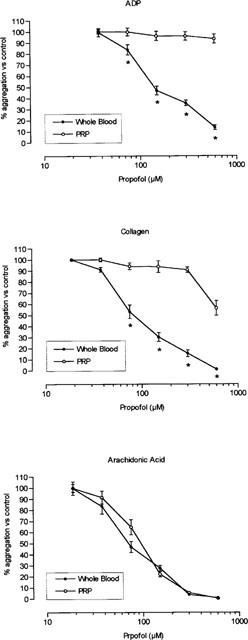
Inhibition of platelet aggregation in whole blood and platelet rich plasma (PRP). Aggregation was induced with 2.5 μM ADP, 1 μg ml−1 collagen, or 0.4 mM arachidonic acid in samples incubated with increasing concentrations of propofol. *P<0.05 in comparison with the value in platelet rich plasma. Each point represents mean±s.e.mean of ten experiments.
Influence of red blood cells and leucocytes
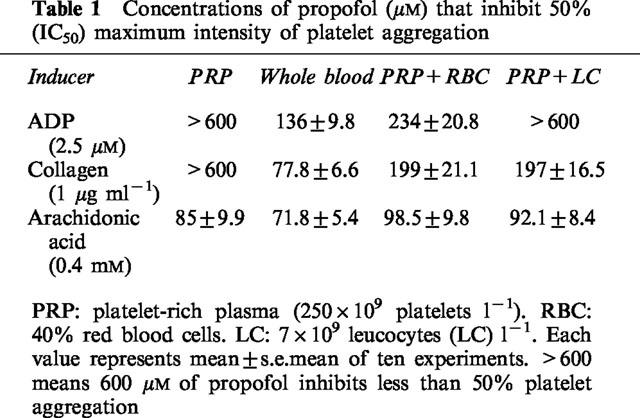
In samples of PRP with RBC or leucocytes at a proportion similar to that found in cell counts of whole blood (263±15.6×109 platelets l−1, 4.6±0.3×1012 RBC l−1, 6.8±0.3×109 leucocytes l−1), the antiaggregatory effect of propofol was greater than in PRP alone. The effect in PRP, RBC or leucocytes separately was less than the effect recorded in the corresponding mixture with whole blood. Table 1 shows the concentrations of propofol that inhibited 50% of the maximum aggregation (IC50) in whole blood, PRP, PRP+RBC and PRP+leucocytes. Intralipid did not modify platelet aggregation in any of the samples analysed.
Table 1.
Concentrations of propofol (μM) that inhibit 50% (IC50) maximum intensity of platelet aggregation
Effect of propofol on platelet granule ATP release
Propofol inhibited granular ATP release in a dose-dependent manner; this effect was greater in whole blood than in PRP (Figure 2). Intralipid did not significantly modify ATP release by platelet granules (maximum inhibition 3.9±0.6% in whole blood, n=7).
Figure 2.
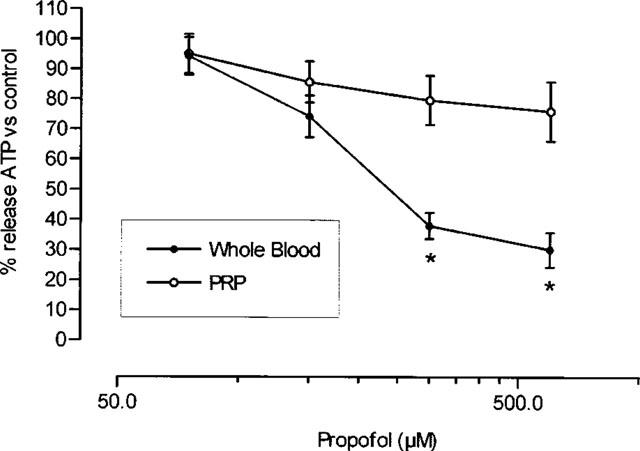
Per cent granular ATP release in whole blood and platelet rich plasma (PRP). Aggregation was induced with 2 μg ml−1 collagen in samples incubated with increasing concentrations of propofol. *P<0.05 in comparison with the value in platelet rich plasma. Each point represents mean±s.e.mean of ten experiments.
Effect of propofol on platelet synthesis of thromboxane B2
Propofol inhibited arachidonic acid-induced production of TxB2 by platelets in a dose-dependent way (Figure 3), with an IC50 of 101±8.53 μM. Intralipid (10 μl from a concentration of 100 mg ml−1) had no significant effect on TxB2 synthesis (maximum inhibition 6.9±1.1%, n=6).
Figure 3.
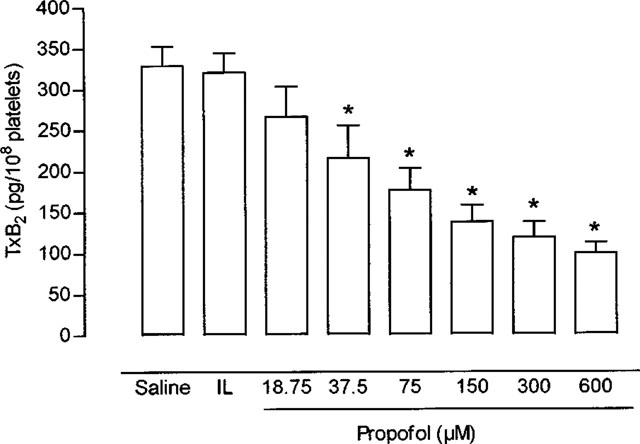
Platelet production of thromboxane B2 in platelet rich plasma. Aggregation was induced with 100 μM arachidonic acid in samples incubated with saline, Intralipid (IL), or increasing concentrations of propofol. *P<0.05 in comparison with the value in saline. Each bar represents mean±s.e.mean of ten experiments.
Effect of propofol on [14C]-adenosine uptake by RBC
Propofol inhibited the uptake of [14C]-adenosine by RBC (Figure 4), in both the rapid phase and the stable uptake phase. Mean uptake time in the rapid phase increased from 1.13±0.09 min in control samples (n=10) to 8.14±0.85 min in samples incubated with 75 μM of propofol (n=10), 9.93±0.71 min in samples incubated with a concentration of 150 μM propofol (n=10), 14.12±0.98 min in samples incubated with 300 μM (n=10), and 14.25±1.08 min with 600 μM propofol (n=10).
Figure 4.
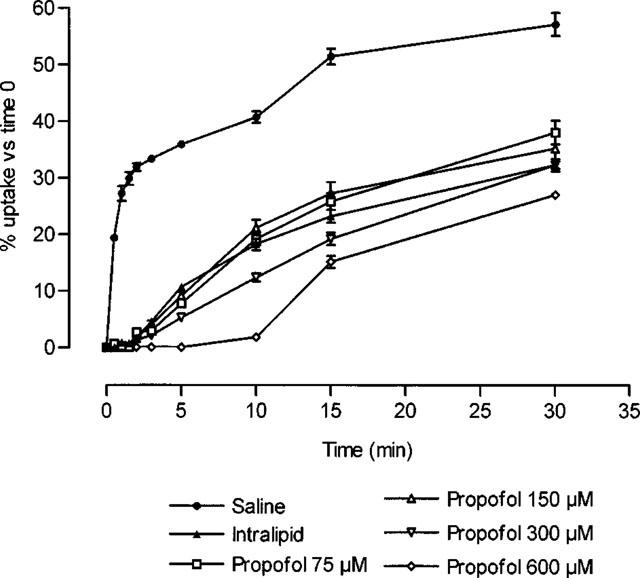
Uptake of [14C]-adenosine by red blood cells (2×1011 cells l−1) after different periods of incubation with saline, Intralipid, or propofol. All values for Intralipid or propofol were significantly different (P<0.05) from those obtained in experiments with saline. Each point represents mean±s.e.mean of ten experiments.
Intralipid significantly inhibited [14C]-adenosine uptake by RBC (Figure 4), with a mean uptake time of 8.24±0.38 min during the rapid phase.
Modification of the adenosine-cyclic AMP axis
The antiaggregatory effect of propofol in PRP was not significantly modified by incubation with inducers or inhibitors of the adenosine-cyclic AMP pathway. Intraplatelet levels of cyclic AMP were not modified by increasing concentrations of propofol or by incubation with Intralipid (maximum increase of 2.1±0.5%, with 600 μM propofol, respect to saline).
Modification of the NO-cyclic GMP axis
These experiments were undertaken in samples of PRP+leucocytes in order to study the effect on NO synthesis. The antiaggregatory effect of propofol was enhanced by incubation together with stimulants of the NO-cyclic GMP pathway (L-arginine and zaprinast), and was inhibited in the presence of blockers of this pathway (L-NAME, haemoglobin and methylene blue) (Figure 5).
Figure 5.
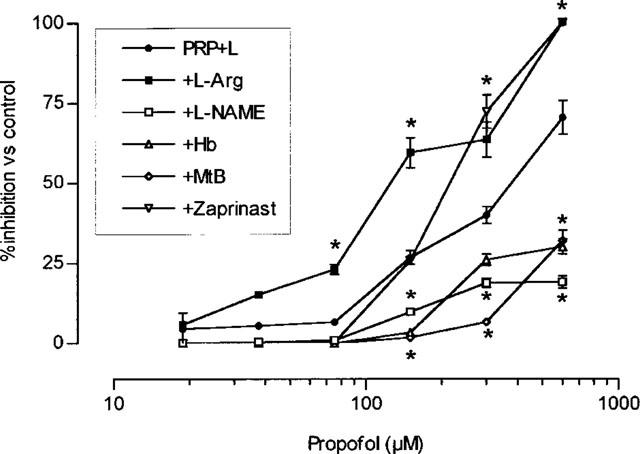
Per cent platelet aggregation in platelet rich plasma plus 7−109 leucocytes l−1 (PRP+L). Aggregation was induced with 1 μg ml−1 collagen in the presence of increasing concentrations of propofol plus saline, 100 μM L-arginine (L-Arg), 300 μM L-NG nitro arginine methyl ester (L-NAME), 10 μM haemoglobin (Hb), 100 μM zaprinast or 10 μM methylene blue (MtB). *P<0.05 in comparison with the value in propofol plus saline. Each point represents mean±s.e.mean of ten experiments.
Intraplatelet levels of cyclic GMP were not affected by propofol or Intralipid in samples of platelets alone, but were increased in a concentration-dependent manner in samples of platelets+leucocytes (Figure 6).
Figure 6.
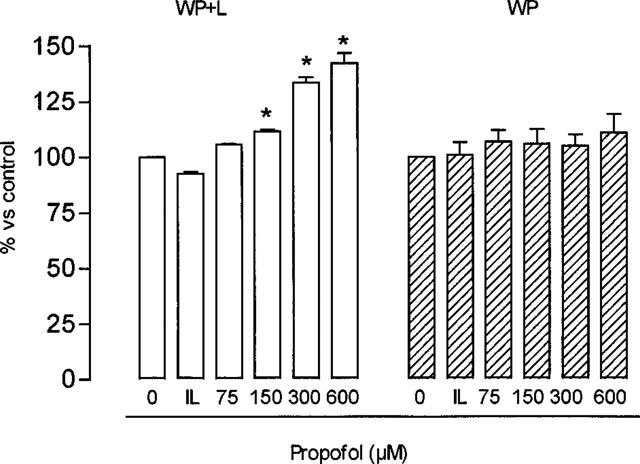
Levels of cyclic GMP in samples of washed platelets (WP) or WP+7×109 leucocytes l−1 (WP+L) incubated with saline (0), Intralipid (IL) or different concentrations of propofol. Cyclic GMP in non-incubated WP was 0.85±0.07 μM and in WP+L was 1.78±0.2 μM. *P<0.05 in comparison with the value in saline. Each bar represents mean±s.e.mean of ten experiments.
Effect of propofol on leucocyte production of nitric oxide
Leucocyte production of NO (measured by electrochemical method and nitrite production) increased in a concentration-dependent manner in the presence of propofol, and showed no significant change in response to Intralipid (Figure 7). Incubating the leucocytes with L-NAME prevented the propofol-induced increase in NO production (90.3±4.6% inhibition respect to each value obtained in propofol-incubated samples).
Figure 7.
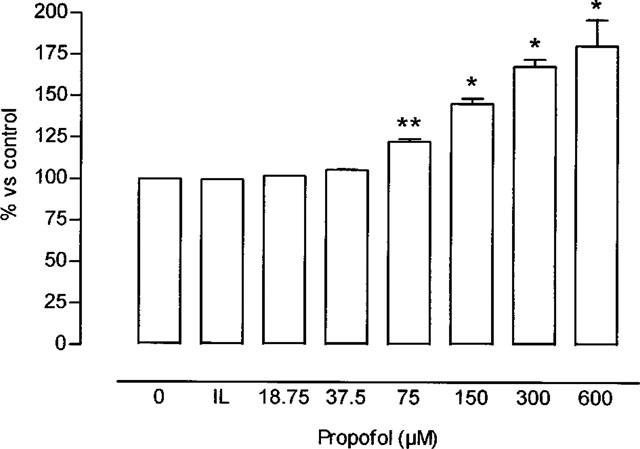
Percentage increase in leucocyte production of nitric oxide, measured by electrochemical method (see Methods) (saline= 1.55±0.09 nmol/106 leucocytes–100%-corresponding to nitrite concentrations of 1.07±0.09 nmol/106 leucocytes) in the presence of Intralipid (IL) or different concentrations of propofol. *P<0.05 in comparison with the value in IL. Each bar represents mean±s.e.mean of ten experiments. Where no error bars are shown error lies within dimensions of histogram.
Discussion
Our results show that propofol inhibits platelet aggregation in whole blood, but was much attenuated in PRP. This finding corroborates earlier results (De La Cruz et al., 1997; Aoki et al., 1998), and is in partial agreement with the findings of Türkan et al. (1996), who found no antiaggregatory effect of propofol in PRP, like our results in the same type of samples.
A further result that supports the findings obtained with aggregometry is the inhibition by propofol of platelet granule ATP release, a phenomenon that defines the process of irreversible platelet aggregation (Feinman et al., 1985). The fact that the effect of propofol was greatest in whole blood is entirely compatible with the greater antiaggregatory effect of propofol in this medium.
An important aspect of our results is the similarity in the antiaggregatory effect seen in whole blood and PRP when aggregation was induced with arachidonic acid. This behaviour is similar to the antiaggregatory effect of acetylsalicylic acid in vitro and ex vivo in whole blood and PRP (De La Cruz et al., 1986; 1987). The principal mechanism of antiaggregatory action of acetylsalicylic acid is the inhibition of thromboxane A2 synthesis, through an irreversible blockade of platelet cyclo-oxygenase. For this reason we determined the production of its metabolite, TxB2, in the presence of propofol. The results show a concentration-dependent inhibition of TxB2, an effect which showed a relation with the inhibition of aggregation induced by arachidonic acid: although the amount of arachidonic acid used to stimulate platelet aggregation and thromboxane production were different (400 and 100 μM, respectively), we found a statistically significant direct linear correlation (r2=0.83, P<0.00001) between these tests, which indicates an influence of propofol in this pathway.
One of the mechanisms by which propofol inhibits platelet aggregation may therefore be by inhibiting platelet thromboxane synthesis. These effects might have been caused by the solvent Intralipid; however, we used controls with the solvent in all experiments, and in no case was the effect on platelet aggregation significant, which is in accordance to the in vivo results of Aoki et al. (1998).
The range of concentrations in which the antiaggregatory effect of propofol was seen in whole blood corresponds to the plasma concentrations reached in humans when propofol is given as an anaesthetic. After an intravenous bolus dose of 2 mg kg−1 (the dose used to induce anaesthesia), plasma concentrations of 40–50 μmol l−1 are achieved (Cockshott, 1985). When these concentrations are interpolated on the curve of inhibition of platelet aggregation in whole blood, the resulting percentage values of inhibition are 10–12% with ADP induction, 20–25% with collagen induction, and 25–30% with arachidonic acid induction.
On the other hand, this study was designed to answer was the reason for the differences in results in whole blood and PRP, with particular attention to possible platelet-RBC and platelet-leucocyte interactions. Our results show that both RBC and leucocytes increase the antiaggregatory effect of propofol, as we reported previously (De La Cruz et al., 1997): when platelets were incubated with increasing concentrations of RBC or with leucocytes, the antiaggregatory effect increased proportionally with the increase in the percentage of RBC or leucocytes in the sample.
With respect to the interaction between RBC and platelets, drugs such as dipyridamole has been found to inhibit platelet aggregation in whole blood by inhibiting the uptake of adenosine by RBC (Roos & Pleger, 1972; Gresele et al., 1983). We found that Intralipid inhibited adenosine uptake by RBC, and that propofol showed an inhibitory effect only at concentrations above those used in human clinical practice. Intralipid is a complex of lipids that may affect the phospholipid composition of the plasma membrane of erythrocytes, and thus interfere with the mechanism of active uptake of adenosine by these cells. However, although propofol/Intralipid can inhibit adenosine uptake by RBC, this effect probably does not contribute significantly to its antiaggregatory effect.
Adenosine is an endogenous substance that acts as an antiaggregatory by binding to membrane receptors and stimulating adenyl cyclase, thus increasing intraplatelet levels of cyclic AMP. We used aggregometry to investigate whether propofol affected this mechanism in functional terms. Our results show that the neither the anaesthetic nor its solvent modified the adenosine-cyclic AMP axis, and that neither substance affected intraplatelet levels of cyclic AMP. Therefore, the solvent Intralipid may have been responsible for at least part of the increased antiaggregatory effect of propofol in the platelet-RBC interaction in comparison with the antiaggregatory effect seen in PRP alone.
In platelet-leucocyte experiments, one of the mechanisms proposed to explain the antiaggregatory effect of propofol is leucocyte production of NO, which stimulates guanyl cyclase activity in platelets and increases the level of cyclic GMP, thus inhibiting aggregation (Radomski et al., 1987). In studies of the NO-cyclic GMP axis, we found that the antiaggregatory effect of propofol increased when NO synthesis was stimulated or cyclic GMP content was increased, whereas the antiaggregatory effect was diminished in the presence of inhibitors of this pathway. Moreover, propofol did not change cyclic GMP levels in PRP, but did increase cyclic GMP levels in samples that contained leucocytes. This supports the hypothesis that propofol stimulates NO synthesis by leucocytes. We found that propofol increased calcium-dependent NO production by leucocytes in a dose-dependent manner, a finding that confirms results reported by Petros et al. (1993), who found that this anaesthetic increased the level of cyclic GMP in cultured endothelial cells.
The two mechanisms that the present findings illustrate (inhibition of thromboxane synthesis and stimulation of NO production) have also been invoked to explain the antiplatelet effect of acetylsalicylic acid (Lopez-Farre et al., 1995; Vane, 1971). This coincidence may be related to the similarities in the chemical structure of these two drugs. In addition, drugs that inhibit thromboxane synthesis in the platelet are also known to show a greater antiaggregatory effect in whole blood (De La Cruz et al., 1986; 1987; 1988); as in our experiments with propofol, this effect is further increased in the presence of leucocytes in the sample.
In conclusion, propofol inhibits platelet aggregation in whole blood, possibly in combination with the effects of the solvent, Intralipid. This effect can be traceable to interference with the leucocyte-platelet interaction, increasing leucocyte NO production, and inhibition of platelet thromboxane synthesis.
Acknowledgments
This study was partially supported by Zeneca Farma S.A., Madrid, Spain. We thank A Pino for his excellent technical assistance and Karen Shashok for translating the original manuscript into English.
Abbreviations
- LC
leucocytes
- NO
nitric oxide
- PRP
platelet-rich plasma
- RBC
red blood cells
- Tx
thromboxane
References
- AOKI H., MIZOBE T., NOZUCHI S., HIRAMATSU N. In vivo and in vitro studies of the inhibitory effect of propofol on human platelet aggregation. Anesthesiology. 1998;88:362–370. doi: 10.1097/00000542-199802000-00015. [DOI] [PubMed] [Google Scholar]
- BOYUM A. Isolation of mononuclear cells and granulocytes from human blood. Scand. J. Clin. Invest. 1968;21 suppl 97:77–89. [PubMed] [Google Scholar]
- CARDINAL D.C., FLOWER R.J. The electronic aggregometer, a novel device for assesing platelet behaviour in blood. J. Pharm. Meth. 1980;3:135–158. doi: 10.1016/0160-5402(80)90024-8. [DOI] [PubMed] [Google Scholar]
- COCKSHOTT ID. Propofol pharmacokinetocs and metabolism-an overview. Postgrad. Med. J. 1985;61 suppl 3:45–50. [PubMed] [Google Scholar]
- DE LA CRUZ J.P., BELLIDO I., CAMARA S., MARTOS F., SANCHEZ DE LA CUESTA F. Effects of acetylsalicylic acid on platelet aggregation in male and female whole blood: an in vitro study. Scand. J. Haematol. 1986;36:394–397. doi: 10.1111/j.1600-0609.1986.tb01755.x. [DOI] [PubMed] [Google Scholar]
- DE LA CRUZ J.P., CAMARA S., BELLIDO I., SÁNCHEZ DE LA CUESTA F. Platelet aggregation in human whole blood after chronic administration of aspirin. Thromb. Res. 1987;46:133–140. doi: 10.1016/0049-3848(87)90213-1. [DOI] [PubMed] [Google Scholar]
- DE LA CRUZ J.P., CARMONA J.A., PAEZ M.V., BLANCO E., SANCHEZ DE LA CUESTA F. Propofol inhibits in vitro platelet aggregation in human whole blood. Anesth. Analg. 1997;84:919–921. doi: 10.1097/00000539-199704000-00040. [DOI] [PubMed] [Google Scholar]
- DE LA CRUZ J.P., PAVIA J., GARCIA ARNES J., SANCHEZ DE LA CUESTA F. Effects of triflusal and aspirin on platelet aggregation in whole blood and platelet-rich plasma of diabetic patients. Eur. J. Haematol. 1988;40:232–236. doi: 10.1111/j.1600-0609.1988.tb00829.x. [DOI] [PubMed] [Google Scholar]
- DE LA CRUZ J.P., VILLALOBOS M.A., SEDEÑO G., SANCHEZ DE LA CUESTA F. Effect of propofol on oxidative stress in an in vitro model of anoxia-reoxygenation in the rat brain. Brain Res. 1998;800:136–144. doi: 10.1016/s0006-8993(98)00516-2. [DOI] [PubMed] [Google Scholar]
- DOGAN I.V., OVALI E., ETI Z., YAYCI A., GÖGUSF Y. The in vitro effect of isofluorane, sevofluorane, and propofol on platelet aggregation. Anesth. Analg. 1999;88:432–436. doi: 10.1097/00000539-199902000-00039. [DOI] [PubMed] [Google Scholar]
- FEINMAN R., DETWILER T., INGERMANWOJENSKI C.The lumi-aggregometer as a research and clinical tool The Platelet: Physiology and Pharmacology 1985London: Academic Press; 429–440.ed. Longenecker, G. pp [Google Scholar]
- GEPTS E., CAMU F., COCKSHOTT I.D., DOUGLAS E.J. Disposition of propofol administered as constant rate intravenous infusion in humans. Anaesth. Analg. 1987;66:1256–1263. [PubMed] [Google Scholar]
- GRESELE P., ZOJA C., DECKMYN H., ARNOUT J., VERMYLEN J., VERSTRAETE M. Dipyridamole inhibits platelet aggregation in whole blood. Thromb. Haemostas. 1983;50:852–860. [PubMed] [Google Scholar]
- LOPEZ-FARRE A., CARAMELO C., ESTEBAN A., ALBEROLA M.L., MILLAS I., MONTON M, CASADO S. Effects of aspirin on platelet-neutrophil interactions. Role of nitric oxide and endothelin-1. Circulation. 1995;91:2080–2088. doi: 10.1161/01.cir.91.7.2080. [DOI] [PubMed] [Google Scholar]
- MUSACCHIO E., RIZZOLI V., BIANCHI M., BINDOLI A., GALZIGNA L. Antioxidant action of propofol on liver microsomes, mitochondria and brain synaptosomes in the rat. Pharmacol. Toxicol. 1991;69:75–77. doi: 10.1111/j.1600-0773.1991.tb00414.x. [DOI] [PubMed] [Google Scholar]
- PETROS A.J., BOGLE R.G., PEARSON A.D. Propofol stimulates nitric oxide release from cultured porcine aortic endothelial cells. Br. J. Pharmacol. 1993;109:6–7. doi: 10.1111/j.1476-5381.1993.tb13523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADOMSKI M.W., PALMER R.M.J., MONCADA S. Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide, and prostacyclin in platelets. Br. J. Pharmacol. 1987;92:181–187. doi: 10.1111/j.1476-5381.1987.tb11310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROOS H., PLEGER K. Kinetics of adenosine uptake, by erithrocytes, and the influence of dipyridamole. Mol. Pharmacol. 1972;8:417–425. [PubMed] [Google Scholar]
- SHIBUKI K. Detection of nitric oxide by an electrochemical microprobe. A companion to methods in neurosciences. Neuroprotocols. 1992;1:151–157. [Google Scholar]
- TÚRKAN H., SÜER A.H., BEYAN C., YALÇIN A. Propofol does not affect platelet aggregation. Eur. J. Anaesth. 1996;13:408–409. [PubMed] [Google Scholar]
- VANE Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]