

Abstract
Chronic inflammatory diseases have been shown to be associated with NF-κB activation and impaired apoptosis of immune cells. The aim of the present study was to investigate if sulfasalazine and its colonic metabolites 5-aminosalicylic acid (5ASA) and sulfapyridine affect NF-κB/Rel activation and viability of T-lymphocytes.
Sulfasalazine inhibits NF-κB/Rel activation in the murine T-lymphocyte cell line RBL5 using electrophoretic mobility shift assays. In transfection assays sulfasalazine treatment for 4 h inhibits κB-dependent transcription with an IC50 value of ∼0.625 mM.
Higher doses or prolonged treatment result in cell death of T-lymphocytes in a dose- and time-dependent manner. Cell death is caused by apoptosis as judged by DNA fragmentation, annexin V and Apo 2.7 staining. Induction of apoptosis is a fast event with 50% apoptotic cells after a 4 h incubation with 2.5 mM sulfasalazine. The ED50 value for apoptosis induction after 24 h treatment was ∼0.625 mM.
In contrast, 5ASA and sulfapyridine neither inhibit NF-κB/Rel activation nor induce apoptosis in T-lymphocytes at doses up to 5.0 mM.
These results demonstrate that sulfasalazine, but not 5ASA or sulfapyridine, strongly inhibits NF-κB activation and potently induces apoptosis in T-lymphocytes. Inhibition of NF-κB/Rel activation and subsequent clearance of activated T-lymphocytes by apoptosis might thus explain the beneficial effects of sulfasalazine in the treatment of chronic inflammatory disorders.
Keywords: Sulfasalazine, 5ASA, sulfapyridine, NF-κB, chronic inflammatory diseases, inflammatory bowel disease, T-lymphocytes
Introduction
Homeostasis of the immune system is maintained as a function of proliferation and cell death. One mechanism known to remove potentially autoreactive cells of the immune system is programmed cell death or apoptosis (Cohen & Duke, 1992). This phenomenon has been most extensively studied in the T-lymphoid lineage, where autoreactive thymocytes undergo T-cell-receptor-mediated apoptosis upon encountering ‘self' antigens in the thymus (MacDonald & Lees, 1990). In addition, apoptosis also plays an important role in limiting the persistence of activated T-cells (Strasser et al., 1995), B-cells (McDonnell et al., 1989; Nisitani et al., 1993), granulocytes (Haslett, 1992), and macrophages (Munn et al., 1995). Chronic inflammatory diseases such as inflammatory bowel disease (IBD) and rheumatoid arthritis have been demonstrated to be associated with abnormally low levels of apoptosis of inflammatory cells (Ina et al., 1995; Boirivant et al., 1999; Salmon et al., 1997; Mountz et al., 1994). It is easily conceivable that failure of programmed cell death is a major mechanism by which initially mild diseases progress to more severe chronic inflammatory stages. Therefore, pharmacological agents inducing apoptosis can be effective in the treatment of autoimmune disorders (Cohen & Duke, 1992; Krammer et al., 1994; Thompson, 1995; Anderson, 1996).
Recently, we have shown that the anti-inflammatory drug sulfasalazine interferes with NF-κB/Rel activation (Wahl et al., 1998). NF-κB/Rel proteins comprise a family of inducible transcription factors which are pivotal regulators of the immune and acute phase response. NF-κB/Rel activates transcription of proinflammatory cytokines such as tumour necrosis factor α (TNFα), interleukin-1 (IL-1), IL-6, IL-8 or adhesion molecules as ICAM (reviewed in Ghosh et al., 1998), which have been shown to be upregulated in the inflamed mucosa of patients with ulcerative colitis or Crohn's disease (Pang et al., 1994; Fiocchi, 1998). Some of these gene products, TNFα and IL-1, are able to induce NF-κB by themselves, leading to a positive autocrine loop and amplified cellular activation and, implicating NF-κB as a central player in the pathogenesis of chronic inflammation (Ghosh et al., 1998; Neurath et al., 1998; Rogler et al., 1998).
There is increasing evidence that inhibition of NF-κB/Rel induces apoptosis, most likely by preventing the expression of anti-apoptotic genes (Ghosh et al., 1998; Wang et al., 1998). Here, we report that sulfasalazine inhibits NF-κB/Rel activation and subsequently induces apoptosis in a murine T-cell line. In contrast, 5ASA or sulfapyridine did not affect NF-κB activation or T-cell viability.
Methods
Cell culture and treatments
From inbred C57BL/6J (B6) mice derived RBL5 T-lymphoma cells (a generous gift from J. Reimann, Ulm) were grown under standard conditions in RPMI 1640 Click's medium (Seromed, Biochrom, Berlin, Germany) supplemented with 5% FCS. Recombinant TNFα was purchased from Sigma (Deisenhofen, Germany). Sulfasalazine, 5ASA, and sulfapyridine (Sigma, Deisenhofen) were freshly dissolved in culture media and added to the cultures at the indicated concentrations and for the indicated time periods.
Nuclear protein extractions and electrophoretic mobility shift assay (EMSA)
Nuclear protein extracts were prepared as described earlier (Schmid et al., 1991). Protein concentrations were determined by the method of Bradford (BioRad, München, Germany). EMSAs were performed as described (Liptay et al., 1994). The following oligonucleotides were used: IgκB, containing the κB site of the mouse immunoglobulin κ-light chain (5′-AG CT TG GG GA CT TT CC AC TA GT AC G-3′), AP1 (5′-A G C T T A C T C A G T A C T A G T A C G-3′), OCT-1 (5′-CTAGTGTCGAATGCAAATCACTAGAAT-3′). Of each probe the sense strand is listed. For competition experiments, unlabelled oligonucleotides were added at a 20 fold molar excess.
DNA transfection and luciferase assay
RBL5 cells were transfected by electroporation as described elsewhere (Oswald et al., 1998). As reporter plasmid the 3xIgκBluc plasmid (Wahl et al., 1998) was used. 16 to 20 h after transfection, cells were treated with control medium, or the indicated concentrations of serially diluted sulfasalazine, 5ASA, or sulfapyridine for 1 h, followed by stimulation with TNFα (150 U ml−1) for 3 additional h. Cells were harvested and assayed for luciferase activity according to the manufacturer's instructions (Promega, Mannheim, Germany) using a Berthold luminometer. All transfections were normalized to the level of total cellular protein (Bradford assay, BioRad). Luciferase activity from transfected cells treated with TNFα and control medium was used as a reference and set to 100% luciferase activity. Experiments were done in duplicate. Data from four independent transfections are presented as mean and standard error of the mean (s.e.mean).
Cell viability and apoptosis assays
MTS-conversion assay were performed according to the manufacturer's instructions (Promega, Mannheim, Germany). 2.5×104 cells were seeded in 100 μl in 96-well plates and treated as indicated. The A490 was measured in an ELISA plate reader. For controls, medium with supplements and without cells was similarly incubated to zero the reaction.
Cell viability was determined by forward/side scatter analysis in a fluorescence-activated cell sorter (FACSCalibur, Becton Dickinson, Heidelberg, Germany) as described (Carbonari et al., 1994; Friesen et al., 1996).
For determination of hypodiploid DNA, cells were harvested by centrifugation and washed in phosphate buffered saline. Cell pellets were resuspended in 1 ml cold 70% ethanol by vigorous vortexing and fixed at 4°C overnight. For staining, cells were centrifuged and resuspended in 1 ml of propidium iodide staining solution containing 50 μg ml−1 propidium iodide, and 100 U ml−1 RNase A in phosphate buffered saline. Cells were incubated in the dark for 30 min at room temperature and kept at 4°C until analysed. Cells were analysed by flow cytometry (FACSCalibur, Becton Dickinson). The percentage of hypodiploid cells were calculated with specific software (Cell Quest, Becton Dickinson).
For analysis of DNA fragmentation, 2×107 cells were treated for 24 h as indicated. Cells were washed, centrifuged and resuspended in 10 mM Tris pH 8.0, 400 mM NaCl, 2 mM EDTA, 1% SDS. Two-hundred μg ml−1 Proteinase K was added and cells were incubated at 50°C overnight. After protein precipitation with NaCl, DNA was precipitated. Twenty μg DNA were run on a 2% agarose gel. DNA was visualized by ethidium bromide staining and UV illumination.
For immunofluorescence microscopy RBL5 cells were incubated with medium or 2.5 mM sulfasalazine for 24 h and cytospins were prepared. Cells were fixed for 1 h using buffered formaldehyde (6%). Unspecific binding was blocked by incubation in TNB-buffer (Tris 50 mM, NaCl 150 mM, 0.5% bovine serum albumin, 0.5% sodium-azide, pH 7.7). Cells were stained for 1 h with biotinylated annexin V (Hölzel Diagnostika, Köln, Germany) diluted 1 : 30 in TBS (in mM: Tris 50, NaCl 150, CaCl2 2, pH 7.4). After washing with TNT (Tris 50 mM, NaCl 150 mM, CaCl2 2 mM, 0.05% Tween 40, pH 7.4) streptavidin-PE (Immunotech, Marseille, France) 1 : 100 in TNB (with 2 mM CaCl2) was added and incubated for 1 h. Counterstaining of the nuclei was performed using bisbenzimide (Hoechst 33258, Sigma, Deisenhofen, Germany). For Apo2.7 staining formaldehyde fixed cytospins were permeabilized with 70% ethanol for 1 h. Anti-Apo2.7 (Immunotech, Marseille, France) 1 : 20 in TNB was added for 2 h. After washing HRP-conjugated rabbit anti-mouse-IgG (DAKO, Hamburg, Germany) 1 : 50 in TNB was added for one additional hour. Cytospins were washed and incubated for 15 min with Tyramide Signal Amplification (TSA-Indirect NEN™, Life Science Products, Boston, U.S.A.) 1 : 40 diluted. Fluorescein-conjugated streptavidin (DAKO, Hamburg, Germany) (1 : 100 in TNB) was added for one additional hour. Finally, nuclei were counterstained with propidium iodide.
For quantitative analysis of annexin V binding RBL5 cells were treated as indicated. Cells were washed once in PBS and resuspended in annexin V binding buffer (in mM: HEPES 10, pH 7.4, NaCl 140, CaCl2 2.5). Fluorescein isothiocyanate (FITC)-conjugated annexin V (Becton Dickenson, Heidelberg, Germany) was added, and samples were analysed by flow cytometry with a FACSCalibur (Becton Dickenson).
Results
Sulfasalazine inhibits NF-κB activation in T-lymphocytes
RBL5 cells, a murine T-lymphocytic cell line were treated with control medium or 1.25 mM sulfasalazine for 1, 2, 4, 8, or 24 h, respectively. After stimulation with 150 U ml−1 TNFα for one additional hour, nuclear extracts were prepared and electrophoretic mobility shift assays performed using a κB specific probe (IgκB) (Figure 1A). Uninduced RBL5 cells showed a weak basal κB-binding activity, complex c1 (lane 1), which was strongly increased after treatment with TNFα, complex c1 and c2 (lane 2). By supershift assays c1 could be identified as NF-κB1 p50 homodimers, whereas c2 contained predominantly RelA p65, NF-κB1 p50, but also traces of NF-κB2 p52, RelB and c-Rel (data not shown). Binding specificity of both complexes was shown by competition with an excess of unlabelled κB-oligonucleotide (lane 8). No competition was observed witih the unrelated Sp1 binding site (lane 9). Pretreatment with sulfasalazine for 1 h completely inhibited TNFα induced NF-κB activation (lane 3). Incubation for longer time periods (2 and 4 h) resulted in the reappearance of NF-κB1 p50 homodimers (complex c1), comparable to κB activity in uninduced cells (compare lanes 4 and 5 to lane 1). Prolonged incubation with sulfasalazine also affected binding of NF-κB1 p50 homodimers (complex C1) (lanes 6–7).
Figure 1.
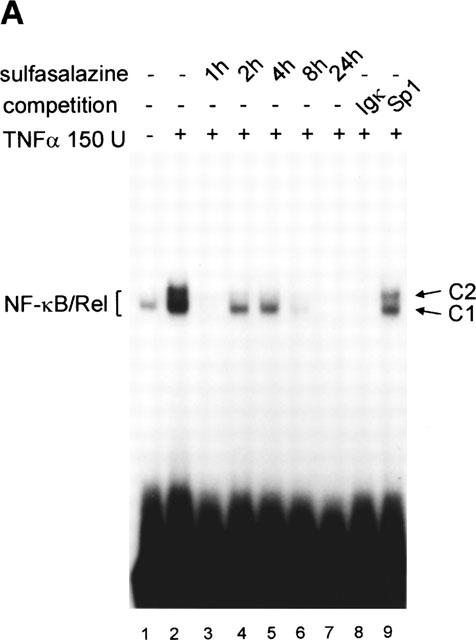
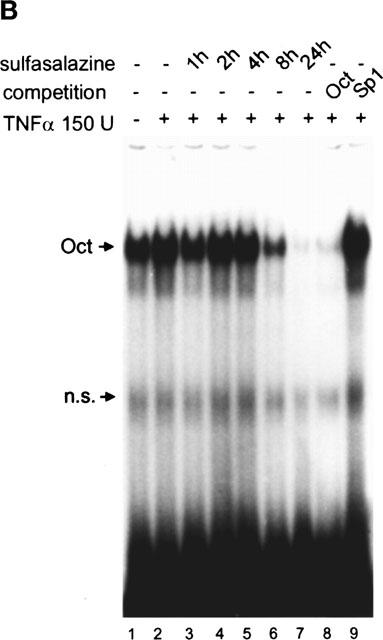
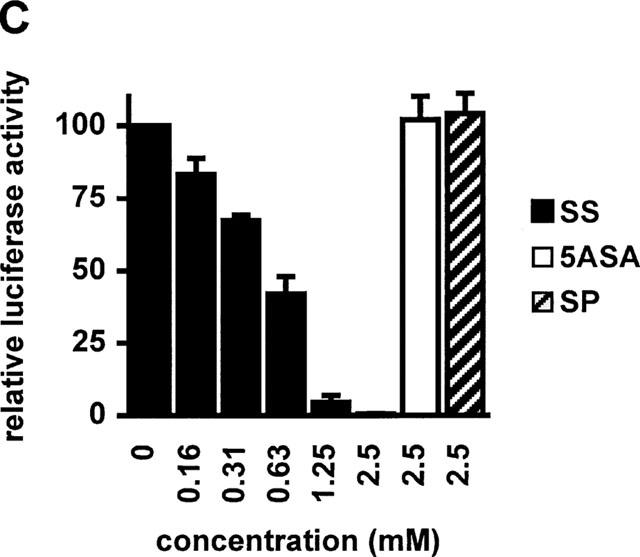
Sulfasalazine is a potent inhibitor of NF-κB activity in RBL5 T-lymphocytes. (A) Rapid inhibition of NF-κB binding activity by treatment with sulfasalazine. RBL5 cells were left untreated (lane 1), stimulated for 1 h with TNFα (lanes 2–9), and pretreated with 1.25 mM sulfasalazine for the indicated time periods (lanes 3–7). Nuclear proteins were extracted and electrophoretic mobility shift assays performed using a κB specific probe (IgκB). For competition experiments a 20 fold molar excess of unlabelled IgκB (lane 8) or Sp1 oligonucleotides (lane 9) were used. Running position of the NF-κB/Rel/DNA complexes c1 and c2 are indicated. One representative experiment is shown (n=4). (B) Sulfasalazine is a specific inhibitor of NF-κB and does not interfere with Oct-1 binding. The same nuclear extracts as in (A) were used in electrophoretic mobility shift assays using an Oct-1 probe. Running positions of the Oct-1 and a non-specific (n.s.) complex are indicated by arrows. One representative experiment is shown (n=3). (C) Sulfasalazine, but not its metabolic moieties 5ASA and sulfapyridine, is a potent inhibitor of NF-κB-dependent transcription. RBL5 cells were transiently transfected with a κB-dependent reporter plasmid (3xIgκBLuc). Cells were pretreated with medium or serial dilutions of sulfasalazine (SS) (black bars), 2.5 mM 5ASA (white bar), or 2.5 mM sulfapyridine (SP) (hatched bar) for 1 h, followed by stimulation with 150 U ml−1 TNFα for three additional hours. Luciferase activity from transfected cells stimulated with TNFα was used as reference and set to 100% luciferase activity. Experiments were done in duplicate. Mean values and standard error of the mean (s.e.mean) of four independent experiments (n=8) are shown.
To test the specificity of sulfasalazine-induced inhibition of NF-κB, nuclear extracts of sulfasalazine treated RBL5 cells were tested for DNA binding activity of other transcription factors. Uninduced RBL5 cells showed the constitutive Oct-1 binding activity, which was not affected by treatment with 1.25 mM sulfasalazine for 1–4 h (Figure 1B). Prolonged treatment for 8 and 24 h decreased Oct-1 binding activity (lanes 6–7) most likely due to nonspecific events caused by decreasing cell viability (see below). Almost identical results were obtained for AP1 binding activity (data not shown).
To examine whether 5-aminosalicylic acid (5ASA) or sulfapyridine, the colonic metabolites of sulfasalazine, showed any effect on κB binding activity RBL5 cells were treated for 1, 4, or 8 h with 2.5 or 5.0 mM 5ASA or sulfapyridine, respectively, followed by stimulation with 150 U ml−1 TNFα for an additional hour. Even at these high concentrations treatment with 5ASA or sulfapyridine did not affect κB binding activity (data not shown).
To examine the effects of sulfasalazine, 5ASA, and sulfapyridine on κB dependent gene expression RBL5 cells were transiently transfected with a plasmid containing three κB sites from the immunoglobulin κ-light chain enhancer upstream of a luciferase reporter gene (3xIgκBLuc). As shown in Figure 1C treatment with sulfasalazine effectively decreased TNFα-induced κB-dependent transcription in a dose-dependent fashion. Half maximal inhibition was detected at approximately 0.625 mM sulfasalazine, whereas 2.5 mM sulfasalazine suppressed luciferase activity to background levels (Figure 1C, black bars). In contrast, no significant changes of luciferase activity were seen after treatment with 2.5 mM 5ASA (white bar) or 2.5 mM sulfapyridine (hatched bar).
Sulfasalazine, but not its metabolites 5ASA or sulfapyridine, causes cell death in RBL5 T-lymphocytes in a dose- and time-dependent manner
To determine if the observed nonspecific loss of DNA binding activity after prolonged sulfasalazine treatment was caused by a decrease in cell viability, RBL5 cells were treated with increasing doses of sulfasalazine for 24 h. Cell viability was analysed by conversion of MTS dye to its formazan product, a reaction depending on living mitochondria. As shown in Figure 2 less than 50% of the cells were viable after incubation with 313 μM sulfasalazine for 24 h. Since the MTS assay can not discriminate between growth inhibition and induction of cell death, sulfasalazine treated RBL5 cells were further analysed for characteristic features of cell death and apoptosis.
Figure 2.
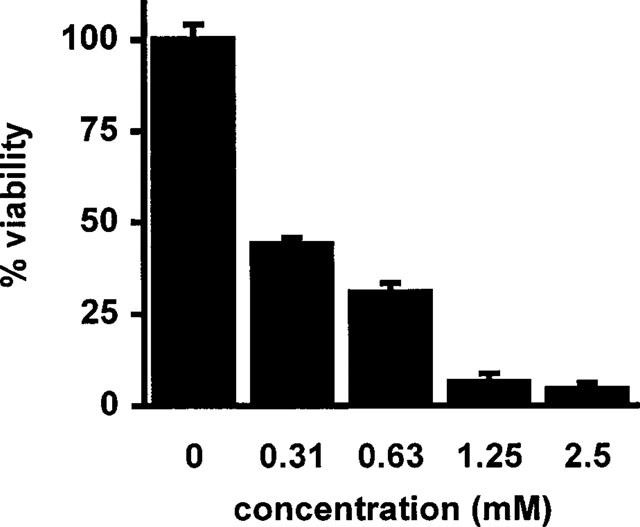
Sulfasalazine decreases cell viability in RBL5 T-lymphocytes. RBL5 cells were treated for 24 h with medium or increasing doses of sulfasalazine as indicated. Cells were analysed for viability by conversion of MTS dye to its formazan product. A490 obtained from control cells were set to 100% viability. Data were obtained from three independent experiments done in triplicate (n=9) and are presented as mean and standard error of the mean (s.e.mean).
RBL5 cells were treated with increasing doses of sulfasalazine for 24 h and analysed by forward scatter/sideward scatter (FSC/SSC) analysis in a fluorescence-activated cell sorter (FACS). Dead lymphocytes were identified on the basis of their characteristic forward/sideward scatter changes (Carbonari et al., 1994). A dose-dependent induction of cell death was found in a concentration ranging from 0.313 to 2.5 mM sulfasalazine, with 50% dead cells between 0.313 and 0.625 mM and almost 100% dead cells at 2.5 mM sulfasalazine (Figure 3A). RBL5 cells were incubated with 1.25 mM sulfasalazine for varying time periods (0, 1, 2, 4, 8, and 24 h) followed by FACS analysis. As shown in Figure 3B induction of cell death started as early as 4 h after addition of sulfasalazine into the culture medium. Incubation between 4 and 8 h resulted in 50% dead cells. To study whether the metabolites of sulfasalazine had similar effects on T-cell viability, RBL5 cells were incubated for 24 h with 5ASA and/or sulfapyridine, respectively. Cell viability was assayed by FACS analysis as above (FSC/SSC). Doses up to 5.0 mM 5ASA (white bars), sulfapyridine (grey bars) or 5ASA plus sulfapyridine (hatched bars) did not show any effect on cell viability compared to control medium (grey bar) (Figure 3C).
Figure 3.
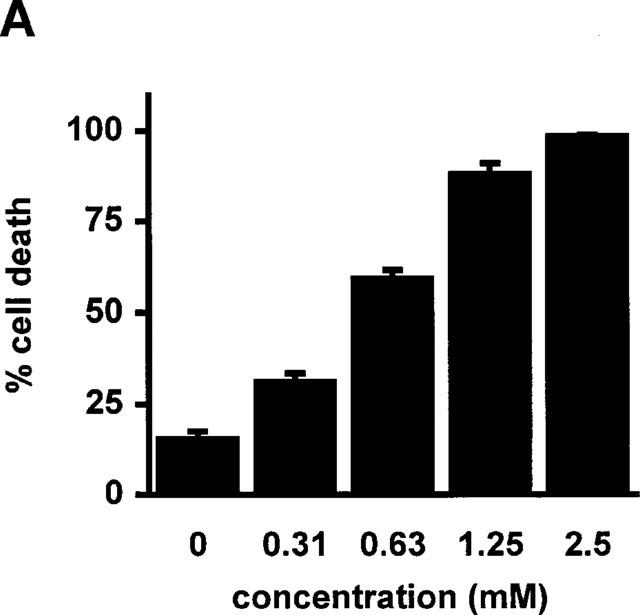
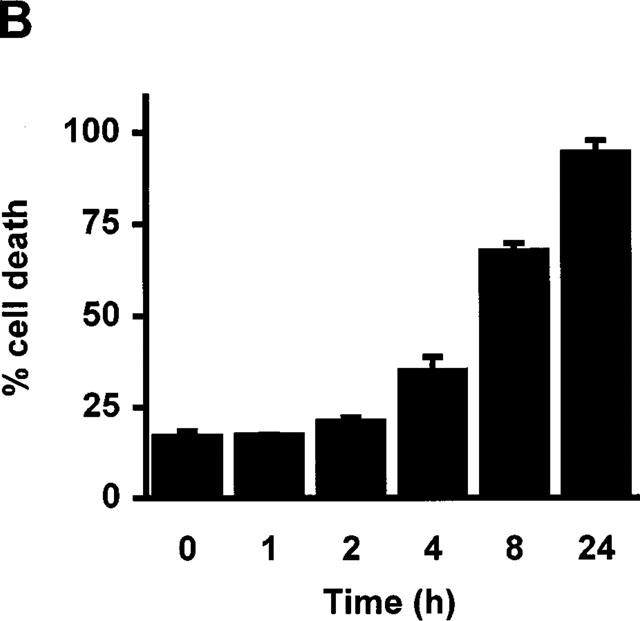
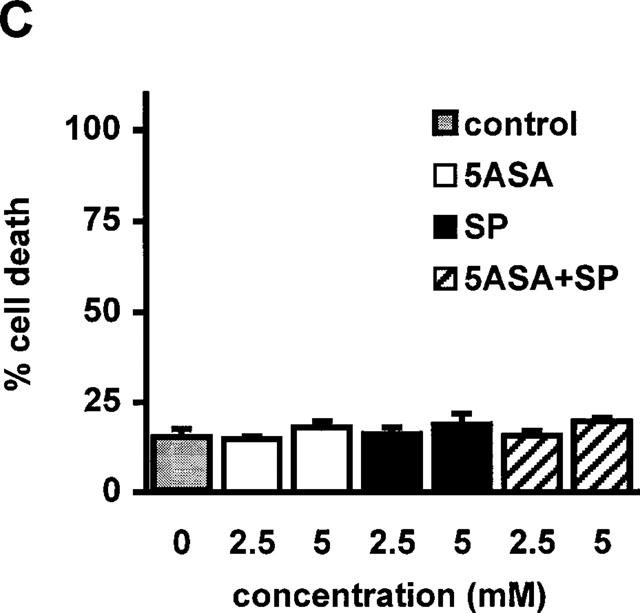
Sulfasalazine induces cell death in RBL5 T-lymphocytes in a dose- and time-dependent manner. (A) RBL5 cells were incubated with the indicated concentrations of sulfasalazine for 24 h, (B) or with 1.25 mM sulfasalazine for the indicated time periods. (C) 5ASA and sulfapyridine, the colonic metabolites of sulfasalazine, have no effect on T-cell viability. RBL5 T-lymphocytes were treated for 24 h either with medium (control) or increasing concentrations of 5ASA (white bars), sulfapyridine (SP) (black bars) or 5ASA plus sulfapyridine (hatched bars) as indicated. The percentage of dead cells was determined by FACS analysis on the basis of their characteristic forward/sideward light scatter changes as described (Carbonari et al., 1994). Data are obtained from three independent experiments done in duplicate (n=6) and are presented as mean and standard error of the mean (s.e.mean).
Sulfasalazine induces apoptosis in RBL5 T-lymphocytes
One characteristic, late event during apoptosis is the internucleosomal cleavage of genomic DNA following activation of cytoplasmic endonucleases (Wyllie, 1980; 1998). DNA-fragmentation can be detected by staining of nuclei with intercalating dyes (Nicoletti et al., 1991) or by extraction and gel electrophoresis of genomic DNA (Wyllie, 1980). To determine if sulfasalazine-induced cell death was due to apoptosis we analysed sulfasalazine treated RBL5 cells for the appearance of hypodiploid DNA. As shown in Figure 4, RBL5 cells were treated either with medium (A), 0.625 mM (B) or 2.5 mM (C) sulfasalazine for 24 h. Cells were harvested, ethanol fixed, stained with propidium iodide and analysed by flow cytometry. In untreated control cells, 11% showed hypodiploid nuclei indicative of apoptosis, whereas treatment with sulfasalazine resulted in 44% at 0.625 mM, and 98% apoptotic cells at 2.5 mM sulfasalazine. No significant increase of hypodiploid DNA was observed after treatment with either 5ASA (2.5 mM) (D) or sulfapyridine (2.5 mM) (E). Furthermore, as shown in Figure 4F sulfasalazine (0.625 mM, lane 6) but not 5ASA (2.5 mM, lane 4) or sulfapyridine (2.5 mM, lane 5) treatment of RBL5 cells resulted in a typical DNA fragmentation pattern on an ethidium bromide stained agarose gel.
Figure 4.
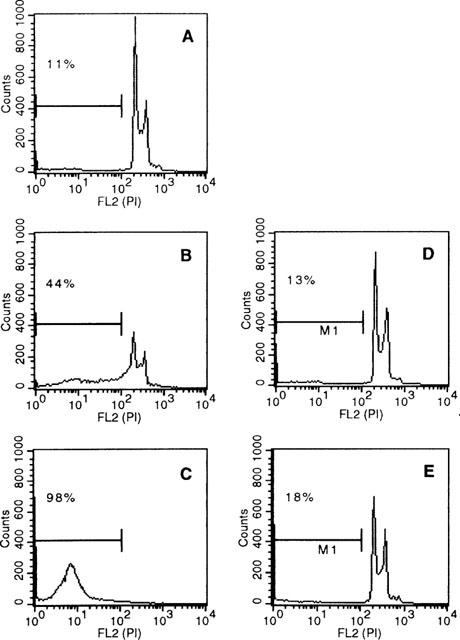
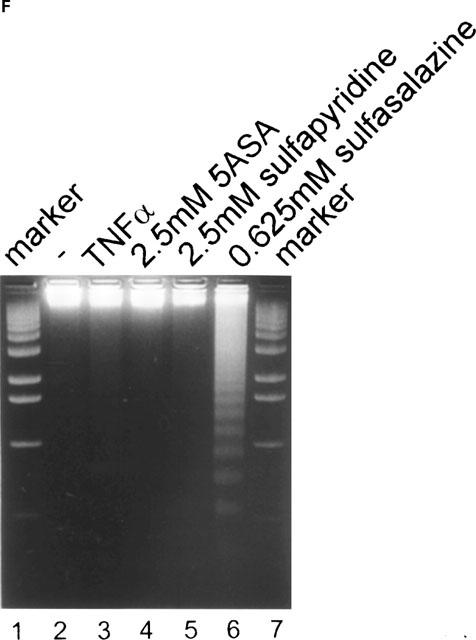
Sulfasalazine induces apoptosis in RBL5 T-lymphocytes as judged by DNA fragmentation. RBL5 cells were treated for 24 h with either (A) medium, (B) 0.625 mM sulfasalazine, (C) 2.5 mM sulfasalazine, (D) 2.5 mM 5ASA, or (E) 2.5 mM sulfapyridine. After ethanol fixation cells were stained with propidium iodide and analysed by FACS for the appearance of hypodiploid DNA. Numbers above the histogram markers indicate the percentage of apoptotic nuclei (broad hypidiploid peak) in a representative experiment (n=3). (F) Analysis of sulfasalazine induced DNA fragmentation pattern. RBL5 cells were treated with medium, 150 U ml−1 TNFα, 2.5 mM 5ASA, 2.5 mM sulfapyridine, or 0.625 mM sulfasalazine for 24 h. Genomic DNA was extracted and analysed on an ethidium bromide stained agarose gel. As molecular weight marker a 100 bp marker was used (Gibco). One representative experiment is shown (n=4).
In living cells the phospholipid phosphatidylserine is situated on the inner leaflet of the plasma membrane. When cell death occurs, phosphatidylserine is translocated to the outer layer of the membrane. This occurs in the early phase of apoptosis during which the cell membrane itself remains intact. Annexin V binds to phosphatidlyserine and is therefore useful to quantitate apoptotic cells (Koopman et al., 1994). Furthermore, apoptotic cells can be detected with Apo 2.7, a monoclonal antibody recognizing specifically apoptotic cells early during apoptosis (Zhang et al., 1996). RBL5 cells were treated with 2.5 mM sulfasalazine for 24 h. Double immunofluorescence microscopy of annexin V binding (red) and DNA staining (blue, Hoechst 33258) is shown in Figure 5A,B, Apo 2.7 expression (yellow-green) and propidium iodide staining (red) is shown in Figure 5C,D. More than 90% of sulfasalazine treated cells were positive for annexin V (Figure 5B) and Apo 2.7 binding (Figure 5D), whereas almost no positive cells could be detected among the untreated control cells (Figure 5A,C). Furthermore, sulfasalazine treated cells display smaller, condensed nuclei, recognized as a morphological characteristic of apoptotic cells.
Figure 5.
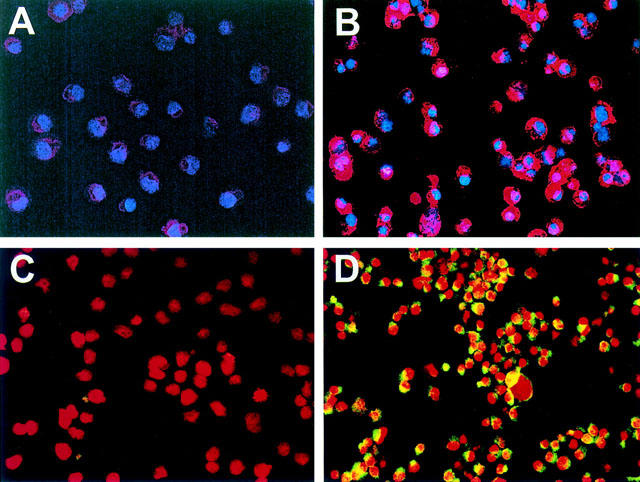
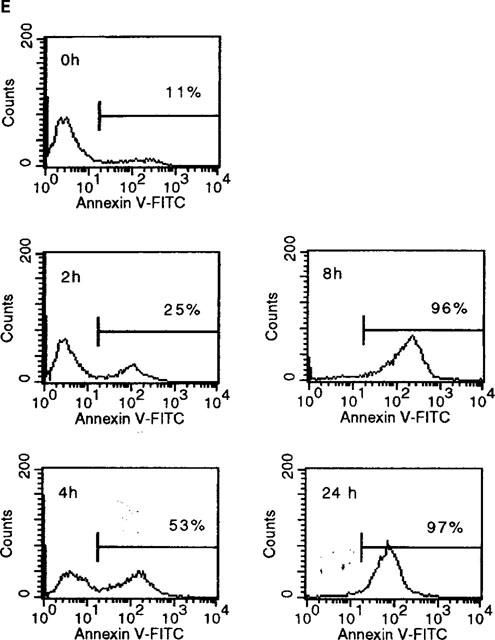
Sulfasalazine induces apoptosis in RBL5 T-lymphocytes as judged by annexin V and Apo 2.7 staining. (A–D) Cells were incubated with medium (A,C) or 2.5 mM sulfasalazine (B,D) for 24 h. Cytospins were prepared and double immunofluorescence of annexin V binding (red) and Hoechst 33258 nuclear staining (blue) (A,B) or Apo 2.7 expression (yellow-green) and propidium iodide staining (red) (C,D) were performed. Fluorescence was observed and photographed with a fluorescence microscope (C. Zeiss, Oberkochen, Germany) equipped with epilumination. Magnification 400×. One representative experiment is shown (n=2). (E) For quantitative analysis of annexin V binding RBL5 cells were incubated with medium or 2.5 mM sulfasalazine for 2, 4, 8, and 24 h. Cells were stained with FITC-conjugated annexin V and analysed by FACS. 10,000 cells were analysed under each condition. Numbers above the histogram markers indicate the percentage of apoptotic cells in a representative experiment (n=3).
Induction of apoptosis by sulfasalazine seems to be a fast event as judged by staining with FITC-labelled annexin V and FACS analysis. RBL5 cells were incubated with medium or 2.5 mM sulfasalazine for 2, 4, 8, and 24 h. After 4 h treatment 50%, after 8 h more than 90% of the cells were positive for annexin V staining (Figure 5E). In summary, these results demonstrate that sulfasalazine induces apoptosis in RBL5 T-lymphocytes.
Discussion
Although inflammatory bowel disease (IBD) and rheumatoid arthritis are distinct entities, similar pharmacological agents are useful for their clinical treatment. Despite intense research, there are still no specific therapies for these diseases. Treatment is directed towards symptomatic improvement and controlling the disease process. Sulfasalazine, 5ASA and corticosteroids are among the commonly used drugs for the treatment of IBD and rheumatoid arthritis (Riis et al., 1973; Azad Khan et al., 1977; Klotz et al., 1980; Gaginella & Walsh, 1992; Taggart et al., 1986; Bird, 1995). However, the molecular mechanisms of action remain unclear (Gaginella & Walsh, 1992).
In this study we demonstrate that sulfasalazine is a specific inhibitor of NF-κB activation in T-lymphocytes. Prolonged treatment with sulfasalazine induces cell death of T-lymphocytes in a time- and dose-dependent manner. Cell death is due to apoptosis as judged by DNA fragmentation, FACS analysis of hypodiploid DNA, Annexin V and Apo 2.7 staining, and nuclear condensation. In contrast, 5ASA and sulfapyridine up to concentrations of 5 mM neither inhibited NF-κB activation nor induced apoptosis. Our data suggest that the anti-inflammatory effect of sulfasalazine in IBD and rheumatoid arthritis might be due to NF-κB inhibition and subsequent induction of apoptosis of T-lymphocytes.
During the last few years evidence has accumulated to show that NF-κB plays a predominant role in the pathogenesis of IBD. Overexpression of NF-κB regulated cytokines as well as increased nuclear levels of NF-κB were demonstrated in the mucosa of patients with active Crohn's disease and ulcerative colitis (Neurath et al., 1996; Jobin et al., 1997; Rogler et al., 1998; Schreiber et al., 1998; Ardite et al., 1998). Most intriguingly, a single application of NF-κB RelA antisense oligonucleotides led to abrogation of chronic intestinal inflammation in experimental colitis in mice (Neurath et al., 1996). Therefore, the observed inhibition of NF-κB activation by sulfasalazine is a good explanation for the long known anti-inflammatory potency of this drug. In addition, it explains previous reports that sulfasalazine inhibits IL-2 synthesis in lymphocytes (Sheldon et al., 1988), as well as TNFα and IL-1 production in macrophages (Fujiwara et al., 1990). In our hands 5ASA and and sulfapyridine in concentrations up to 5 mM did not effect NF-κB activation. However, previous studies have shown that salicylates including aspirin and a derivative of 5ASA (mesalamine) block cytokine-induced activation of NF-κB as well as other signalling cascades such as ERK1/ERK2 and JNK/SAPK kinases at a concentration of 20 mM (Kopp & Ghosh, 1994; Frantz & O'Neill, 1995; Kaiser et al., 1999). Moreover, high doses of mesalamine inhibit IL-1-stimulated phosphorylation of the NF-κB subunit RelA but does not affect IκBα degradation (Egan et al., 1999). These data suggest that both sulfasalazine and 5ASA, suppress NF-κB activation by different mechanisms, although with different efficiency.
Apoptosis, or programmed physiological cell death, is a central regulatory check-point which limits the intensity of immune responses. Failure of apoptosis may be a major mechanism by which initially mild diseases progress to severe chronic inflammatory disorders (Cohen & Duke, 1992; Thompson, 1995; Anderson, 1996). Lack of apoptosis is seen in patients with chronic inflammatory conditions, although the cause of impaired apoptosis is unknown. It has been suggested that in rheumatoid arthritis the inflammatory T-cell infiltrate persists because apoptosis is actively suppressed by the synovial microenvironment (Salmon et al., 1997). Lamina propria T-cells from patients with Crohn's disease and ulcerative colitis exhibit increased proliferation, cytokine production and decreased rate of apoptosis (Boirivant et al., 1999). In addition, mucosal T-cells of patients with Crohn's disease are resistant to apoptosis induced by deprivation of IL-2 in vitro (Ina et al., 1995).
During apoptosis cells display characteristic morphological changes: cells shrink, rapidly display an altered plasma membrane, and undergo nuclear condensation prior to endonuclease-dependent DNA-fragmentation. Early changes in the plasma membrane seem to be particularly important since they signal macrophages to phagocytose dying cells before toxic breakdown products can injure the surrounding tissue (Savill et al., 1989). For this reason, therapeutic induction of apoptosis holds the attraction of rapidly and safely killing activated effector cells of the immune system without secondary tissue damage. In contrast, necrosis releases all cellular contents into the surrounding tissue leading to ongoing inflammation.
This data provides strong evidence that sulfasalazine induces apoptosis in T-lymphocytes. No such effect was seen with 5ASA or sulfapyridine. Therefore sulfasalazine and its metabolic moiety 5ASA act differently, thus the beneficial effects of both reagents are not explained by the same mechanisms. These findings are of particular interest since there is still a debate about the pharmacological active moiety of sulfasalazine (Azad Khan et al., 1977; Klotz et al., 1980; Sutherland et al., 1993; Taggart et al., 1986; Bird, 1995). Our study suggests that sulfasalazine is not only a prodrug, but moreover an active pharmacological agent by itself with distinct and unique actions not shared by its metabolites 5ASA and sulfapyridine.
The concentration of sulfasalazine achieved in inflamed tissue is not known. However, it is reported that serum levels of sulfasalazine after an average oral dose of 3–6 g per day in patients with IBD are 10–15 μg ml−1, equivalent to 0.025–0.038 mM (Das et al., 1973). Stool concentrations are in the order of 1.25–2.0 mM and interstitial concentrations may be as high as 0.5–1.0 mM (Peppercorn & Goldman, 1973). These concentrations are comparable to those used in our study.
In conclusion, our data clearly demonstrate that sulfasalazine inhibits NF-κB activation and induces apoptosis in T-lymphocytes. This may explain why treatment with sulfasalazine leads to clearance of inflammatory cells and therefore can break the cycle of unrelenting cellular activation and tissue damage in chronic inflammation. Thus, both inhibition of NF-κB activation as well as the clearance of activated T-lymphocytes are the mechanisms of action of sulfasalazine.
Acknowledgments
We are indebted to Sabine Schirmer, Ester Rüber and Janet Köhler for excellent technical assistance. We thank Sonja Aigner for preparing the manuscript. This work was supported by grants from the Bundesministerium für Bildung und Forschung and the Deutsche Krebshilfe Mildred Scheel Stiftung to S. Liptay and R.M. Schmid.
Abbreviations
- 5ASA
5-aminosalicylic acid
- EMSA
electrophoretic mobility shift assay
- ERK
extracellular signal-regulated kinase
- FACS
fluorescence-activated cell sorter
- FSC
forward scatter
- IBD
inflammatory bowel diseases
- ICAM
intercellular adhesion molecule
- Ig
immunoglobulin
- IκBα
inhibitor of κB α
- IL-1
interleukin-1
- IL-2
interleukin-2
- IL-6
interleukin-6
- IL-7
interleukin-7
- IL-8
interleukin-8
- JNK
jun-N-terminal kinase
- MTS
3-(4,5-dimethythiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NF-κB
Nuclear factor κB
- TNFα
tumour necrosis factor α
- SAPK
stress activated protein kinase
- SCID
severe combined immunodeficiency
- SSC
sideward scatter
References
- ANDERSON G.P. Resolution of chronic inflammation by therapeutic induction of apoptosis. TIPS. 1996;17:438–442. doi: 10.1016/s0165-6147(96)01004-8. [DOI] [PubMed] [Google Scholar]
- ARDITE E., PANÉS J., MIRANDA M., SALAS A., ELIZALDE J.I., SANS M., ARCE Y., BORDAS J.M., FERNÁNDEZ-CHECA J.C., PIQUÉ J.M. Effects of steroid treatment on activation of nuclear factor κB in patients with inflammatory bowel disease. Br. J. Pharmacol. 1998;124:431–433. doi: 10.1038/sj.bjp.0701887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZAD KHAN A.K., PIRIS J., TRUELOVE S.C. An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet. 1977;2:892–895. doi: 10.1016/s0140-6736(77)90831-5. [DOI] [PubMed] [Google Scholar]
- BIRD H.A. Sulphasalazine, sulphapyridine or 5-aminosalicylic acid–which is the active moiety in rheumatoid arthritis. Br. J. Rheumatol. 1995;34 suppl. 2:16–19. [PubMed] [Google Scholar]
- BOIRIVANT M., MARINI M., DI FELICE G., PRONIO A.M., MOTESANI C., TERSIGNI R., STROBER W. Lamina propria T cells in Crohn's disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology. 1999;116:557–565. doi: 10.1016/s0016-5085(99)70177-0. [DOI] [PubMed] [Google Scholar]
- CARBONARI M., CIBATI M., CHERCHI M., SBARIGIA D., PESCE A.M., DELL'ANNA L., MODICA A., FIORILLI M. Detection and characterization of apoptotic peripheral blood lymphocytes in human immunodeficiency virus infection and cancer chemotherapy by a novel flow immunocytometric method. Blood. 1994;83:1268–1277. [PubMed] [Google Scholar]
- COHEN J.J., DUKE R.C. Apoptotis and programmed cell death in immunity. Annu. Rev. Immunol. 1992;10:267–293. doi: 10.1146/annurev.iy.10.040192.001411. [DOI] [PubMed] [Google Scholar]
- DAS K.M., EASTWOOD M.A., MCMANUS J.P.A., SIRCUS W. The metabolism of salicylazosulphapyridine in ulcerative colitis. I. The relationship between metabolites and the response to treatment in inpatients. Gut. 1973;14:631–641. doi: 10.1136/gut.14.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EGAN L.J., KEAN D.J.M., MAYS D.C., BELL M., HUNTOON C., PIKE M.G., SANDBORN W.J., LIPSKY J.J. Mesalamine regulates nuclear factor kappa B (NF-κB) activity in intestinal epithelial cells by a novel mechanism: inhibition of inducible phosphorylation of RelA (p65) Gastroenterology. 1999;116:G3064. [Google Scholar]
- FIOCCHI C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- FRANTZ B., O'NEILL E.A. The effect of sodium salicylate and aspirin on NF-κB. Science. 1995;279:2017–2018. doi: 10.1126/science.270.5244.2017. [DOI] [PubMed] [Google Scholar]
- FRIESEN C., HERR I., KRAMMER P.H., DEBATIN K.M. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat. Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- FUJIWARA M., MITSUI K., YAMAMOTO I. Inhibition of proliferative responses and interleukin 2 production by salazosulfapyridine and its metabolites. Jpn. J. Pharmacol. 1990;54:121–132. doi: 10.1254/jjp.54.121. [DOI] [PubMed] [Google Scholar]
- GAGINELLA T.S., WALSH R.E. Sulfasalazine: multiplicity of action. Dig. Dis. Sci. 1992;37:801–812. doi: 10.1007/BF01300376. [DOI] [PubMed] [Google Scholar]
- GHOSH S., MAY M.J., KOPP E.B. NF-κB and Rel proteins. Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- HASLETT C. Resolution of acute inflammation and the role of apoptosis in the tissue fate of granulocytes. Clin. Sci. (Lond) 1992;83:639–648. doi: 10.1042/cs0830639. [DOI] [PubMed] [Google Scholar]
- INA K., BINION D.G., WEST G.A., DOBREA G.M., FIOCCHI C. Secretion of soluble factors and phagocytosis by intestinal fibroblasts regulate T-cell apoptosis. Gastroenterology. 1995;108:A841. [Google Scholar]
- JOBIN C., HASKILL S., MAYER L., PANJA A., SARTOR R.B. Evidence for altered regulation of IκBα degradation in human colonic epithelial cells. J. Immunol. 1997;158:226–234. [PubMed] [Google Scholar]
- KAISER G.C., YAN F., POLK D.B. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor κB activation in mouse colonocytes. Gastroenterology. 1999;116:602–609. doi: 10.1016/s0016-5085(99)70182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLOTZ U., MAIER K., FISCHER C., HEINKEL K. Therapeutic efficacy of sulfasalazine and its metabolites in patients with ulcerative colitis and Crohn's disease. N. Engl. J. Med. 1980;303:1499–1502. doi: 10.1056/NEJM198012253032602. [DOI] [PubMed] [Google Scholar]
- KOPP E., GHOSH S. Inhibition of NF-κB by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- KOOPMAN G., REUTELINGSPERGER C.P.M., KUIJTEN G.A.M., KEEHNEN R.M.J., PALS S.T., VAN OERS M.H.J. Annexin V for flow cytometric detection of phosphatidyserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- KRAMMER P.H., DHEIN J., WALCZAK H., BEHRMANN I., MARIANI S., MATIBA B., FATH M., DANIEL P.T., KNIPPING E., WESTENDORP M.O., STRICKER K., BÄUMLER C., HELLBARDT S., GERMER M., PETER M.E., DEBATIN K.M. The role of APO-I-mediated apoptosis in the immune system. Immunol. Rev. 1994;142:175–191. doi: 10.1111/j.1600-065x.1994.tb00889.x. [DOI] [PubMed] [Google Scholar]
- LIPTAY S., SCHMID R.M., NABEL E.G., NABEL G.J. Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Mol. Cell Biol. 1994;14:7695–7703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDONALD H.R., LEES R.K. Programmed death of autoreactive thymocytes. Nature. 1990;343:642–644. doi: 10.1038/343642a0. [DOI] [PubMed] [Google Scholar]
- MCDONNELL T.J., DEANE N., PLATT F.M., NUNEZ G., JAEGER U., MCKEARN J.P., KORSMEYER S.J. Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- MOUNTZ J.D., WU J., CHENG J., ZHOU T. Autoimmune disease. A problem of defective apoptosis. Arthritis Rheum. 1994;37:1415–1420. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- MUNN D.H., BEALL A.C., SONG D., WRENN R.W., THROCKMORTON D.C. Activation-induced apoptosis in human macrophages: developmental regulation of a novel cell death pathway by macrophage colony-stimulating factor and interferon γ. J. Exp. Med. 1995;181:127–136. doi: 10.1084/jem.181.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEURATH M.F., BECKER C., BARBULESCU K. Role of NF-κB in immune and inflammatory response in the gut. Gut. 1998;43:856–860. doi: 10.1136/gut.43.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEURATH M.F., PETTERSSON S., MEYER ZUM BÜSCHENFELD K.H., STROBER W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-κB abrogates established experimental colitis in mice. Nat. Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- NICOLETTI I., MIGLIORATI G., PAGLIACCI M.C., GRIGNANI F., RICCARDI C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- NISITANI S., TSUBATA T., MURAKAMI M., OKAMOTO M., HONJO T. The bcl-2 gene product inhibits clonal deletion of self-reactive B lymphocytes in the periphery but not in the bone marrow. J. Exp. Med. 1993;178:1247–1254. doi: 10.1084/jem.178.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSWALD F., LIPTAY S., ADLER G., SCHMID R.M. NF-κB2 is a putative target gene of activated notch-1 via RBP-Jκ. Mol. Cell Biol. 1998;18:2077–2088. doi: 10.1128/mcb.18.4.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANG G., COUCH L., BATEY R., CLANCY R., CRIPPS A. GM-CSF., IL-1α, IL-1β, IL-6, IL-8, IL-10, ICAM-1 and VCAM-1 gene expression and cytokine production in human duodenal fibroblasts stimulated by lipopolysaccharide, IL-1α and TNF-α. Clin. Exp. Immunol. 1994;96:437–443. doi: 10.1111/j.1365-2249.1994.tb06048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEPPERCORN M.A., GOLDMAN P. Distribution studies of salicylazosulfapyridine and its metabolites. Gastroenterology. 1973;64:240–245. [PubMed] [Google Scholar]
- RIS P., ANTHONSIEN P., WULFF H.R., FOLKENBORG O., BONNEVIE O., BINDER V. The prophylactic effect of salazosulphapyridine in ulcerative colitis during long-term treatment: a double-blind trial on patients asymptomatic for one year. Scand. J. Gastroenterol. 1973;8:71–74. [PubMed] [Google Scholar]
- ROGLER G., BRAND K., VOGL D., PAGE S., HOFMEISTER R., ANDUS T., KNUECHEL R., BAEUERLE P.A., SCHÖLMERICH J., GROSS V. Nuclear factor κB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- SALMON M., SCHEEL-TOELLNER D., HUISSOON A.P., PILLING D., SHAMSADEEN N., DUPUY D'ANGEAC A., HYDE H., BACON P.A., EMERY P., AKBAR A.N. Inhibition of T cell apoptosis in the rheumatoid synovium. J. Clin. Invest. 1997;99:439–446. doi: 10.1172/JCI119178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAVILL J.S., WYLLIE A.H., HENSON J.E., WALPORT M.L., HENSON P.M., HASLETT C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMID R.M., PERKINS N.D., DUCKETT C.S., ANDREWS P.C., NABEL G.J. Cloning of a NF-κB subunit which stimulates HIV transcription in synergy with p65. Nature (London) 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S., NICOLAUS S., HAMPE J. Activation of nuclear factor κB in inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHELDON P.J., WEBB C., GRINDULIS K.A. Effect of sulphasalazine and its metabolites on mitogen induced transformation of lymphocytes–clues to its clinical action. Br. J. Rheumatol. 1988;27:344–349. doi: 10.1093/rheumatology/27.5.344. [DOI] [PubMed] [Google Scholar]
- STRASSER A., HARRIS A.W., HUANG D.C., KRAMMER P.H., CORY S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUTHERLAND L.R., MAY G.R., SHAFFER E.A. Sulfasalazine revisited: a meta-analysis of 5-aminosalicylic acid in the treatment of ulcerative colitis. Ann. Int. Med. 1993;118:540–549. doi: 10.7326/0003-4819-118-7-199304010-00009. [DOI] [PubMed] [Google Scholar]
- TAGGART A.J., NEUMANN V.C., HILL J., ASTBURY C., LE GALLEZ P., DIXON J.S. 5-aminosalicylic acid or sulphapyridine, which is the active moiety of sulphasalazine in rheumatoid arthritis. Drugs. 1986;32 suppl. 1:27–34. doi: 10.2165/00003495-198600321-00006. [DOI] [PubMed] [Google Scholar]
- THOMPSON C.B. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- WAHL C., LIPTAY S., ADLER G., SCHMID R.M. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 1998;101:1163–1174. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG C.Y., MAYO M.W., KORNELUK R.G., GOEDDEL D.V., BALDWIN A.S., JR NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- WYLLIE A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- WYLLIE A.H. Apoptosis. An endonuclease at last. Nature. 1998;391:20–21. doi: 10.1038/34040. [DOI] [PubMed] [Google Scholar]
- ZHANG C., AO Z., SETH A., SCHLOSSMAN S.F. A mitochondrial membrane protein defined by a novel monoclonal antibody is preferentially detected in apoptotic cells. J. Immunol. 1996;157:3980–3987. [PubMed] [Google Scholar]