

Abstract
Effects of indomethacin, the selective cyclo-oxygenase (COX)-2 inhibitors NS-398 and DFU, and dexamethasone on gastric damage induced by 30 min ischaemia followed by 60 min reperfusion (I-R) were investigated in rats. Modulation of gastric levels of COX-1 and COX-2 mRNA by I-R was evaluated using Northern blot and reverse transcription-polymerase chain reaction.
I-R-induced gastric damage was dose-dependently aggravated by administration of indomethacin (1–10 mg kg−1), NS-398 (0.4–4 mg kg−1) or DFU (0.02–2 mg kg−1) as assessed macroscopically and histologically.
Likewise, administration of dexamethasone (1 mg kg−1) significantly increased I-R damage.
Low doses of 16,16-dimethyl-prostaglandin(PG)E2, that did not protect against ethanol-induced mucosal damage, reversed the effects of the selective COX-2 inhibitors, indomethacin and dexamethasone.
I-R had no effect on gastric COX-1 mRNA levels but increased COX-2 mRNA levels in a time-dependent manner. Dexamethasone inhibited the I-R-induced expression of COX-2 mRNA.
I-R was not associated with a measurable increase in gastric mucosal formation of 6-keto-PGF1α and PGE2. PG formation was substantially inhibited by indomethacin (10 mg kg−1) but was not significantly reduced by NS-398 (4 mg kg−1), DFU (2 mg kg−1) or dexamethasone (1 mg kg−1).
The findings indicate that selective COX-2 inhibitors and dexamethasone markedly enhance gastric damage induced by I-R. Thus, whereas COX-2 has no essential role in the maintenance of gastric mucosal integrity under basal conditions, COX-2 is rapidly induced in a pro-ulcerogenic setting and contributes to mucosal defence by minimizing injury. This suggests that in certain situations selective COX-2 inhibitors may have gastrotoxic effects.
Keywords: Cyclo-oxygenase-1, cyclo-oxygenase-2, ischaemia-reperfusion, NS-398, DFU, dexamethasone, indomethacin, gastric mucosal damage
Introduction
Cyclo-oxygenase (COX) catalyzes the bis-oxygenation of free arachidonic acid to generate the intermediate prostaglandin (PG) H2. Cell-specific enzymes convert PGH2 to the physiologically active PGs or thromboxane A. Two isoforms of COX, referred to as COX-1 and COX-2, have been identified, which are encoded by different genes (Hla & Neilson, 1992). The two isoforms have similar amino acid sequences and catalytic activities but differ in their role in physiological and pathological events (DuBois et al., 1998). COX-1 is expressed constitutively in most tissues and cells (O'Neill & Ford-Hutchinson, 1993). In contrast, levels of COX-2 are low or undetectable in peripheral tissues under basal conditions (Kargman et al., 1996). COX-2 mRNA rises rapidly in response to inflammatory and mitogenic stimuli (Raz et al., 1989; Kujubu et al., 1991) and increased expression of COX-2 mRNA and protein occurs at sites of inflammation (for review see Mitchell et al., 1995), in tumours (Eberhart et al., 1994), and in ulcerated gastric tissue (Mizuno et al., 1997; Schmassmann et al., 1998). This led to the supposition that COX-1 functions as a house-keeping enzyme catalyzing the formation of PGs that are involved in physiological functions and homeostasis reactions, whereas COX-2-derived PGs mediate pathological reactions such as pro-inflammatory effects and tumour growth (Vane & Botting, 1995). Up-regulation of COX-2 is prevented by glucocorticoids, that have no effect on the expression of constitutive COX-1 (Kujubu & Herschman, 1992). In contrast, non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the activity of COX-1 and COX-2. While most conventional NSAIDs show little selectivity and inhibit both isoenzymes to a comparable extent (Meade et al., 1993), recently compounds have been developed, that selectively block the activity of COX-2 without affecting COX-1 (for review see Hawkey, 1999).
Non-selective COX inhibitors damage the gastrointestinal mucosa and gastrointestinal injury represents the clinically most significant side effect of chronic NSAID use (Soll et al., 1991). In contrast, selective COX-2 inhibitors do not induce gastric lesions in rats (Futaki et al., 1993; Masferrer et al., 1994; Riendeau et al., 1997; Schmassmann et al., 1998) and cause less gastrointestinal side effects than conventional NSAIDs in humans (for review see Hawkey, 1999). From these findings it was suggested that inhibition of COX-2 underlies the anti-inflammatory and anti-tumour effects of NSAIDs, whereas the gastrointestinal side effects are related to blockade of COX-1 (Vane & Botting, 1995). Hence, selective COX-2 inhibitors were expected to be efficacious anti-inflammatory and chemopreventive drugs devoid of gastrointestinal toxicity.
Recently, evidence has accumulated that, in contrast to the initial concept, selective COX-2 inhibitors interfere with important physiological functions including the resistance of the gastric mucosa against injury. Thus, work by our group indicates that in rats, selective COX-2 inhibitors antagonize the adaptive gastroprotection induced by the mild irritant 20% ethanol (Gretzer et al., 1998), partially counteract the protection evoked by perfusion of the gastric lumen with peptone (Ehrlich et al., 1998) and promote acid-related gastric mucosal damage after inhibition of nitric oxide synthase or depletion of neuropeptides (Maricic et al., 1999). Furthermore, selective COX-2 inhibitors delay the healing of chronic gastric ulcers in mice (Mizuno et al., 1997) and rats (Schmassmann et al., 1998) and cause exacerbation of inflammation-associated colonic injury in rats (Reuter et al., 1996).
The aim of the present study was to compare the effects of the selective COX-2 inhibitors N-[2-(cyclohexyloxy)-4-nitrofenyl]-methanesulphonamide (NS-398) (Futaki et al., 1994) and 5.5-dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl)phenyl-2(5II)-furanone (DFU) (Riendeau et al., 1997) as well as the corticosteroid, dexamethasone on gastric mucosal damage induced by ischaemia-reperfusion (I-R) with that of the non-selective COX inhibitor indomethacin and to determine the influence of concurrent PG administration. To substantiate a role of COX-2-derived PGs, the effect of I-R on the expression of COX-1 and COX-2 mRNA levels in the gastric mucosa of rats subjected to I-R has been assessed.
A preliminary account of this work has been presented in abstract form (Maricic et al., 1998).
Methods
Male Wistar rats (180–220 g) were fasted overnight with free access to water. All experimental protocols were approved by the Animal Care Committee of the University of Bochum.
Ischaemia-reperfusion damage of the gastric mucosa
The rats were anaesthetized (pentobarbital, 50 mg kg−1, i.p.) and tracheotomized. The pylorus was ligated, the celiac artery clamped and 1 ml of 0.1 N HCl injected into the gastric lumen. Reperfusion was established 30 min later by removal of the clamp. After a 60 min reperfusion period, the stomach was excised and gross mucosal damage was assessed in a blinded manner by calculation of a lesion index using a 0–3 scoring system based on the number and severity factor of the lesions as described in detail previously (Stroff et al., 1996). The severity factor was defined according to the length of lesions. Severity factor 0=no lesions visible; 1=lesions <1 mm; 2=lesions 2-4 mm; and 3=lesions>4 mm. The lesion index was calculated as the total number of lesions multiplied by their respective severity factor. In sham operated rats, the abdomen was opened and the celiac artery manipulated without clamping.
For histological study, a strip of the stomach wall parallel to the limiting ridge was processed using routine methods, stained with H&E and examined under a light microscope in a randomized blinded fashion. Two grades of histological injury were assessed: grade 1, superficial damage confined to the surface epithelium; grade 2, deep damage extending beyond the surface epithelium into the region of pits and glands as described before (Gretzer et al., 1998). The length of mucosal areas showing superficial and deep damage was determined and expressed as % of the total section length studied.
Effect of inhibition of COX-1 and COX-2 on ischaemia-reperfusion damage
To assess the role of COX-1 and COX-2, groups of rats were pretreated with indomethacin (1–10 mg kg−1, s.c.) or the selective COX-2 inhibitors NS-398 (0.4–4 mg kg−1, s.c.) and DFU (0.02–2 mg kg−1, s.c.) 60 min before, or dexamethasone (1 mg kg−1, p.o.) 2 h before clamping the celiac artery. In additional experiments, rats received i.v. injections of DFU (2 mg kg−1), indomethacin (10 mg kg−1) or dexamethasone (1 mg kg−1) immediately before or 30 min after removing the arterial clamp. Indomethacin was dissolved in 3% NaHCO3, NS-398 in absolute ethanol and DFU in dimethylsulphoxide. Dexamethasone was suspended in 0.25% methylcellulose for oral administration and dissolved in saline for i.v. injection. Controls received the corresponding vehicle.
Effect of prostaglandin administration
To examine whether the effects of COX inhibitors and dexamethasone are related to suppression of endogenous PGs, rats received concurrent treatment with 16,16-dimethyl-PGE2 (dissolved in 70% ethanol). Rats treated with NS-398 (4 mg kg−1), DFU (2 mg kg−1) or dexamethasone (1 mg kg−1) received s.c. injections of 4 ng kg−1 16,16-dimethyl-PGE2 65 and 2 min before clamping the celiac artery. In rats treated with indomethacin (10 mg kg−1), 15 ng kg−1 16,16-dimethyl-PGE2 was injected s.c. 65 and 2 min before induction of ischaemia.
To exclude a general protective effect, the PG analogue (15 ng kg−1) was injected s.c. 95 min and 32 min before oral instillation of 1 ml of 70% ethanol. Control rats received the vehicle 95 and 32 min before challenge with 70% ethanol. This time schedule was used as pilot experiments had shown that I-R damage did not occur during ischaemia but developed during the reperfusion period.
Northern blot analysis of COX-1 and COX-2
Three groups of rats were studied: (1) Sham operated rats; (2) Rats subjected to ischaemia for 30 min followed by reperfusion for 60 min; and (3) Rats subjected to ischaemia for 30 min and reperfusion for 5.5 h. Aliquots (20 μg) of total RNA extracted from gastric mucosal tissues using Rneasy kit and quantified by measuring the absorbance at 260 nm were fractionated by 1.2% agarose-formaldehyde gel electrophoresis in 1× (N-morpholino)-propanesulphonic acid (MOPS). Transfer onto a nylon membrane was performed by vacuum blotting. After u.v. crosslinking, blots were prehybridized for 4 h and hybridized for 16 h at 42°C in 50% formamide, 0.9 M NaCl, 90 μM sodium citrate, 0.4% sodium dodecyl sulphate (SDS), 5× Denhard's solution and 200 μg ml−1 salmon sperm DNA. The blots were sequentially hybridized with COX-1 (murine 1.2 kb cDNA fragment), COX-2 (murine 1.2 kb cDNA fragment) and with glyceraldehyde-3 phosphate dehydrogenase (GAPDH; human 1.0 kb cDNA fragment) probes, labelled with [32P]-dCTP (3000 Ci mmol−1) by random priming. The RNA blots were washed twice in 150 mM NaCl, 15 mM sodium citrate and 0.1% SDS for 30 min and twice in 30 mM NaCl, 3 mM sodium citrate, 0.1% SDS at 50°C for 10 min. Blots were visualized by autoradiography using reflection film and intensifying screen at −70°C. The expression of the mRNAs was determined by densitometry using computerized image analysis system (Imaging Research Inc., Canada). The ratios of COXs/GAPDH expression were calculated and in sham operated rats were set to 100%.
Reverse transcription-polymerase chain reaction (RT–PCR)
Reverse transcription was performed on 0.8 μg RNA with avian myoblastosis virus reverse transcriptase and an oligo (dT)15 primer according to the manufacturer's protocol. A volume of 2 μl for COX-2 and 1 μl for COX-1 and for GAPDH of the reverse transcribed products were amplified using specific primers. PCR was carried out according to the manufacturer's protocol using 2.5 mM MgCl2, 0.2 mM desoxynucleotide triphosphates, 50 pmol primers and 0.25 U Taq polymerase. Samples were denatured for 2 min at 95°C and specific cDNA amplified using a cycling program: 95°C for 1 min, 59°C for 1 min, and 72°C for 2 min. Final extension time was 7 min at 72°C. Thirty cycles were performed for COX-2 and for COX-1 and 26 for GAPDH amplification. Ten μl volumes of the PCR-product were separated by electrophoresis in agarose gel stained with ethidiumbromide. Specific primers for COX-2 (Beiche et al., 1998) yield an amplification product of 279 bp; for COX-1 (Beiche et al., 1998) give rise to a 447 bp fragment and for GAPDH of a 450 bp fragment. The quantitation of the mRNA expression was performed as described above.
Assessment of gastric mucosal prostaglandin formation
Rats were sham operated or pretreated with vehicle or drugs (each at the highest dose tested) as described above. Mucosal fragments were excised from the glandular part of the stomach 90 min after sham operation or after I-R (30/60 min) and 2 aliquots (40 mg) were incubated in oxygenated Tyrode solution at 37°C for 10 min. The medium was analysed for the content of 6-keto-PGF1α and PGE2 by radioimmunoassays (RIA) (Gretzer et al., 1998). The RIAs used are highly specific for the PG analysed with less than 0.1% crossreaction by other eicosanoids or related compounds.
Statistical analysis
All data are expressed as mean±s.e.mean of n values. Comparisons between groups were made using Student's t-test for paired or unpaired data as appropriate or the Wilcoxon rank test for non-parametric data. A P value of <0.05 was considered significant.
Materials
DFU was a generous gift from Dr A. W. Ford-Hutchinson (Merck-Frosst Canada, Montreal, Canada). NS-398, COX-1 and COX-2 probes were from Cayman Chemical Co. (Ann Arbor, MI, U.S.A.). Rneasy kit was from Qiagen (Hilden, Germany). Salmon sperm DNA was from stratagene (Heidelberg, Germany), GAPDH from Clontech (Palo Alto, CA, U.S.A.). Reverse Transcription System and PCR Core kit were obtained from Promega, Mannheim, Germany. Nylon membranes (Hybond N), readyprime DNA labelling system and [32P]-dCTP for labelling probes were from Amersham (Little Chalfont, U.K.). All other chemicals including MOPS and SDS were purchased from Sigma Chemicals Co. (St. Louis, MO, U.S.A.). [3H]-6-keto-PGF1α and [3H]-PGE2 were from New England Nuclear Co. (Dreieich, Germany).
Results
Effect of inhibition of COX-1 and COX-2 on ischaemia-reperfusion damage
No lesions were observed in the gastric mucosa of sham operated rats. Ischaemia for 30 min followed by reperfusion for 60 min resulted in minor gastric mucosal damage (lesion index 10±1, n=14). Pretreatment (60 min) with the non-selective COX inhibitor indomethacin (1–10 mg kg−1) or the selective COX-2 inhibitors NS-398 (0.4–4 mg kg−1) and DFU (0.02–2 mg kg−1) increased gastric mucosal damage induced by I-R in a dose-dependent manner. At the highest doses of indomethacin, NS-398 and DFU used, the lesion index was 42±3, 42±4 and 39±4, respectively (P<0.001 vs I-R alone, n=6 each group). Likewise, pretreatment with dexamethasone (1 mg kg−1) 2 h before clamping the celiac artery markedly aggravated gastric mucosal damage (lesion index 40±3, P<0.001 vs I-R alone, n=7) (Figure 1). A similar increase of mucosal injury compared to controls (lesion index 4±1, n=5) was observed, when indomethacin (10 mg kg−1), DFU (2 mg kg−1) or dexamethasone (1 mg kg−1) were administered immediately before or during the reperfusion period. Thus, the lesion index was 37±8 (P<0.01) and 37±9 (P<0.002) for indomethacin; 33±8 (P<0.01) and 41±5 (P<0.001) for DFU; and 33±8 (P<0.001) and 44±1 (P<0.001) for dexamethasone, respectively, when the drugs were injected immediately before or 30 min after opening the arterial clamp, (n=3–5 each group). Mucosal damage in rats treated with the various vehicles before or during I-R was not different from that in rats treated with I-R alone. Furthermore, treatment with the highest doses each of indomethacin, NS-398, DFU (for 60 min) or dexamethasone (for 2 h) did not induce damage to the gastric mucosa 90 min after sham operation or 90 min after pylorus ligation and intragastric instillation of HCl in the absence of I-R (data not shown).
Figure 1.
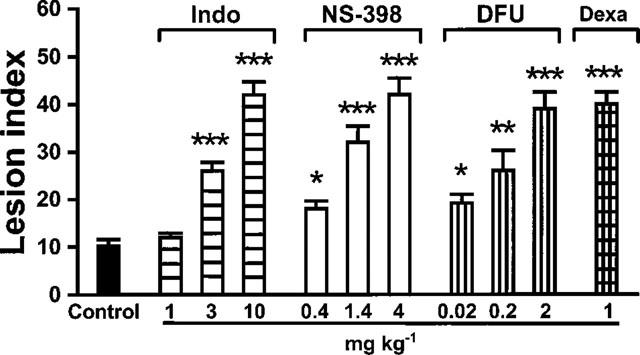
Effect of COX inhibitors on I-R-induced gastric mucosal damage. Pretreatment with the non-selective COX inhibitor indomethacin (Indo), the COX-2-selective inhibitors NS-398 and DFU or dexamethasone (Dexa) aggravated I-R-induced mucosal damage (Control) in a dose-dependent manner. Each bar represents the mean±s.e.mean of 6–7 rats. As the various vehicles did not modify mucosal damage induced by I-R, values of vehicle-treated controls were combined (n=14); *P<0.05; **P<0.01; ***P<0.001 vs I-R alone.
Effect of prostaglandin administration
Treatment with 16,16-dimethyl-PGE2 (15 ng kg−1, administered 5 min before dosing indomethacin and 2 min before induction of ischaemia) completely reversed the damage-augmenting effect of indomethacin (10 mg kg−1) (P<0.001 vs indomethacin alone, n=6). The damage-aggravating effects of NS-398 (4 mg kg−1) and DFU (2 mg kg−1) were abolished by concurrent treatment with 16,16-dimethyl-PGE2 at a dose of 4 ng kg−1 administered 5 min before injection of the COX-2 inhibitors and 2 min before induction of ischaemia (P<0.001 vs selective COX-2 inhibitors alone, n=6 each group). Likewise, concurrent treatment with 16,16-dimethyl-PGE2 (4 ng kg−1) injected 65 and 2 min before clamping the celiac artery reversed the effect of dexamethasone (1 mg kg−1) (P<0.001 vs dexamethasone alone, n=8) (Figure 2).
Figure 2.
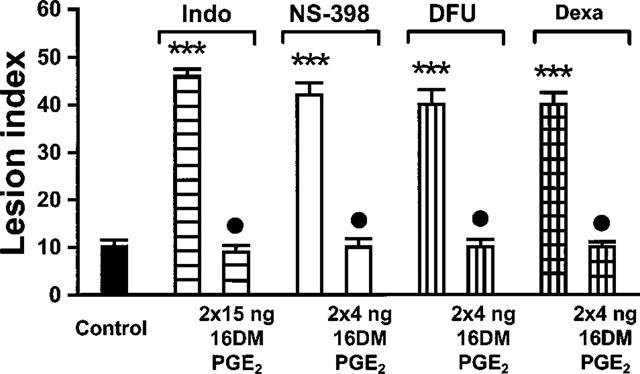
Effect of PG administration on the aggravation of I-R damage induced by suppression of COX activity or induction. Pretreatment with 16,16-dimethyl-PGE2 (16DM PGE2) reversed the damage-aggravating effect of indomethacin (Indo), NS-398, DFU and dexamethasone (Dexa). Rats treated with indomethacin (10 mg kg−1) received concurrent treatment with 16,16-dimethyl-PGE2 at a dose of twice 15 ng kg−1, rats treated with NS-398 (4 mg kg−1), DFU (2 mg kg−1) or dexamethasone (1 mg kg−1) at a dose of twice 4 ng kg−1. Bars represent the mean±s.e.mean of 6–8 rats; vehicle-treated controls were combined (n=14). ***P<0.001 vs controls subjected to I-R alone; •P<0.001 vs COX inhibitor or dexamethasone without concurrent treatment with the PG analogue.
To exclude the possibility that 16,16-dimethyl-PGE2 at the doses used has general gastroprotective activity, we assessed the effect of the PG analogue against ethanol-induced gastric mucosal injury. Oral instillation of 1 ml of 70% ethanol resulted in severe mucosal damage (lesion index 40±1, n=6), that was not reduced by pretreatment with the PG analogue at a dose of twice 15 ng kg−1 (lesion index 40±2, n=4).
Histological injury
Sham operated rats showed no histological injury of the gastric mucosa (results not shown). Deep histological mucosal injury extending beyond the surface epithelium into the region of pits and glands was negligible in rats subjected to I-R alone (1±1% of section length) but was significantly increased in rats pretreated with indomethacin (10 mg kg−1), NS-398 (4 mg kg−1), DFU (2 mg kg−1) or dexamethasone (1 mg kg−1). Concurrent administration of 16,16-dimethyl-PGE2 (twice 4 ng kg−1) prevented the damage-aggravating effect of DFU and dexamethasone (Figure 3). Effects of the various treatments on the extent of grade 1 superficial damage were not significantly different (data not shown).
Figure 3.
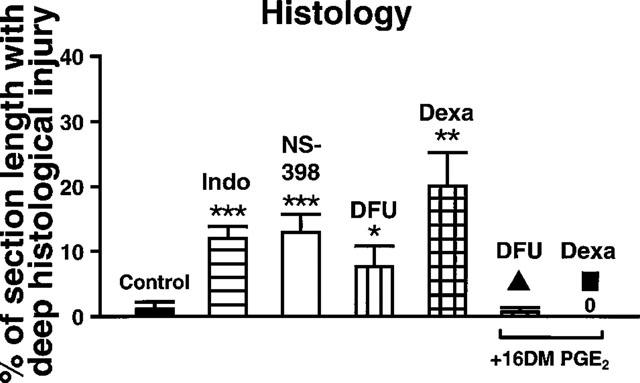
Effects of COX inhibitors and dexamethasone on histological damage induced by I-R. The length of mucosal areas that showed deep mucosal injury reaching beyond the epithelial surface into the area of pits and glands was determined and expressed as % of the total section length studied per stomach. Rats treated with indomethacin (Indo, 10 mg kg−1), NS-398 (4 mg kg−1), DFU (2 mg kg−1) or dexamethasone (Dexa, 1 mg kg−1) before induction of I-R were compared with rats subjected to I-R only (Control). In addition, groups of rats received treatment with 16,16-dimethyl-PGE2 (16DM PGE2, twice 4 ng kg−1) concurrent with DFU (2 mg kg−1) or dexamethasone (1 mg kg−1). All values are the mean±s.e.mean of 5–8 experiments in each group; vehicle-treated controls were combined. *P<0.05, **P<0.01, ***P<0.001 vs controls; ▴P<0.02, ▪P<0.01 vs the corresponding compound alone.
Gastric mucosal expression of COX-1 and COX-2
COX-1 mRNA as assessed by Northern blot analysis was readily detectable in the gastric mucosa of sham operated rats, with no apparent change in the expression of COX-1 for up to 6 h after induction of ischaemia. In contrast, COX-2 mRNA was just above the detection limit in the gastric mucosa of sham operated rats but after ischaemia for 30 min and reperfusion for 60 min a substantial up-regulation was observed. Six hours after induction of ischaemia (with prolongation of the reperfusion period to 5.5 h), levels of COX-2 mRNA were further increased. A Northern blot analysis and the corresponding COX/GAPDH ratios as assessed by densitometry are shown in Figure 4A,B. RT–PCR confirmed induction of COX-2 after 30 min of ischaemia and 60 min of reperfusion and showed that pretreatment with dexamethasone (1 mg kg−1) attenuated the up-regulation of COX-2 but not levels of COX-1 mRNA (ratio of COX-2/GAPDH: sham operation 100, I-R 656, dexamethasone before I-R 440; ratio of COX-1/GAPDH: sham operation 100, I-R 95, dexamethasone before 1-R 100).
Figure 4.
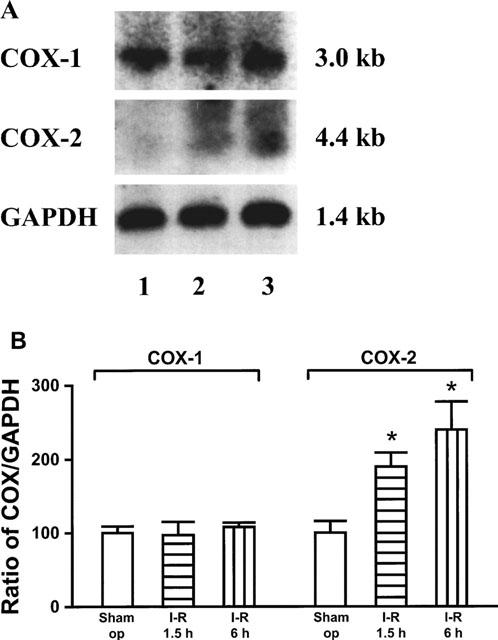
Effect of I-R on mRNA expression of COX-1 and COX-2. Total RNA was extracted from gastric mucosa of sham operated rats (Sham op, lane 1), rats subjected to 30 min ischaemia and 60 min reperfusion (1.5 h, lane 2) or 30 min ischaemia and 5.5 h reperfusion (6 h, lane 3). The autoradiograph shows a representative Northern blot analysis hybridized with cDNAs specific for COX-1, COX-2 and GAPDH (as internal standard) (A). Relative signal intensities of COX-1 and COX-2 to that of GAPDH were quantified and the ratio of sham operated rats was set to 100% (B). The values are expressed as mean±s.e.mean (n=3); *P<0.05 vs sham operated rats.
Gastric mucosal formation of prostaglandins
Gastric mucosal fragments obtained from sham operated rats released 518±107 pg mg−1 10 min−1 6-keto-PGF1α and 468±51 pg mg−1 10 min−1 PGE2 (n=6 per group) during incubation in vitro. Release of 6-keto-PGF1α and PGE2 was not significantly different after 30 min of ischaemia followed by 60 min of reperfusion. In rats subjected to I-R, indomethacin (10 mg kg−1) suppressed gastric mucosal release of 6-keto-PGF1α by 93% and PGE2 by 96% (P<0.001 each). In contrast, pretreatment with DFU (2 mg kg−1) or dexamethasone (1 mg kg−1) did not cause a measurable reduction of gastric mucosal release of 6-keto-PGF1α and PGE2 in rats subjected to I-R. Results are shown in Figure 5.
Figure 5.
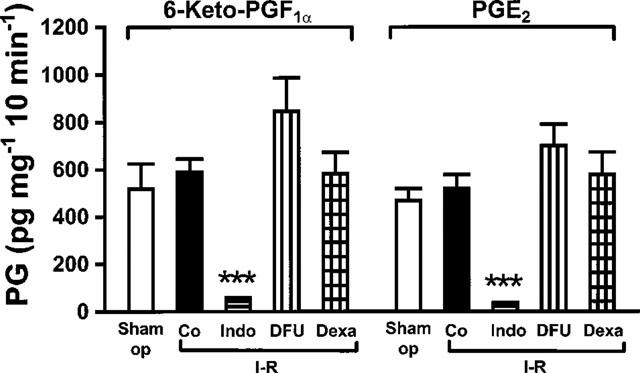
Effect of indomethacin, DFU and dexamethasone on gastric mucosal PGs. Mucosal fragments were incubated in Tyrode solution at 37°C for 10 min and release of PGs into the medium was measured by RIA. Rats were sham operated (Sham op) or subjected to ischaemia for 30 min followed by reperfusion for 60 min (Co). Additional groups of rats were pretreated with indomethacin (Indo, 10 mg kg−1), DFU (2 mg kg−1) or dexamethasone (Dexa, 1 mg kg−1) before I-R. Bars represent the mean±s.e.mean of 6–8 experi-ments; vehicle-treated rats were combined. ***P<0.001 vs I-R alone.
Discussion
Hypoxia followed by reperfusion leads to tissue injury. Several mechanisms seem to contribute to I-R damage. Considerable evidence has accumulated that reactive oxygen metabolites produced by invading neutrophils mediate the microvascular and parenchymal injury associated with I-R (Hernandez et al., 1987). Neutrophil adherence and activation is brought about by potent chemoattractants such as platelet-activating factor (Kubes et al., 1990) and leukotriene B4 (Zimmerman et al., 1990), that are generated following lipid peroxidation and phospholipase A 2 activation during I-R (Otamiri & Tagesson, 1989).
Whereas there is a substantial body of experimental data characterizing the factors that promote injury, tissue defence reactions that counterbalance the noxious effects of I-R are less well understood. This study investigates the role of COX-1 and COX-2 in gastric mucosal resistance against damage induced by I-R. Gastric mucosal levels of COX-1 mRNA were high in sham operated rats and did not change during the course of I-R. In contrast, in sham operated rats, levels of COX-2 mRNA were low confirming previous findings in normal rat gastric mucosa (Kargman et al., 1996; Ferraz et al., 1997). Ischaemia for 30 min followed by reperfusion for 60 min resulted in a marked increase in COX-2 mRNA expression which was further pronounced when the reperfusion period was prolonged to 5.5 h. Dexamethasone diminished the induction of COX-2 but did not affect levels of COX-1 in the gastric mucosa during I-R. Up-regulation of COX-2 but not COX-1 mRNA associated with I-R in the rat stomach has also been reported by Kishimoto et al. (1998). Furthermore, in human vascular endothelial cells in culture, hypoxia was found to increase the expression of the COX-2 gene (Schmedtje et al., 1997). Finally, Davies et al. (1997) described a significant increase in COX-2 mRNA and immunoreactivity in the rat gastric mucosa 3 h after oral administration of aspirin. Taken together, these findings show that the gastric mucosa has the capacity to rapidly up-regulate COX-2 in response to various stimuli.
The cellular localization of COX-2 induced by I-R remains to be identified. Using immunohistochemistry, in the rat stomach COX-2 protein was found in surface mucus cells in normal gastric mucosa (Iseki, 1995) and in monocytes, macrophages, fibroblasts and endothelial cells in ulcerated mucosa (Schmassmann et al., 1998). COX-2 could be up-regulated in these cells during I-R. In addition, neutrophils activated by proinflammatory stimuli express increased levels of COX-2 in vitro (Pouliot et al., 1998) and in vivo (Tomlinson et al., 1994). Activated neutrophils invading into I-R mucosa could thus represent a major source of the increased COX-2 expression.
To evaluate whether the up-regulation of COX-2 observed during I-R is functionally relevant as a mechanism contributing to gastric mucosal defense, we studied the effects of a non-selective NSAID, two selective inhibitors of COX-2 and dexamethasone on injury induced by I-R. Rats subjected to I-R alone showed only minor gastric mucosal injury. Pretreatment with indomethacin, which inhibits both COX-1 and COX-2 (Meade et al., 1993), aggravated I-R-induced gastric mucosal damage in a dose-dependent manner. With the highest dose of indomethacin tested (10 mg kg−1), the gastric mucosa of rats subjected to I-R showed severe damage resulting in a 4 fold increase in the lesion index. Similarly, pretreatment with the selective COX-2 inhibitors NS-398 and DFU dose-dependently augmented gastric mucosal injury induced by I-R. The doses of indomethacin, NS-398 and DFU that maximally increased I-R damage exert only submaximal (40–60%) inhibition of inflammation and pain (Futaki et al., 1993; Riendeau et al., 1997). In addition, dexamethasone, which inhibited the up-regulation of COX-2 associated with I-R, enhanced gastric mucosal damage. The deleterious effects of the selective COX-2 inhibitors and dexamethasone were comparable to that elicited by indomethacin as assessed by gross mucosal damage and histology. The damage-aggravating effect of indomethacin, DFU and dexamethasone was also observed when the drugs were administered at the beginning or during the reperfusion period. This is in keeping with the finding that mucosal damage does not occur during ischaemia but during reperfusion and drug-induced aggravation of injury develops within 10 min (data not shown). Thus, a very short period of suppressed COX-2 activity is sufficient to markedly increase mucosal damage associated with I-R. Two-and-a-half hours after administration, the compounds studied did not induce any lesions in the stomach of rats not subjected to I-R indicating that the aggravation of I-R injury was not the consequence of a general ulcerogenic activity.
Gastric mucosal formation of 6-keto-PGF1α and PGE2 as determined during an ex vivo incubation was not different in sham operated rats and rats subjected to I-R. In vivo pretreatment with indomethacin abolished gastric mucosal formation of both PGs during incubation ex vivo. In contrast, under identical conditions neither DFU nor dexamethasone caused measurable suppression of gastric mucosal synthesis of 6-keto-PGF1α and PGE2. This indicates that the aggravation of I-R damage evoked by NS-398 and DFU is not mediated by inhibition of COX-1. Lack of inhibitory action of selective COX-2 inhibitors on gastric mucosal PG formation in normal rats has been reported previously (Masferrer et al., 1994; Mizuno et al., 1997; Riendeau et al., 1997; Schmassmann et al., 1998). Our findings show that selective inhibition of COX-2-derived PGs cannot be demonstrated even when gastric mucosal expression of COX-2 is increased by I-R. This result suggests that even after induction of COX-2 mRNA during I-R formation of gastric PGs ex vivo is still catalyzed mainly by COX-1. COX-2-derived PGs obviously represent only such a small part of the total PG pool that isolated inhibition of PG biosynthesis via the COX-2 pathway does not result in a substantial and therefore measurable reduction of the total amount of PGs generated in the gastric mucosa. This does not exclude the possibility that under in vivo conditions COX-2 expression may prevail at sites relevant for mucosal defence and thus contribute to essential physiological functions.
Despite the failure to demonstrate inhibition of gastric mucosal PG formation by selective COX-2 inhibitors and dexamethasone, the findings of this study clearly indicate that concurrent treatment with the PG analogue 16,16-dimethyl-PGE2 completely prevents the aggravation of I-R injury induced by indomethacin, NS-398, DFU, and dexamethasone. The PG analogue was effective at very low doses that did not protect against ethanol-induced mucosal injury suggesting that the reversal of the effect of COX inhibition on I-R damage is not the result of general mucosal protection.
Although, at present, a mechanism independent of suppression of endogenous PG cannot be excluded with certainty, the reversal of the damage-aggravating effects of selective COX-2 inhibitors and dexamethasone by PG administration supports the proposition that COX-2-derived PGs strengthen the resistance of the gastric mucosa against I-R-induced injury. In addition to mucosal protection (Robert, 1979), PGs exert various effects that could counteract I-R damage. Thus, PGs of the I, E and D series have been found to inhibit the generation of reactive oxygen metabolites, respiratory burst and/or chemotaxis in activated neutrophils (Wong & Freund, 1981; Gryglewski et al., 1987; Kainoh et al., 1990). E-type PGs inhibited the release of leukotriene B4 from activated neutrophils (Ham et al., 1983). In human vascular smooth muscle cells, the selective COX-2 inhibitor L-745,337 significantly increased the interleukin-1-induced expression of the intercellular adhesion molecule (ICAM)-1, an effect which was reversed by PGE2 (Bishop-Bailey et al., 1998). Expression of adhesion molecules represents an essential step in the invasion of neutrophils into tissues during I-R (Hernandez et al., 1987).
Apoptosis was suggested to be a major mode of cell death in the destruction of rat small intestinal epithelial cells induced by I-R (Ikeda et al., 1998). Non-selective NSAIDs and selective COX-2 inhibitors caused apoptosis and induced COX-2 in various cell types (Hanif et al., 1996; Liu et al., 1998). However, in contrast to the aggravation of I-R damage evoked by COX inhibitors in our experiments, NSAID-promoted induction of apoptosis was not antagonized by exogenous PGE2 and was not mimicked by dexamethasone (Lu et al., 1995). Enhanced induction of apoptosis does, therefore, probably not mediate the aggravating effects of COX inhibitors and dexamethasone on I-R injury.
In normal rats, treatment with indomethacin caused severe injury to the gastric mucosa. The lesions took several hours to occur and were fully developed 5 h after drug administration (Schmassmann et al., 1998). Ulcerogenicity of indomethacin in experimental animals has been reported previously (Whittle, 1981) and similar effects occur with most available NSAIDs. In contrast, in normal rats, selective inhibitors of COX-2 did not damage the gastric mucosa (Futaki et al., 1993; Masferrer et al., 1994; Riendeau et al., 1997; Schmassmann et al., 1998). In humans, NSAID-induced injury to the gastric mucosa is well established (Soll et al., 1991). First clinical trials in patients with osteoarthritis, rheumatoid arthritis and post-surgical dental pain suggest that selective COX-2 inhibitors at anti-inflammatory and analgesic doses cause less gastrointestinal side effects than conventional NSAIDs (for review see Hawkey, 1999). Thus, there is a clear-cut difference in the ulcerogenic potency between conventional NSAIDs and selective COX-2 inhibitors when the drugs are administered in the absence of a potentially noxious stimulus suggesting that COX-2 does not play an important role in the maintenance of gastric mucosal integrity under resting conditions. In the face of pending injury, however, COX-2 appears to gain relevance as a mediator of mucosal defence. In this situation, levels of COX-2 expression rapidly increase and COX-2-derived PGs seem to strengthen mucosal resistance and minimize mucosal damage. When the up-regulation of COX-2 expression is prevented by dexamethasone or the activity of the COX-2 enzyme is blocked by a non-selective NSAID or a selective COX-2 inhibitor, frank ulceration occurs. Reuter et al. (1996) demonstrated a marked increase in COX-2 mRNA expression in the colon of rats after induction of colitis and exacerbation of colonic inflammation following treatment with selective COX-2 inhibitors. Together with our findings in the stomach this suggests a generalized role for COX-2 in gastrointestinal mucosal defence.
In the human forearm and calf, conventional NSAIDs markedly reduced the reactive hyperaemia that occurs during reperfusion after arterial occlusion. Basal blood flow or exercise-induced hyperaemia was not affected by NSAIDs (Nowak & Wennmalm, 1979; Carlsson & Wennmalm, 1983; Carlsson et al., 1987). These findings suggest an appreciable contribution of endogenous PG to post-occlusive vasodilation in man. Whether reduction of post-occlusion hyperaemia is also induced by selective COX-2 inhibitors and whether inhibition of COX-1 and/or COX-2 aggravates I-R tissue damage in man is not known. In the clinical setting, I-R may occur in various disorders such as shock and resuscitation, short-term mesenteric infarction, non-occlusive intestinal ischaemia, surgery or cardiovascular disease.
Our findings thus demonstrate that in rats selective COX-2 inhibitors and dexamethasone substantially aggravate gastric mucosal damage following I-R. As I-R is associated with increased expression of COX-2 this could reflect protective effects of COX-2-derived PGs in the gastric mucosa during I-R. While COX-2 does not play a major role in the maintenance of gastric mucosal integrity in the absence of a noxious stimulus, the enzyme may have an important function in minimizing damage to the gastric mucosa in a pro-ulcerogenic setting.
Acknowledgments
This work was supported by grant Pe-215/6 from the Deutsche Forschungsgemeinschaft.
Abbreviations
- COX
cyclo-oxygenase
- GAPDH
glyceraldehyde-3 phosphate dehydrogenase
- I-R
ischaemia-reperfusion
- MOPS
(N-morpholino)-propanesulphonic acid
- NSAID
non-steroidal anti-inflammatory drug
- PG
prostaglandin
- RIA
radioimmunoassay
- SDS
sodium dodecyl sulphate
References
- BEICHE F., BRUNE K., GEISSLINGER G., GOPPELT-STRUEBE M. Expression of cyclooxygenase isoforms in the rat spinal cord and their regulation during adjuvant-induced arthritis. Inflamm. Res. 1998;47:482–487. doi: 10.1007/s000110050362. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., BURKE-GAFFNEY A., HELLEWELL P.G., PEPPER J.R., MITCHELL J.A. Cyclo-oxygenase-2 regulates inducible ICAM-1 and VCAM-1 expression in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998;249:44–47. doi: 10.1006/bbrc.1998.8966. [DOI] [PubMed] [Google Scholar]
- CARLSSON I., SOLLEVI A., WENNMALM A. The role of myogenic relaxation, adenosine and prostaglandins in human forearm reactive hyperaemia. J. Physiol. 1987;389:147–161. doi: 10.1113/jphysiol.1987.sp016651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARLSSON I., WENNMALM A. Effect of different prostaglandin synthesis inhibitors on post-occlusive blood flow in human forearm. Prostaglandins. 1983;26:241–252. doi: 10.1016/0090-6980(83)90092-8. [DOI] [PubMed] [Google Scholar]
- DAVIES N.M., SHARKEY K.A., ASFAHA S., MACNAUGHTON W.K., WALLACE J.L. Aspirin causes rapid up-regulation of cyclo-oxygenase-2 expression in the stomach of rats. Aliment. Pharmacol. Ther. 1997;11:1101–1108. doi: 10.1046/j.1365-2036.1997.00247.x. [DOI] [PubMed] [Google Scholar]
- DUBOIS R.N., ABRAMSON S.B., CROFFORD L., GUPTA R.A., SIMON L.S., VAN DE PUTTE L.B.A., LIPSKY P.E. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- EBERHART C.E., COFFEY R.J., RADHIKA A., GIARDIELLO F.M., FERRENBACH S., DUBOIS R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- EHRLICH K., PLATE S., STROFF T., GRETZER B., RESPONDEK M., PESKAR B.M. Peptidergic and cholinergic neurons and mediators in peptone-induced gastroprotection: role of cyclooxygenase-2. Am. J. Physiol. 1998;274:G955–G964. doi: 10.1152/ajpgi.1998.274.5.G955. [DOI] [PubMed] [Google Scholar]
- FERRAZ J.G.P., SHARKEY K.A., REUTER B.K., ASFAHA S., TIGLEY A.W., BROWN M.L., MCKNIGHT W., WALLACE J.L. Induction of cyclooxygenase 1 and 2 in the rat stomach during endotoxemia: role in resistance to damage. Gastroenterology. 1997;113:195–204. doi: 10.1016/s0016-5085(97)70095-7. [DOI] [PubMed] [Google Scholar]
- FUTAKI N., TAKAHASHI S., YOKOYAMA M., ARAI I., HIGUCHI S., OTOMO S. NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins. 1994;47:55–59. doi: 10.1016/0090-6980(94)90074-4. [DOI] [PubMed] [Google Scholar]
- FUTAKI N., YOSHIKAWA K. , HAMASAKA Y., ARAI I., HIGUCHI S., IISUKA H., OTOMO S. NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen. Pharmacol. 1993;24:105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- GRETZER B., EHRLICH K., MARICIC N., LAMBRECHT N., RESPONDEK M., PESKAR B.M. Selective cyclo-oxygenase-2 inhibitors and their influence on the protective effect of a mild irritant in the rat stomach. Br. J. Pharmacol. 1998;123:927–935. doi: 10.1038/sj.bjp.0701673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., SZCZEKLIK A., WANDZILAK M. The effect of six prostaglandins, prostacyclin and iloprost on generation of superoxide anions by human polymorphonuclear leukocytes stimulated by zymosan or formyl-methionyl-leucyl-phenylalanine. Biochem. Pharmacol. 1987;36:4209–4213. doi: 10.1016/0006-2952(87)90660-5. [DOI] [PubMed] [Google Scholar]
- HAM E.A., SODERMAN D.D., ZANETTI M.E., DOUGHERTY H.W., MCCAULEY E., KUEHL F.A., Jr Inhibition by prostaglandins of leukotriene B4 release from activated neutrophils. Proc. Natl. Acad. Sci. U.S.A. 1983;80:4349–4353. doi: 10.1073/pnas.80.14.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANIF R., PITTAS A., FENG Y., KOUTSOS M.I., QIAO L., STAIANO-COICO L., SHIFF S.I., RIGAS B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- HAWKEY C.J. COX-2 inhibitors. Lancet. 1999;353:307–314. doi: 10.1016/s0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]
- HERNANDEZ L.A., GRISHAM M.B., TWOHIG B., ARFORS K.E., HARLAN J.M., GRANGER D.N. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am. J. Physiol. 1987;253:H699–H703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- HLA T., NEILSON K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IKEDA H., SUZUKI Y., SUZUKI M., KOIKE M., TAMURA J., TONG J., NOMURA M., ITOH G. Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut. 1998;42:530–537. doi: 10.1136/gut.42.4.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISEKI S. Immunocytochemical localization of cyclooxygenase-1 and cyclooxygenase-2 in the rat stomach. Histochem. J. 1995;27:323–328. doi: 10.1007/BF00398975. [DOI] [PubMed] [Google Scholar]
- KAINOH M., IMAI R., UMETSU T., HATTORI M., NISHIO S. Prostacyclin and beraprost sodium as suppressors of activated rat polymorphonuclear leukocytes. Biochem. Pharmacol. 1990;39:477–484. doi: 10.1016/0006-2952(90)90053-n. [DOI] [PubMed] [Google Scholar]
- KARGMAN S., CHARLESON S., CARTWRIGHT M., FRANK J., RIENDEAU D., MANCINI J., EVANS J., O'NEILL G. Characterization of prostaglandin G/H synthase 1 and 2 in rat, dog, monkey, and human gastrointestinal tracts. Gastroenterology. 1996;111:445–454. doi: 10.1053/gast.1996.v111.pm8690211. [DOI] [PubMed] [Google Scholar]
- KISHIMOTO Y., WADA K., NAKAMOTO K., KAWASAKI H., HASEGAWA J. Levels of cyclooxygenase-1 and -2 mRNA expression at various stages of acute gastric injury induced by ischemia-reperfusion in rats. Arch. Biochem. Biophys. 1998;352:153–157. doi: 10.1006/abbi.1997.0572. [DOI] [PubMed] [Google Scholar]
- KUBES P., IBBOTSON G., RUSSELL J., WALLACE J.L., GRANGER D.N. Role of platelet-activating factor in ischemia/reperfusion-induced leukocyte adherence. Am. J. Physiol. 1990;259:G300–G305. doi: 10.1152/ajpgi.1990.259.2.G300. [DOI] [PubMed] [Google Scholar]
- KUJUBU D.A., FLETCHER B.S., VARNUM B.C., LIM R.W., HERSCHMAN H.R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- KUJUBU D.A., HERSCHMAN H.R. Dexamethasone inhibits mitogen induction of the TIS10 prostaglandin synthase/cyclooxygenase gene. J. Biol. Chem. 1992;267:7991–7994. [PubMed] [Google Scholar]
- LIU X.H., YAO S., KIRSCHENBAUM A., LEVINE A.C. NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res. 1998;58:4245–4249. [PubMed] [Google Scholar]
- LU X., XIE W., REED D., BRADSHAW W.S., SIMMONS D.L. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7961–7965. doi: 10.1073/pnas.92.17.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARICIC N., EHRLICH K., SCHULIGOI R., PESKAR B.M. Cyclooxygenase-2-derived prostaglandins interact with nitric oxide and afferent neurons in the maintenance of rat gastric mucosal integrity. Gastroenterology. 1999;116:A280–281. [Google Scholar]
- MARICIC N., EHRLICH K., SCHULIGOI R., RESPONDEK M., PESKAR B.M. Cyclooxygenase-2-derived prostaglandins contribute to mucosal resistance to ischemia/reperfusion injury in the rat stomach. Gastroenterology. 1998;114:A215. [Google Scholar]
- MASFERRER J.L., ZWEIFEL B.S., MANNING P.T., HAUSER S.D., LEAHY K.M., SMITH W.G., ISAKSON P.C., SEIBERT K. Selective inhibition of inducible cyclooxygenase-2 in vivo is antiinflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEADE E.A., SMITH W.L., DE WITT D.L. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- MITCHELL J.A., LARKIN S., WILLIAMS T.J. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem. Pharmacol. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-x. [DOI] [PubMed] [Google Scholar]
- MIZUNO H., SAKAMOTO C., MATSUDA K., WADA K., UCHIDA T., NOGUCHI H., AKAMATSU T., KASUGA M. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112:387–397. doi: 10.1053/gast.1997.v112.pm9024292. [DOI] [PubMed] [Google Scholar]
- NOWAK J., WENNMALM A. A study on the role of endogenous prostaglandins in the development of exercise-induced and post-occlusive hyperemia in human limbs. Acta Physiol. Scand. 1979;106:365–369. doi: 10.1111/j.1748-1716.1979.tb06411.x. [DOI] [PubMed] [Google Scholar]
- O'NEILL G.P., FORD-HUTCHINSON A.W. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett. 1993;330:156–160. doi: 10.1016/0014-5793(93)80263-t. [DOI] [PubMed] [Google Scholar]
- OTAMIRI T., TAGESSON C. Role of phospholipase A2 and oxygenated free radicals in mucosal damage after small intestinal ischemia and reperfusion. Am. J. Surg. 1989;157:562–565. doi: 10.1016/0002-9610(89)90699-5. [DOI] [PubMed] [Google Scholar]
- POULIOT M., GILBERT C., BORGEAT P., POUBELLE P.E., BOURGOIN S., CREMINON C., MACLOUF J., MCCOLL S.R., NACCACHE P.H. Expression and activity of prostaglandin endoperoxide synthase-2 in agonist-activated human neutrophils. FASEB J. 1998;612:1109–1123. doi: 10.1096/fasebj.12.12.1109. [DOI] [PubMed] [Google Scholar]
- RAZ A., WYCHE A., NEEDLEMAN P. Temporal and pharmacological division of fibroblast cyclooxygenase expression into transcriptional and translational phases. Proc. Natl. Acad. Sci. U.S.A. 1989;86:1657–1661. doi: 10.1073/pnas.86.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REUTER B.K., ASFAHA S., BURET A., SHARKEY K.A., WALLACE J.L. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J. Clin. Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIENDEAU D., PERCIVAL M.D., BOYCE S., BRIDEAU C., CHARLESON S., CROMLISH W., ETHIER D., EVANS J., FALGUEYRET J.P., FORD-HUTCHINSON A.W., GORDON R., GREIG G., GRESSER M., GUAY J., KARGMAN S., LÉGER S., MANCINI J.A., O'NEILL G., OUELLET M., RODGER I.W., THÉRIEN M., WANG Z., WEBB J.K., WONG E., XU L., YOUNG R.N., ZAMBONI R., PRASIT P., CHAN C.C. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br. J. Pharmacol. 1997;121:105–117. doi: 10.1038/sj.bjp.0701076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERT A. Cytoprotection by prostaglandins. Gastroenterology. 1979;77:761–767. [PubMed] [Google Scholar]
- SCHMASSMANN A., PESKAR B.M., STETTLER C., NETZER P., STROFF T., FLOGERZI P., HALTER F. Effects of inhibition of prostaglandin endoperoxide synthase-2 in chronic gastro-intestinal ulcer models in rats. Br. J. Pharmacol. 1998;123:795–804. doi: 10.1038/sj.bjp.0701672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMEDTJE J.F., Jr, JI Y.S., LIU W.L., DUBOIS R.N., RUNGE M.S. Hypoxia induces cyclooxygenase-2 via the NF-KB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- SOLL A.H., WEINSTEIN W.M., KURATA J., MCCARTHY D. Nonsteroidal antiinflammatory drugs and peptic ulcer disease. Ann. Intern. Med. 1991;114:307–319. doi: 10.7326/0003-4819-114-4-307. [DOI] [PubMed] [Google Scholar]
- STROFF T., PLATE S., EBRAHIM J.S., EHRLICH K., RESPONDEK M., PESKAR B.M. Tachykinin-induced increase in gastric mucosal resistance: role of afferent neurons, CGRP, and NO. Am. J. Physiol. 1996;271:G1017–G1027. doi: 10.1152/ajpgi.1996.271.6.G1017. [DOI] [PubMed] [Google Scholar]
- TOMLINSON A., APPLETON I., MOORE A.R., GILROY D.W., WILLIS D., MITCHELL J.A., WILLOUGHBY D.A. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. Br. J. Pharmacol. 1994;113:693–698. doi: 10.1111/j.1476-5381.1994.tb17048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE J.R., BOTTING R.M. New insights into the mode of action of anti-inflammatory drugs. Inflamm. Res. 1995;44:1–10. doi: 10.1007/BF01630479. [DOI] [PubMed] [Google Scholar]
- WHITTLE B.J.R. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology. 1981;80:94–98. [PubMed] [Google Scholar]
- WONG K., FREUND K. Inhibition of the n-formylmethionyl-leucyl-phenylalanine induced respiratory burst in human neutrophils by adrenergic agonists and prostaglandins of the E series. Can. J. Physiol. Pharmacol. 1981;59:915–920. doi: 10.1139/y81-141. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN B.J., GUILLORY D.J., GRISHAM M.B., GAGINELLA T.S., GRANGER D.N. Role of leukotriene B4 in granulocyte infiltration into the postischemic feline intestine. Gastroenterology. 1990;99:1358–1363. doi: 10.1016/0016-5085(90)91162-y. [DOI] [PubMed] [Google Scholar]