

Abstract
Potent and highly selective small molecule antagonists have recently been developed by us for C5a receptors (C5aR) on human polymorphonuclear leukocytes (PMN). In this study we compared a new cyclic antagonist, F-[OPdChaWR], with an acyclic derivative, MeFKPdChaWr, for their capacities to bind to C5aR on human PMN and human umbilical artery membranes. We also compared their inhibition of myeloperoxidase (MPO) secretion from human PMNs and their inhibition of human umbilical artery contraction induced by human recombinant C5a.
In both PMNs and umbilical artery, the cyclic and acyclic C5a antagonists displayed insurmountable antagonism against C5a. There were differences in selectivities for the C5aR with F-[OPdChaWR] (pKb 8.64±0.21) being 30 times more potent than MeFKPdChaWr (pKb 7.16±0.11, P<0.05) in PMNs, but of similar potency (pKb 8.19±0.38 vs pKb 8.28±0.29, respectively) in umbilical artery. This trend was also reflected in their relative binding affinities, both antagonists having similar affinities (−logIC50 values) for C5aR in umbilical artery membranes (F-[OPdChaWR], 7.00±0.46; MeFKPdChaWr, 7.23±0.17), whereas in PMN membranes the C5aR affinity of the cycle F-[OPdChaWR] (7.05±0.06) was four times higher than that of acyclic MeFKPdChaWr (6.43±0.24, P<0.05).
In summary, the results reveal that these antagonists are insurmountable in nature against C5a for C5aR on at least two human cell types, and the differences in relative receptor binding affinities and antagonistic potencies against C5a are consistent with differences in receptors within these cell types. The nature of these differences is yet to be elucidated.
Keywords: C5a, C5a antagonist, C5a receptor, insurmountable antagonism
Introduction
The peptide inflammatory mediator, C5a, is released following activation of the serum complement system. The complement system is activated during host defence, but inappropriate or prolonged activation is thought to contribute to the pathogenesis of many immuno-inflammatory disorders (Whaley, 1987). C5a causes chemotaxis and degranulation of polymorphonuclear leukocytes (PMNs) and the contraction of smooth muscle via release of secondary mediators such as eicosanoids (Gerard & Gerard, 1994; Taylor et al., 1994) and the release of cytokines from macrophages and monocytes (Clancy et al., 1985; Ember et al., 1994; Morgan et al., 1992), these actions contributing to the pro-inflammatory effects of C5a. Many non-myeloid cell types have recently been reported to express C5a receptors (C5aR) (Floreani et al., 1998; Gasque et al., 1997; Haviland et al., 1995; Werfel et al., 1996), although the precise functions of these receptors are largely unclear.
The C-terminus of C5a contains within it the sole requirements for receptor activation (Ember et al., 1992; DeMartino et al., 1994; Siciliano et al., 1994), and has been used for the design of receptor antagonists of C5a (Konteatis et al., 1994). Only one short peptide, MeFKPdChaWr, has been demonstrated to antagonize C5a-induced myeloperoxidase release, calcium flux, chemotaxis and the stimulation of GTPase activity in PMNs (Konteatis et al., 1994). We found evidence for a turn conformation in its solution structure (Wong et al., 1998), and have since reported more potent cyclic antagonists (Finch et al., 1999). Their superior C5aR affinities and antagonist potencies on human PMNs (Wong et al., 1998), stem from restraining the antagonist structure to a turn conformation that is preorganized for receptor-binding.
Potent and selective antagonists of C5aRs would be very valuable probes for determining and confirming pathophysiological roles for C5a. Until recently, no potent or selective small molecule antagonists have been available to evaluate therapeutic effects of blocking C5aRs. As a step towards this goal, we recently reported that F-[OPdChaWR], inhibits C5a- and lipopolysaccharide-induced neutropenia in rats (Short et al., 1999).
Although C5aR antagonists are now available for testing and further development, to date, no studies have reported on either the pharmacological nature of the antagonism or on their activities on cells other than PMNs. The present study therefore aimed to elucidate the characteristics of the interaction between C5a and the antagonists, and to determine if there were cell or tissue differences. There have been no reports of C5aR subtypes so far, partly because only one C5aR gene has been described (Gerard et al., 1993; Gerard & Gerard, 1991) and because a variety of C5a antagonists have not been available to assist in receptor classification. To address these issues, we now compare the pharmacological properties of the acyclic peptide, MeFKPdChaWr, with the cyclic peptide, F-[OPdChaWR] (Finch et al., 1999; Short et al., 1999) on human PMNs and umbilical artery C5aR.
Methods
The peptides used were synthesized as previously described (Short et al., 1999; Wong et al., 1998). Human recombinant C5a was from Sigma (MO, U.S.A.) or produced as C5aWT (see below). Vector pME10 containing the C5a cDNA was a generous gift from Wilfried Bautsch (Medizinische Hochschule, Hannover, Germany). The Qia expressionist kit was obtained from Qiagen (U.K.).
Production of C5aWT
C5a fragment (aa1-74) was amplified by polymerase chain reaction (PCR) from the clone pME10 (Bautsch et al., 1992) then cloned into BamHI and KpnI of the bacterial expression pQE 30 (Qiagen) resulting in the clone pQE C5a. The expression vector pQE 30 was expressed and purified from the E. coli strain M15[pREP4] (Qiagen), essentially as instructed by the manufacturer. The frozen cell pellet was thawed on ice and resuspended in 6 M Gu-HCl, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH 8.0, in 5 ml gram−1 wet weight of cells. The cells were lysed at room temperature and the lysate centrifuged at 10,000×g for 20–30 min at room temperature to pellet cellular debris. One to two ml of 50% Ni-NTA slurry (Qiagen) was added to 10–15 ml of lysate and mixed gently on an orbital mixer for 15–60 min at room temperature. The lysate-resin mixture was loaded into a disposable column. The column was then washed with 50 ml 8 M Urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, 1 M NaCl, pH 6.3, followed by 20 ml 8 M Urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, 1 M NaCl, 10 mM imidazole, pH 5.9. The recombinant protein was eluted with 1–2 ml fractions of 8 M Urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, 1 M NaCl, pH 4.5. The purified C5a fragments were then refolded by dialysis overnight on 1000× volume of 0.1 M Tris-Cl pH 8.0, 2 mM reduced glutathione, 0.2 mM oxidized glutathione, 0.005% Tween 80.
Myeloperoxidase release from PMNs
Whole blood was collected from healthy volunteers, added to heparin (2 U ml−1), and layered over histopaque (density 1.007 g ml−1, Sigma). After centrifugation (400×g, 30 min, 22°C), the top layers were removed, leaving a layer of erythrocytes and PMNs. Erythrocyte lysis with water (4°C) for 40 s was followed by the addition of phosphate buffered saline to restore isotonicity. The mixture was centrifuged (700×g, 4°C, 20 min) and most of the supernatant carefully removed. The lysis and centrifugation steps were performed twice more until a pellet of PMNs visibly free from erythrocytes was obtained.
To investigate the nature of the antagonism caused by these new C5a antagonists, PMNs (4×106 cells ml−1 Hank's balanced salt solution and 0.1% gelatin) were pre-incubated with cytochalasin B (10 μg ml−1) for 10 min at 37°C . The cells were then incubated with increasing concentrations of antagonists for 10 min at 37°C, following which concentration-response curves to C5a were performed by incubating the cells for a further 10 min at 37°C with increasing concentrations of C5a. The level of release of myeloperoxidase was determined as the end point as follows: 50 μl of phosphate buffer (0.1 M, pH 6.8) was added to each well, followed by the addition of 25 μl of a fresh 1 : 1 mixture of dimethoxybenzidine (5.7 mg ml−1) and H2O2 (0.51%). The reaction was stopped at 20 min by addition of 2% sodium azide (25 μl). Absorbances were measured at 450 nm in a Tecan plate reader and corrected for control values (no peptide). Results were expressed as a percentage of the maximal response of the agonist alone.
Umbilical artery spasmogenesis
Umbilical artery strips were prepared as described previously (Sanderson et al., 1994; Taylor et al., 1994). Human umbilical cords were obtained from the Mater Mothers' Hospital, South Brisbane, Queensland, from pregnant women at full term within 15 min of vaginal delivery or Caesarian section. Approximately 20 cm of cord was cut from the mid-section of the umbilical cord, gently squeezed to expel the blood, and then transported in ice-cold Krebs-Ringers solution to the Organ Bath Laboratory in the Department of Physiology and Pharmacology, University of Queensland.
The umbilical arteries were excised, and slit to yield longitudinal strips. The intimal surface of the vessel was gently rubbed with a cotton bud to remove the endothelium. Strips, approximately 4 mm×2 cm, were set up in 2 ml organ baths, with a resting tension of 20 mN, in Krebs-Ringer solution. All tissues were maintained at 37°C and solutions were gassed with 95%O2/5%CO2, pH 7.4 Isometric tensions were measured using strain-gauge transducers (Grass, FT-03), and the incoming signal monitored by means of a computerized recording system (MacLab/8). Tissues were equilibrated for 1–2 h prior to testing. The strips were preincubated with antagonists for 10 min and then challenged with C5aWT. When the contraction reached a plateau, a supramaximal concentration of histamine (10 μM) was added. The results were expressed as a percentage of the maximum contraction to C5a in the absence of antagonists.
Receptor binding assays
Binding assays were performed on human PMN membranes prepared as previously described (Rollins et al., 1988; Siciliano et al., 1990). Membranes (0.5–1 μg protein) were incubated with 125I-C5a (∼50 pM, New England Nuclear, MA, U.S.A.) and antagonists in buffer (in mM): (HEPES 50, CaCl2 1, MgCl2 5, bovine serum albumin 0.5%, bacitracin 0.1%, phenylmethylsulphonyl fluoride 100 μM) for 60 min at 4°C in Millipore multiscreen 96-well plates (GV, 0.22 μm). After filtration and washing, the filters were dried and radioactivity counted in a gamma counter (LKB). Non-specific binding (in the presence of 100 nM C5a or 1 mM peptide) was routinely 10–20% of the total binding.
Umbilical artery membranes were prepared as follows. Arteries from umbilical cords were dissected free of connective tissue, and the endothelium removed. The tissues were stored in Kreb's solution at −20°C until the day of preparation. The tissues were homogenized in liquid N2 in a Waring blender in a pulsatile manner. Buffer (in mM): (HEPES 50, CaCl2 1, MgCl2 5, pH 7.4) was added and the suspension was centrifuged (2000×g, 4°C, 15 min). The resulting supernatant was centrifuged (40,000×g, 4°C, 30 min). The pellet was resuspended in buffer and washed (40,000×g, 4°C, 30 min). The final pellet was homogenized with a glass–glass, hand-held homogenizer. An aliquot was taken for protein concentration analysis using the Lowry procedure (Sigma, MO, U.S.A.), and the remainder aliquotted and frozen (−70°C) until required.
Binding assays included 200 μg protein/tube, which resulted in non-specific binding (in the presence of 100 nM C5a or 1 mM peptide) of approximately 30% of the total binding. Membrane preparation, 125I-C5a (∼50 pM) and antagonists were incubated in buffer as for the PMN membrane binding assay, for 60 min at 4°C. The mixture was filtered onto Millipore GV 0.22 μm, 2.5 cm diameter filters. The filters were then washed, dried and counted. The total amount of specific binding of 125I-C5a under these conditions ranged from 400–1000 c.p.m./tube.
Data analysis
To determine the equilibrium dissociation constant (Kb) of the antagonists, equieffective concentrations of the agonist before and after treatment with antagonist were plotted in a double reciprocal manner from each individual experiment for both PMN myeloperoxidase release and umbilical artery strip contraction. The Kb was calculated from the equation Kb=[antagonist]/slope-1 (Kenakin, 1997). This was obtained from the linear regression of 1/[A] versus 1/[A′] where A is the agonist concentration and A′ is the equiactive concentration of agonist in the presence of antagonist. Kenakin (1997) stated that more accurate estimates of Kb are derived from experiments in which the maximal response to the agonist is depressed by the antagonist to less than 50% of the maximal response, so this calculation was performed using a concentration of antagonist which gave at least 50% inhibition. Probit analysis was performed for the agonist curves in the presence and absence of antagonist, solving for a number of points between 30–50% of the maximum response. The reciprocal inverse log of these concentrations was then used to solve for Kb=[antagonist]/slope-1.
Binding data was analysed by non-linear regression analysis (Graphpad Prism 2.0), to calculate IC50 values for each experiment. Arithmetic means±standard error were calculated for pKb and −log IC50 values. One way ANOVA followed by Newman Keuls post tests or Student's t-tests were also performed on these values, as indicated in the results, to determine statistically significant differences (P<0.05).
Results
Effects of C5a antagonists in PMNs
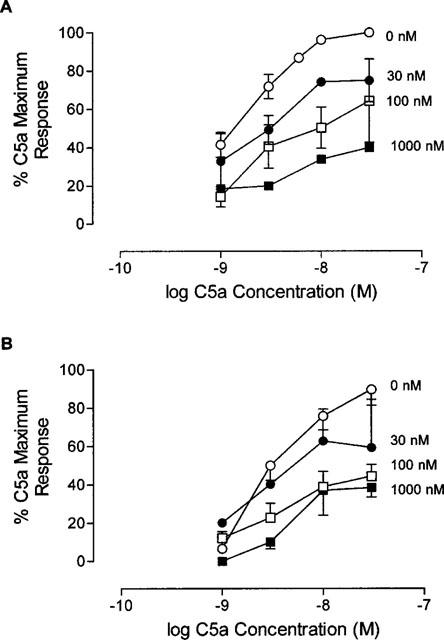
The effects of MeFKPdChaWr and F-[OPdChaWR] were first determined in PMNs using C5a as the stimulus to release myeloperoxidase. Concentration response curves to C5a were performed following pretreatment of the PMNs with increasing concentrations of the antagonists (Figure 1). The results showed that both C5a antagonists inhibited C5a in an insurmountable fashion, for increasing concentrations of the antagonists reduced the maximal response achievable in a dose-dependent manner (Figure 1). The cyclic antagonist, F[OPdChaWR], was 30 times more potent than the linear antagonist, MeFKPdChaWr, as reflected in their pKb values (8.64±0.21 (n=4) and 7.16±0.11 (n=5), respectively (P<0.05, Table 1). The human recombinant C5a (referred to as C5aWT, see Methods), was also tested for its activity in PMNs (Table 1), and no difference in pKb values was apparent compared to those obtained with the commercially available C5a.
Figure 1.

Inhibition of C5a by linear and cyclic C5a antagonists in PMNs. Concentration response curves for C5a-induced MPO release in PMNs were performed after a 10 min incubation with MeFKPdChaWr (A) or F-[OPdChaWR] (B). Data for each point is expressed as a percentage for the maximum C5a response for each experiment. The mean value is shown±s.e. (n=3–6).
Table 1.
Antagonism of C5aWT-induced activity

Effects of C5a antagonists in umbilical artery
C5aWT was used for the determination of Kb values in umbilical artery. Because C5aWT was produced with six histidine residues tagged at its N-terminus, it was necessary to compare the affinity and activity of this compound to that of the commercially available recombinant C5a (Sigma) that was used in the other experiments. The binding affinities for the C5aR in PMNs of C5aWT and Sigma C5a were first obtained. C5aWT, with an IC50=0.2 nM (−log IC50=9.69±0.23, n=3), had an affinity that was not significantly different to Sigma C5a; IC50=0.4 nM (−log IC50=9.37±0.12, n=3; P>0.05). There was also no difference in the ability of C5a and C5aWT to stimulate the release of myeloperoxidase from PMNs; pD2 (−log EC50)=7.98±0.21 (n=3), and 8.02±0.08 (n=3), respectively. In umbilical artery, C5aWT and Sigma C5a displayed similar spasmogenic potencies, for their pD2 values of 8.32±0.23 (n=5) and 8.13±0.13 (n=7) respectively, were not significantly different (P>0.05). The ready availability of C5aWT allowed the determination of the nature of the interaction of the C5a antagonists to be carried out in spasmogenesis assays.
Both C5a antagonists inhibited the spasmogenic effects of C5aWT in umbilical artery (Figure 2), demonstrating insurmountable antagonism, as was seen against C5a in PMNs (Figure 1). In contrast to the results obtained in PMNs, there was no significant difference in potencies (pKb values) of the antagonists against C5aWT (P>0.05, Table 1) in umbilical artery.
Figure 2.
Inhibition of C5a-induced spasmogenesis by linear and cyclic C5a antagonists. Concentration response curves for C5aWT in human umbilical artery were performed after a 10 min incubation with MeFKPdChaWr (A) or F-[OPdChaWR] (B). Force of contraction relative to a supramaximal concentration of histamine was then measured. Data for each point is expressed as a percentage of the maximum C5a response of each day. The mean value is shown±s.e. (n=3–6).
Receptor affinities of C5a antagonists
The affinities of the antagonists for the C5aR were determined in membranes prepared from PMNs and umbilical artery. Competition curves with 125I-C5a resulted in similar affinities for Sigma C5a in both preparations (Table 2, Figure 3). In PMN membranes, the cyclic antagonist had a 4 fold higher affinity for the C5aR than MeFKPdChaWr (P<0.05), whereas the C5aR affinities of the two antagonists in umbilical arterial membranes were similar (Table 2). The C5aR affinities therefore reflected the relative potencies of the antagonists in both preparations. Since the pKb values in umbilical artery were performed with C5aWT, it was necessary to show that the C5aWT had similar affinity as Sigma C5a for the C5aRs in this tissue. The values for C5aWT were −logIC50=10.14± 0.16±0.14 nM, n=3), which was similar to Sigma C5a (IC50 0.12 nM, Table 2). Thus, a comparison of antagonist activity and receptor binding affinities indicated that Sigma C5a and C5aWT behaved identically in both systems employed in this study.
Table 2.
Inhibition of 125I-C5a binding
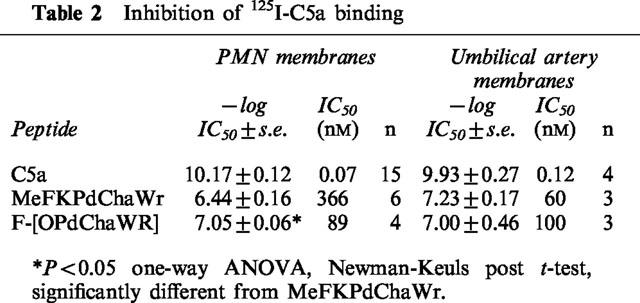
Figure 3.
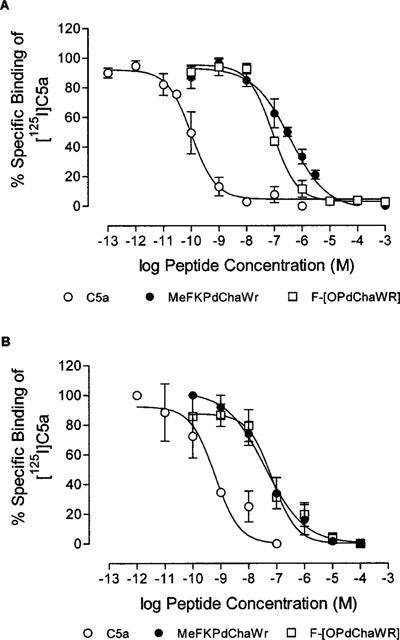
Inhibition of [125I]C5a binding by linear and cyclic C5a antagonists. Peptide, membrane preparation and [125I]C5a were incubated (4°C, 60 min), before filtration and counting. (A) PMN membranes. (B) Umbilical artery membranes. Points are expressed as per cent specific binding of each experiment and the mean±s.e. is shown. (n=3–5).
Discussion
C5a binds to its receptor at multiple sites, and there have been numerous studies using site-directed mutagenesis of C5a which have attempted to identify the residues binding to the receptor (Bubeck et al., 1994; Federwisch et al., 1993; Mollison et al., 1989). Whilst these studies have suggested a number of interaction points between C5a and C5aR, a hypothesis involving two major domains of C5a interacting with the receptor in two distinct regions or sites has evolved (Chenoweth & Hugli, 1980; Siciliano et al., 1994). In this hypothesis, site 1 is the extracellular domain of the C5aR that interacts with the four-helix bundle of C5a, while site two consists of a second, possibly intramembranous region of C5aR, to which the C-terminal region of C5a binds and activates the receptor. Because the C-terminal region of C5a alone contains the receptor-activating sequence, this region has been used as a template for analogue development by a number of research groups, in efforts to develop a C5a receptor antagonist, as well as selective agonists of C5a (Drapeau et al., 1993; Ember et al., 1992; Kawai et al., 1991; Konteatis et al., 1994; Sanderson et al., 1994; 1995).
A specific C5a antagonist derived from the C-terminal region of C5a was first reported by Konteatis et al. (1994). This compound, the linear peptide MeFKPdChaWr, inhibited the effects of C5a in human PMNs, but had only 0.04% of the apparent affinity for the PMN C5aR, compared to C5a. We have confirmed the antagonism of this peptide in the present study, and also examined the nature of the antagonism for this agent, and a new, more potent C5a antagonist recently developed in our laboratories (Short et al., 1999; Wong et al., 1998; Finch et al., 1999), in two systems we have used to characterize C5a and C5a agonists, viz, human PMNs and umbilical artery (Ember et al., 1992; Sanderson et al., 1994; 1995; Finch et al., 1997). In both assay systems, the antagonism of C5a by C5a antagonists was insurmountable in nature, with increasing concentrations of antagonists causing a progressive reduction in the maximum C5a response. In PMNs, the cyclized antagonist had a higher affinity for the PMN C5aR than the linear antagonist, as well as increased antagonistic potency. Conversely, in umbilical artery, there was no difference in the antagonistic potencies or receptor affinities between the two antagonists. The cyclic antagonist exhibited similar affinities for the C5aR in the two systems, while the linear antagonist had a lower C5aR affinity in PMNs than in umbilical artery. The results therefore show functional similarities in the mode of pharmacological antagonism of these peptides, but differences in their relative affinities for the receptors in the different tissues. Insurmountable antagonism can occur by a number of mechanisms, such as pseudoirreversible binding, allosteric binding, receptor internalization or the activation state of the receptor (Kenakin, 1997). Further work is underway to determine the mechanism of action for these C5a antagonists.
Our previous experiments with C5a agonists have shown marked tissue selectivity between umbilical artery and PMNs (Finch et al., 1997; Sanderson et al., 1994; 1995; Vogen et al., 1999). The present results now also demonstrate tissue selectivity for two C5a antagonists. Currently, there is no explanation for these selectivities, as only a single human C5aR has thus far been cloned, with no evidence for receptor subtypes from molecular biological studies (Boulay et al., 1991; Gerard & Gerard, 1991; Haviland et al., 1995; McCoy et al., 1995). However, the C5aR is promiscuous in its coupling with G proteins; that is, it can couple to different G proteins in a cell type-specific manner and can couple to more than one type of G protein in a given cell (Arai & Charo, 1996; Monk et al., 1994; Shum et al., 1995). Also, since different ratios of G proteins exist in different tissues (Monk et al., 1994; Shum et al., 1995), this may alter the conformational or activation state of the receptors in different tissues and hence affect ligand selectivity. Further work is necessary to determine the mechanism(s) for the selectivity of C5a agonists and antagonists. Even if there is only one gene coding for C5aRs, it is possible that C5aRs may eventually need to be classified on the basis of agonist/antagonist selectivity.
The cell type mediating the contractile effect of C5a in umbilical artery has not yet been positively identified. The spasmogenic effect of C5a and C5a agonists in this tissue is abolished by inhibitors of cyclo-oxygenase (Marceau et al., 1987; Taylor et al., 1994), suggesting that vasoconstrictor prostanoids released from an intramural cell stimulate smooth muscle contraction. The umbilical artery is devoid of both mast cells and neurones, but contains macrophages, which is the likely locus of C5aR involved in the spasmogenic effects of C5a agonists (Marceau et al., 1987). In the present study, we have for the first time prepared membranes from this artery, and these displayed specific binding of C5a, with an affinity similar to that seen in PMN membranes. This enabled the determination of the binding affinities of the C5aR antagonists to compare directly with their antagonistic activities in both the artery and PMNs. The binding data confirmed the pharmacological results, demonstrating significant differences between the C5aRs or their associated transducing sytems in the two tissues.
The development of selective C5a agonists and antagonists may have therapeutic applications. Recently, we showed that a specific immune response could be elicited from mice using the agonist YSFKPMPLaR as an immunoadjuvant (Tempero et al., 1997). The development of C5a agonists and antagonists that target differing cell types containing C5aRs may result in specific therapeutic uses. For example, an antagonist that is selective for PMN receptors could be used to inhibit acute inflammatory actions of C5a, while sparing immune processing cells and thereby reducing potential immunosuppressant effects arising from chronic administration of C5a antagonists. The results of the present study indicate the feasibility of such a notion.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, Arthritis and Rheumatism Campaign fellowship (M0543) and a British Heart Foundation project grant (PG/95119) to PNM.
Abbreviations
- C5a
complement factor 5a
- C5aR
C5a receptor
- C5aWT
wild type complement factor 5a
- Kb
equilibrium dissociation constant
- MPO
myeloperoxidase
- PCR
polymerase chain reaction
- pKb
−log Kb
- PMN
polymorphonuclear leukocytes
References
- ARAI H., CHARO I.F. Differential regulation of G-protein-mediated signaling by chemokine receptors. J. Biol. Chem. 1996;271:21814–21819. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- BAUTSCH W., EMDE M., KRETZSCHMAR T., KOHL J., SUCKAU D., BITTER S.D. Human C5a anaphylatoxin: gene cloning and expression in Escherichia coli. Immunobiol. 1992;185:41–52. doi: 10.1016/S0171-2985(11)80316-5. [DOI] [PubMed] [Google Scholar]
- BOULAY F., MERY L., TARDIF M., BROUCHON L., VIGNAIS P. Expression cloning of a receptor for C5a anaphylatoxin on differentiated HL-60 cells. Biochem. 1991;30:2993–2999. doi: 10.1021/bi00226a002. [DOI] [PubMed] [Google Scholar]
- BUBECK P., GROTZINGER J., WINKLER M., KOHL J., WOLLMER A., KLOS A., BAUTSCH W. Site-specific mutagenesis of residues in the human C5a anaphylatoxin which are involved in possible interaction with the C5a receptor. Eur. J. Biochem. 1994;219:897–904. doi: 10.1111/j.1432-1033.1994.tb18571.x. [DOI] [PubMed] [Google Scholar]
- CHENOWETH D.E., HUGLI T.E. Human C5a and C5a analogs as probes of the neutrophil C5a receptor. Mol. Immunol. 1980;17:151–161. doi: 10.1016/0161-5890(80)90067-x. [DOI] [PubMed] [Google Scholar]
- CLANCY R.M., DAHINDEN C.A., HUGLI T.E. Complement-mediated arachidonate metabolism. Prog. Biochem. Pharmacol. 1985;20:120–131. [PubMed] [Google Scholar]
- DEMARTINO J.A., VAN RIPER G., SICILIANO S.J., MOLINEAUX C.J., KONTEATIS Z.D., ROSEN H., SPRINGER M.S. The amino terminus of the human C5a receptor is required for high affinity C5a binding and for receptor activation by C5a but not C5a analogs. J. Biol. Chem. 1994;269:14446–14450. [PubMed] [Google Scholar]
- DRAPEAU G., BROCHU S., GODIN D., LEVESQUE L., RIOUX F., MARCEAU F. Synthetic C5a receptor agonists. Pharmacology, metabolism and in vivo cardiovascular and hematologic effects. Biochem. Pharmacol. 1993;45:1289–1299. doi: 10.1016/0006-2952(93)90282-2. [DOI] [PubMed] [Google Scholar]
- EMBER J.A., SANDERSON S.D., HUGLI T.E., MORGAN E.L. Induction of interleukin-8 synthesis from monocytes by human C5a anaphylatoxin. Am. J. Pathol. 1994;144:393–403. [PMC free article] [PubMed] [Google Scholar]
- EMBER J.A., SANDERSON S.D., TAYLOR S.M., KAWAHARA M., HUGLI T.E. Biologic activity of synthetic analogues of C5a anaphylatoxin. J. Immunol. 1992;148:3165–3173. [PubMed] [Google Scholar]
- FEDERWISCH M., WOLLMER A., EMDE M., STUHMER T., MELCHER T., KLOS A., KOHL J., BAUTSCH W. Tryptophan mutants of human C5a anaphylatoxin: a fluorescence anisotropy decay and energy transfer study. Biophys. Chem. 1993;46:237–248. doi: 10.1016/0301-4622(93)80017-d. [DOI] [PubMed] [Google Scholar]
- FINCH A., VOGEN S., SHERMAN S., KIRNARSKY L., TAYLOR S., SANDERSON S. Biological active conformer of the effector region of human C5a and modulatory effects of N-terminal receptor binding determinants on activity. J. Med. Chem. 1997;40:877–884. doi: 10.1021/jm960727r. [DOI] [PubMed] [Google Scholar]
- FINCH A.M., WONG A.K., PACZKOWSKI N.J., WADI S.K., CRAIK D.J., FAIRLIE D.P., TAYLOR S.M. Low molecular weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J. Med. Chem. 1999;42:1965–1974. doi: 10.1021/jm9806594. [DOI] [PubMed] [Google Scholar]
- FLOREANI A.A., HEIRES A.J., WELNIAK L.A., MILLER-LINDHOLM A., CLARK-PIERCE L., RENNARD S.I., MORGAN E.L., SANDERSON S.D. Expression of receptors for C5a anaphylatoxin (CD88) on human bronchial epithelial cells: enhancement of C5a-mediated release of IL-8 upon exposure to cigarette smoke. J. Immunol. 1998;160:5073–5081. [PubMed] [Google Scholar]
- GASQUE P., SINGHRAO S.K., NEAL J.W., GOTZE O., MORGAN B.P. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am. J. Pathol. 1997;150:31–41. [PMC free article] [PubMed] [Google Scholar]
- GERARD C., GERARD N.P. C5a anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 1994;12:775–808. doi: 10.1146/annurev.iy.12.040194.004015. [DOI] [PubMed] [Google Scholar]
- GERARD N.P., GERARD C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349:614–617. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- GERARD N.P., BAO L., XIAO P.H., EDDY R.J., SHOWS T.B., GERARD C. Human chemotaxis receptor genes cluster at 19q13.3-13.4. Characterization of the human C5a receptor gene. Biochem. 1993;32:1243–1250. doi: 10.1021/bi00056a007. [DOI] [PubMed] [Google Scholar]
- HAVILAND D.L., MCCOY R.L., WHITEHEAD W.T., AKAMA H., MOLMENTI E.P., BROWN A., HAVILAND J.C., PARKS W.C., PERLMUTTER D.H., WETSEL R.A. Cellular expression of the C5a anaphylatoxin receptor (C5aR): demonstration of C5aR on nonmyeloid cells of the liver and lung. J. Immunol. 1995;154:1861–1869. [PubMed] [Google Scholar]
- KAWAI M., QUINCY D.A., LANE B., MOLLISON K.W., LULY J.R., CARTER G.W. Identification and synthesis of a receptor binding site of human anaphylatoxin C5a. J. Med. Chem. 1991;34:2068–2071. doi: 10.1021/jm00111a022. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.P. Pharmacologic analysis of drug-receptor interaction. New York: Lippincott-Raven; 1997. [Google Scholar]
- KONTEATIS Z.D., SICILIANO S.J., VAN R.G., MOLINEAUX C.J., PANDYA S., FISCHER P., ROSEN H., MUMFORD R.A., SPRINGER M.S. Development of C5a receptor antagonists. Differential loss of functional responses. J. Immunol. 1994;153:4200–4205. [PubMed] [Google Scholar]
- MARCEAU F., LUNDBERG C., HUGLI T.E. Effects of the anaphylatoxins on circulation. Immunopharmacol. 1987;14:67–84. doi: 10.1016/0162-3109(87)90031-2. [DOI] [PubMed] [Google Scholar]
- MCCOY R., HAVILAND D.L., MOLMENTI E.P., ZIAMBARAS T., WETSEL R.A., PERLMUTTER D.H. N-formylpeptide and complement C5a receptors are expressed in liver cells and mediate hepatic acute phase gene regulation. J. Exp. Med. 1995;182:207–217. doi: 10.1084/jem.182.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLLISON K.W., MANDECKI W., ZUIDERWEG E.R., FAYER L., FEY T.A., KRAUSE R.A., CONWAY R.G., MILLER L., EDALJI R.P., SHALLCROSS M.A. Identification of receptor-binding residues in the inflammatory complement protein C5a by site-directed mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 1989;86:292–296. doi: 10.1073/pnas.86.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONK P.N., PEASE J.E., BARKER M.D. C5a stimulus-secretion coupling in rat basophilic leukaemia (RBL-2H3) cells transfected with the human C5a receptor is mediated by pertussis and cholera toxin-sensitive G proteins. Biochem. Mol. Biol. Int. 1994;32:13–20. [PubMed] [Google Scholar]
- MORGAN E.L., SANDERSON S., SCHOLZ W., NOONAN D.J., WEIGLE W.O., HUGLI T.E. Identification and characterization of the effector region within human C5a responsible for stimulation of IL-6 synthesis. J. Immunol. 1992;148:3937–3942. [PubMed] [Google Scholar]
- ROLLINS T.E., SICILIANO S., SPRINGER M.S. Solubilization of the functional C5a receptor from human polymorphonuclear leukocytes. J. Biol. Chem. 1988;263:520–526. [PubMed] [Google Scholar]
- SANDERSON S.D., KIRNARSKY L., SHERMAN S.A., EMBER J.A., FINCH A.M., TAYLOR S.M. Decapeptide agonists of human C5a: the relationship between conformation and spasmogenic and platelet aggregatory activities. J. Med. Chem. 1994;37:3171–3180. doi: 10.1021/jm00045a023. [DOI] [PubMed] [Google Scholar]
- SANDERSON S.D., KIRNARSKY L., SHERMAN S.A., VOGEN S.M., PRAKASH O., EMBER J.A., FINCH A.M., TAYLOR S.M. Decapeptide agonists of human C5a: the relationship between conformation and neutrophil response. J. Med. Chem. 1995;38:3669–3675. doi: 10.1021/jm00018a028. [DOI] [PubMed] [Google Scholar]
- SHORT A., WONG A.K., FINCH A.M., HAAIMA G., SHIELS I.A., FAIRLIE D.P., TAYLOR S.M. Effects of a new C5a receptor antagonist on C5a- and endotoxin- induced neutropenia in the rat. Br. J. Pharmacol. 1999;126:551. doi: 10.1038/sj.bjp.0702338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHUM J.K., ALLEN R.A., WONG Y.H. The human chemoattractant complement C5a receptor inhibits cyclic AMP accumulation through Gi and Gz proteins. Biochem. Biophys. Res. Commun. 1995;208:223–239. doi: 10.1006/bbrc.1995.1327. [DOI] [PubMed] [Google Scholar]
- SICILIANO S.J., ROLLINS T.E., DEMARTINO J., KONTEATIS Z., MALKOWITZ L., VAN R.G., BONDY S., ROSEN H., SPRINGER M.S. Two-site binding of C5a by its receptor: an alternative binding paradigm for G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1214–1218. doi: 10.1073/pnas.91.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SICILIANO S.J., ROLLINS T.E., SPRINGER M.S. Interaction between the C5a receptor and Gi in both the membrane-bound and detergent-solubilized states. J. Biol. Chem. 1990;265:19568–19574. [PubMed] [Google Scholar]
- TAYLOR S.M., FINCH A.M., HERON A.E., BROWN L.C., FLORIN T.H. Reversibility of tachyphylaxis to C5a in guinea pig tissues, perfused human placental lobule, and umbilical artery. Inflammation. 1994;18:645–657. doi: 10.1007/BF01535262. [DOI] [PubMed] [Google Scholar]
- TEMPERO R.M., HOLLINGSWORTH M.A., BURDICK M.D., FINCH A.M., TAYLOR S.M., VOGEN S.M., MORGAN E.L., SANDERSON S.D. Molecular adjuvant effects of a conformationally biased agonist of human C5a anaphylatoxin. J. Immunol. 1997;158:1377–1382. [PubMed] [Google Scholar]
- VOGEN S.M., FINCH A.M., WADI S.K., THATCHER J., MONK P.N., TAYLOR S.M., SANDERSON S.D. The influence of Lys68 in decapeptide agonists of C5a on C5a receptor binding, activation and selectivity. J. Pep. Res. 1999;53:8–17. doi: 10.1111/j.1399-3011.1999.tb01612.x. [DOI] [PubMed] [Google Scholar]
- WERFEL T., ZWIRNER J., OPPERMANN M., SIEBER A., BEGEMANN G., DROMMER W., KAPP A., GOTZE O. CD88 antibodies specifically bind to C5aR on dermal CD117+ and CD14+ cells and react with a desmosomal antigen in human skin. J. Immunol. 1996;157:1729–1735. [PubMed] [Google Scholar]
- WHALEY K. Complement in health & disease. MTP Press Ltd. Lancaster; 1987. [Google Scholar]
- WONG A.K., FINCH A.M., PIERENS G.K., CRAIK D.J., TAYLOR S.M., FAIRLIE D.P. Small molecular probes for G-protein-coupled C5a receptors: conformationally constrained antagonists derived from the C terminus of the human plasma protein C5a. J. Med. Chem. 1998;41:3417–3425. doi: 10.1021/jm9800651. [DOI] [PubMed] [Google Scholar]