

Abstract
The brain constituents β-phenylethylamine (PEA) and tryptamine enhance the impulse propagation mediated transmitter release (exocytosis) from the catecholaminergic and serotoninergic neurons in the brain (‘catecholaminergic/serotoninergic activity enhancer, CAE/SAE, effect'). (−)Deprenyl (Selegiline) and (−)1-phenyl-2-propylaminopentane [(−)PPAP] are amphetamine derived CAE substances devoid of the catecholamine releasing property.
By changing the aromatic ring in PPAP we developed highly potent and selective CAE/SAE substances, structurally unrelated to the amphetamines. Out of 65 newly synthetized compounds, a tryptamine derived structure, (−)1-(benzofuran-2-yl)-2-propylaminopentane [(−)BPAP] was selected as a potential follower of (−)deprenyl in the clinic and as a reference compound for further analysis of the CAE/SAE mechanism in the mammalian brain.
(−)BPAP significantly enhanced in 0.18 μmol 1−1 concentration the impulse propagation mediated release of [3H]-noradrenaline and [3H]-dopamine and in 36 nmol 1−1 concentration the release of [3H]-serotonin from the isolated brain stem of rats. The amount of catecholamines and serotonin released from isolated discrete rat brain regions (dopamine from the striatum, substantia nigra and tuberculum olfactorium, noradrenaline from the locus coeruleus and serotonin from the raphe) enhanced significantly in the presence of 10−12–10−14 M (−)BPAP. BPAP protected cultured hippocampal neurons from the neurotoxic effect of β-amyloid in 10−14 M concentration. In rats (−)BPAP significantly enhanced the activity of the catecholaminergic and serotoninergic neurons in the brain 30 min after acute injection of 0.1 μg kg−1 s.c. In the shuttle box, (−)BPAP in rats was about 130 times more potent than (−)deprenyl in antagonizing tetrabenazine induced inhibition of performance.
Keywords: (−)deprenyl (selegiline); (−)1-(benzofuran-2-yl)-2-propylaminopentane [(−)BPAP], (−)1-phenyl-2-propylaminopentane [(−)PPAP]; catecholaminergic activity enhancer (CAE) effect; serotoninergic activity enhancer (SAE) effect
Introduction
PEA and tyramine, the endogenous indirectly acting sympathomimetic amines, are primarily enhancers of the impulse propagation mediated transmitter release from the catecholaminergic neurons in the brain (CAE effect) and only induce release of catecholamines from their storage sites at a higher concentration. Amphetamine and methamphetamine, PEA analogues with a long lasting effect, act like their parent compound, the (−)enantiomers being the more potent CAE substances and the (+)enantiomers are the more potent releasers of catecholamines (Knoll et al., 1996b). All the unwanted effects of the amphetamines which seriously restricted their use in therapy are due to the catecholamine releasing property of these substances.
(−)Deprenyl (Selegiline), the N-propargyl analogue of methamphetamine (Knoll et al., 1965) is at present the only clinically used amphetamine derivative devoid of the catecholamine releasing effect. As an enhancer of catecholaminergic activity in the brain (−)deprenyl is more potent than (+)deprenyl (Knoll et al., 1996a). The unique so called ‘neuroprotective' effect of (−)deprenyl, which prolongs the life of rats, slows the functional decline of otherwise untreated subjects with early Parkinson's disease, and slows the progression of Alzheimer's disease, can reasonably be attributed to the CAE effect of the drug (for review see Knoll, 1998).
Deprenyl was however also the first described highly selective, potent inhibitor of B-type monoamine oxidase, MAO-B (Knoll & Magyar, 1972). Because of the great practical importance of this property in exploring the nature of MAO-B, attention was primarily focused on this effect of the drug and consequently thousands of papers have been published on (−)deprenyl. To furnish direct evidence that the peculiar enhancement of catecholaminergic activity in the brain in animals treated with a small dose of (−)deprenyl is unrelated to MAO inhibition, we developed in the early '90s a family of deprenyl analogues devoid of the MAO-B inhibitory effect (Knoll et al., 1992). The selected reference substance, (−)1-phenyl-2-propylaminopentane [(−)PPAP] enhances the impulse propagation mediated release of catecholamines in the brain like (−)deprenyl, but in contrast to its parent compound, it is not metabolized to amphetamines (Knoll et al., 1996a).
The discovery that also tryptamine, the indole analogue of PEA, is a potent enhancer of the impulse propagation mediated release of catecholamines in the brain (Knoll, 1994), prompted us to start a structure-activity relationship study aiming to develop new, potent and selective CAE substances, structurally unrelated to PEA and the amphetamines. Out of 65 newly synthetized compounds we selected (−)1-(benzofuran-2-yl)-2-propylaminopentane, (−)BPAP, as a potential follower of (−)deprenyl in the clinic and as a promising reference substance to analyse the CAE mechanism in the mammalian brain. Figure 1 shows the chemical structure and pharmacological spectrum of the most representative CAE substances and illustrates the course of development from the introduction of amphetamine until the synthesis of (−)BPAP, the peculiar pharmacological spectrum of which is now analysed in this paper.
Figure 1.
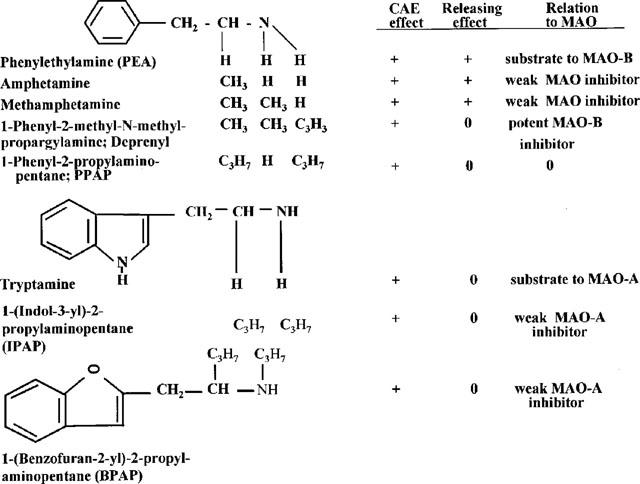
The chemical structure and pharmacological spectrum of PEA and tryptamine derived CAE substances.
Methods
Measurement of the performance of rats in the shuttle box
The method was described in a previous paper (Knoll et al., 1996a). The acquisition of a two-way conditioned avoidance reflex (CAR) was analysed in the shuttle box during five consecutive days. The instrument consists of six boxes, each one separated inside by a barrier with a small gate in the middle. Animals were trained to cross the barrier under the influence of a conditioned stimulus (CS) (light flash). If they failed to do so, they were punished with a footshock (1 mA), i.e. an unconditioned stimulus (US). If the rat failed to respond within 5 s to US, it was noted as escape failure (EF). The rats were trained with 100 trials per day. One trial consisted of 15 s intertrial interval, followed by 15 s CS. The last 5 s of CS overlapped the 5 s of US. At each learning session, the number of CARs, EFs and intersignal reactions (IRs) were automatically counted and evaluated by multi-way analysis of variance (ANOVA). Performance of rats was inhibited by the subcutaneous administration of 1 mg kg−1 tetrabenazine 1 h prior to their testing in the shuttle box. For measuring the antagonists effect of a compound against tetrabenazine induced inhibition of performance, the test substance was injected subcutaneously, concurrently with tetrabenazine.
Measurement of the release of radiolabelled noradrenaline, dopamine or serotonin from the isolated brain stem of rats
The method used was described in a previous paper (Knoll et al., 1996b). To measure drug effects on transmitter-release from the brain stem we incorporated either [3H]-noradrenaline (1-[7,8-3H]-noradrenaline; specific activity: 30–50 Ci mM−1), [3H]-dopamine ([2,5,6-[3H]-dopamine; specific activity: 5–15 Ci mM−1) or [3H]-serotonin (5-hydroxy-[G-3H]-tryptamine creatinine sulphate; specific activity: 10–20 Ci mM−1) (Amersham, Buckinghamshire, U.K.), respectively, into the transmitter stores of the brain stem slices by preincubation. The brain stem was stimulated with rectangular pulses (3 Hz, 1 ms, 60 V) for 3 min. At the beginning of the experiment three consecutive 3-min resting periods preceded the first stimulation. Thereafter seven resting periods were allotted between stimulations.
Measurement of the release of noradrenaline from the locus coeruleus, dopamine from the substantia nigra, striatum and tuberculum olfactorium and serotonin from the raphe
The release of noradrenaline, dopamine or serotonin was measured from selected brain stem areas by HPLC with electrochemical detection. After incubation of the quickly removed brain samples for 20 min, the tissue was soaked for 20 min in fresh Krebs solution and the amount of the biogenic amine released during this period of time was estimated as described in a previous paper (Knoll & Miklya, 1995).
Measurement of β-amyloid induced neurotoxicity in cultured hippocampal neurons
Primary rat embryonic hippocampal cultures were established according to Watt et al. (1994) with some modification. The hippocampus was dissected from the brains of embryonic day 18 rat embryos, incubated in 0.25% trypsin (GIBCO, St. Louis, MO, U.S.A.) and 0.1% Dnase 1 (Boehringer, Mannheim, Germany) at 37°C (20 min), dissociated by gentle tituration, and cultured at a density of 1.5×105 cells per 16 mm polyethylenimine-coated tissue culture glass coverslips (Matsunami Glass, Osaka, Japan) in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% (v v−1) bovine calf serum (GIBCO). After 24 h the medium was changed to DMEM with transferrin-insulin-progesterone, and cultured for 10 days. Medium was changed within 2–3 days. Determination of viable pyramidal neurons were performed in tissue culture with four cover glasses using immunohistochemistry. Hippocampal cultures grown on polyethylenimine-coated tissue culture glass coverslips were fixed with 4% paraformaldehyde, preincubated with 10% goat serum in phosphate buffer saline, and then incubated with the antimicro-associated protein (MAP-2) monoclonal antibody (Amersham) diluted at 1 : 1000 for 24 h at 4°C, then cells were incubated with DAKO kit. At the 10th day the cultured cells were injured by exposing them for 3 days to a 20 μM concentration of β-amyloid 25–35 fragment (Aβ) (Peptide Inst., Osaka, Japan). This concentration of β-amyloid decreased the survival of the neurons (control=100%) to 22.4±7.20% (mean±s.d.). For testing the neuroprotective effect of BPAP, concurrently with β-amyloid a selected concentration of the compound was present in the culture well and the change in survival of the cells was calculated. Statistical significance of data was evaluated by Dunnett's t-test. Significance level was set at P<0.05.
Measurement of tyramine-induced contractions on the pulmonary artery strip of the rabbit
Helical strips of the artery were prepared and set up in an organ bath of 5 ml capacity in Krebs solution at 37°C, bubbled with 5% carbon dioxide in oxygen. The effect of tyramine and the antagonistic effect of (−)BPAP against tyramine induced contraction were measured in the resting state as follows: 0.4; 0.8; 1.6 and 3.2 mg 1−1 tyramine was given in a cumulative manner to the organ bath until the contraction of the strip reached or surpassed the 5 g level. Thereafter tyramine was washed out from the organ and 30 min later the same amounts of tyramine as before were given again in a cumulative manner in the presence of 2 mg 1−1 (−)BPAP in the organ bath.
Measurement of MAO activity
Enzyme activity was measured in brain homogenate of mice and rats as described by Tipton (1985). The radiolabelled substrates (New England Nuclear, U.S.A.) [2-14C]-5-hydroxytryptamine binoxalate (for MAO-A), and [1-14C]-β-phenylethylamine hydrochloride (for MAO-B), specific activity 41.1 mCi mM−1, were used in this study. IC50 values and the 95% confidence interval were determined by Probit analysis.
Assay of radioligand binding to catecholamine and serotonin receptors
Maximum total binding and non-specific binding were determined. Non-specific binding was defined as the proportion of total binding not displaced by unlabelled ligand specific for the receptor. Specific binding was defined as the proportion of total binding that was displaced by unlabelled ligand. Table 1 summarizes the target receptors, their material sources and the experimental conditions of each assay. To determine binding to α-adrenoceptors and serotonin receptors membranes were prepared from brain tissue by standard techniques. Binding to β-adrenoceptors and dopamine receptors utilized membranes prepared from CHO cells expressing the appropriate recombinant human receptor.
Table 1.
Material sources and the experimental conditions of the assay of radioligand binding to catecholamine and serotonin receptors

Results
Effect of (−)BPAP on tetrabenazine induced inhibition of learning performance in the shuttle box
The ability of (−)deprenyl and (−)PPAP to normalize shuttle box performance of rats treated with 1 mg kg−1 tetrabenazine, was of crucial importance in realizing that these PEA derivatives act by enhancing the impulse propagation mediated release of the catecholaminergic transmitter in the brain (Knoll et al., 1996a).
In order to find the minimum dose of a compound which antagonized significantly the effect of tetrabenazine in the shuttle box we started the experiments with 1 and 5 mg kg−1 and changed the dose according to the need as follows: 25; 15; 10; 2.5; 0.5; 0.25; 0.1; 0.05 and 0.025 mg kg−1. Tetrabenazine induced depression was fully antagonized by 5 mg kg−1 (−)deprenyl, 2.5 mg kg−1 (−)PPAP and 0.05 mg kg−1 (−)BPAP. Considering the molecular weight difference between (−)BPAP and (−)deprenyl, (−)BPAP is about 130 times more potent than (−)deprenyl in antagonizing tetrabenazine induced depression in the shuttle-box, thus, it appears that (−)deprenyl and (−)PPAP are relatively weak CAE substances.
For the sake of comparison we also measured the effect of a number of drugs (amantadine, bromocryptine, clomipramine, clorgyline, desmethylimipramine, fluoxetine, lazabemide, levodopa, norzimelidine, pergolide, yohimbine) known to act by diverse mechanisms on catecholaminergic and/or serotoninergic neurons in the brain. With the exception of the CAE substances, only the MAO-A inhibitor clorgyline (0.5 mg kg−1) and the dopamine receptor agonists bromocryptine and pergolide (0.25 mg kg−1) were fully effective antagonists of the inhibition of performance caused by tetrabenazine treatment. Amantadine and levodopa (10 mg kg−1) antagonized the effect of tetrabenazine on CARs but the percentage of EFs remained unchanged. Clomipramine, desmethylimipramine, fluoxetine, lazabemide, norzimelidine and yohimbine were ineffective.
Effect of (−)BPAP on electrical stimulation induced release of [3H]-noradrenaline, [3H]-dopamine and [3H]-serotonin from the isolated brain stem of rats
The brain amines, PEA and tryptamine, enhance the electrical stimulation induced release of [3H]-noradrenaline, [3H]-dopamine and [3H]-serotonin from the isolated brain stem of rats. (−)Methamphetamine, its derivative, (−)deprenyl, and the amphetamine analogue, (−)PPAP, act similarly and are more potent in this respect than the (+)enantiomers (Knoll et al., 1996a,1996b). (−)PPAP strongly enhanced electrical stimulation-induced release of [3H]-noradrenaline and [3H]-dopamine at 2.5 mg l−1 (10 mM l−1) concentration and that of [3H]-serotonin at 10 mg l−1 (40 mM l−1) concentration. (−)BPAP potently enhanced nerve stimulation induced release of noradrenaline (Figure 2), dopamine (Figure 3) and serotonin (Figure 4) from the brain stem. For comparison the effect of PEA and tryptamine are also shown in these figures.
Figure 2.
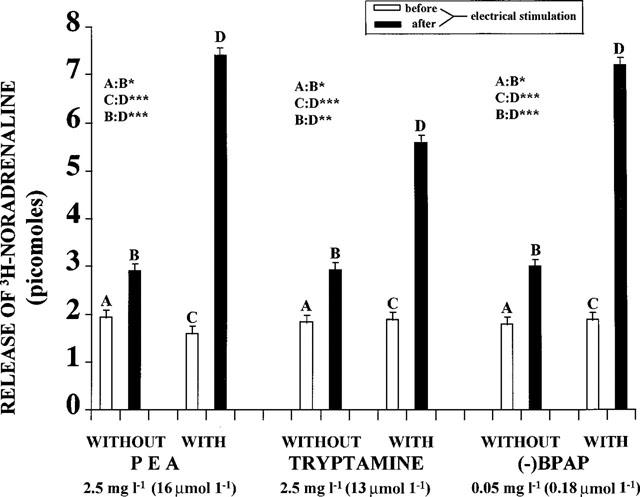
Release of [3H]-noradrenaline from isolated rat brain stem before and after electrical stimulation, in the absence and presence of PEA, tryptamine and (−)BPAP, respectively. (N=8). Each column represents the amount of [3H]-noradrenaline in picomoles released in a 3 min collection period. Vertical lines show s.e.m. Paired Student's t-test. *P<0.05, **P<0.02, ***P<0.01, ****P<0.001.
Figure 3.
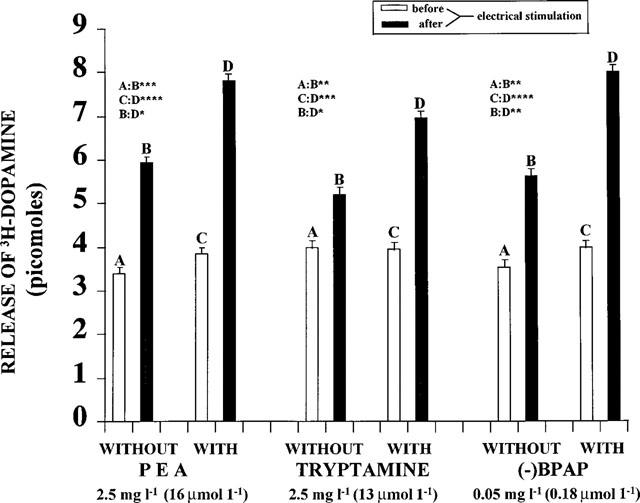
Release of [3H]-dopamine from isolated rat brain stem before and after electrical stimulation, in the absence and presence of PEA, tryptamine and (−)BPAP, respectively (N=8). Each column represents the amount of [3H]-dopamine in picomoles released in a 3 min collection period. Vertical lines show s.e.m. Paired Student's t-test. *P<0.05, **P<0.02, ***P<0.01, ****P<0.001.
Figure 4.
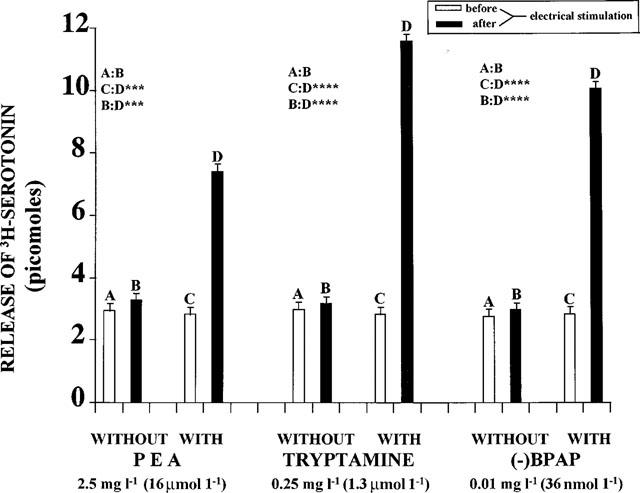
Release of [3H]-serotonin from isolated rat brain stem before and after electrical stimulation, in the absence and presence of PEA, tryptamine and (−)BPAP, respectively. (N=8). Each column represents the amount of [3H-]-serotonin in picomoles released in a 3 min collection period. Vertical lines show s.e.m. Paired Student's t-test. *P<0.05, **P<0.02, ***P<0.01, ****P<0.001.
Effect of the treatment of rats with (−)BPAP on the release of noradrenaline, dopamine and serotonin from selected discrete brain regions
Treatment of rats with low single or repeated doses of (−)deprenyl or (−)PPAP enhanced significantly the release of noradrenaline from the locus coeruleus, dopamine from the striatum, substantia nigra and tuberculum olfactorium and decreased the release of serotonin from the raphe (Knoll & Miklya, 1994; 1995).
Table 2 shows the effect of single dose treatment with (−)BPAP on the release of biogenic amines from selected discrete brain regions. We tested the effect of (−)BPAP in four doses, from 0.0001 to 0.05 mg kg−1. A single injection of (−)BPAP with each of the applied doses caused a highly significant (P<0.001) enhancement of the release of dopamine from the substantia nigra and the tuberculum olfactorium. The amount of noradrenaline released from the locus coeruleus and that of serotonin from the raphe was also significantly enhanced by the two lower concentrations.
Table 2.
The release of catecholamines and serotonin from selected discrete brain regions isolated from the brain of male rats treated with (−)BPAP

Table 2 shows also the effect of a 3-week treatment with (−)BPAP. The rats were killed 24 h after the last injection. The amount of dopamine released from the substantia nigra was significantly enhanced with each of the four doses applied. Only the lowest dose enhanced the release of noradrenaline from the locus coeruleus. The release of serotonin from the raphe was significantly enhanced in the rats treated with 0.1 and 0.5 μg kg−1 (−)BPAP and decreased in the rats treated with the higher doses.
(−)BPAP exerted its CAE/SAE effect in ex vivo experiments too. We performed three series of experiments and Table 3 shows the effect of (−)BPAP on the release of biogenic amines when added to the freshly excised brain tissue samples in a concentration range from 10−4 to 10−14 M. (−)BPAP enhanced significantly the release of noradrenaline and dopamine even in the lowest, 10−14 M concentration. We observed a peculiar concentration dependency regarding the enhancing effect of (−)BPAP on the release of noradrenaline from the locus coeruleus. We found two peaks, one at 10−6 M concentration and one at the 10−13 M concentration. The (−)BPAP induced enhancement of the release of serotonin from the raphe reached its maximum at the 10−10–10−12 M concentration. In the high concentration range (10−4–10−6 M) (−)BPAP did not enhance the release of serotonin.
Table 3.
The ex vivo effect of (–)BPAP on the release of catecholamines and serotonin from selected discrete brain regions isolated from the brain of male rats

Effect of BPAP on β-amyloid induced neurotoxicity in cultured hippocampal neurons
Figure 5 shows that BPAP is protecting the hippocampal neurons from the neurotoxic effect of β-amyloid and this was exerted with the same peculiar concentration dependency (one peak at 10−14 M and another one at 10−8 M) as the (−)BPAP induced enhancement of the release of noradrenaline from the locus coeruleus.
Figure 5.
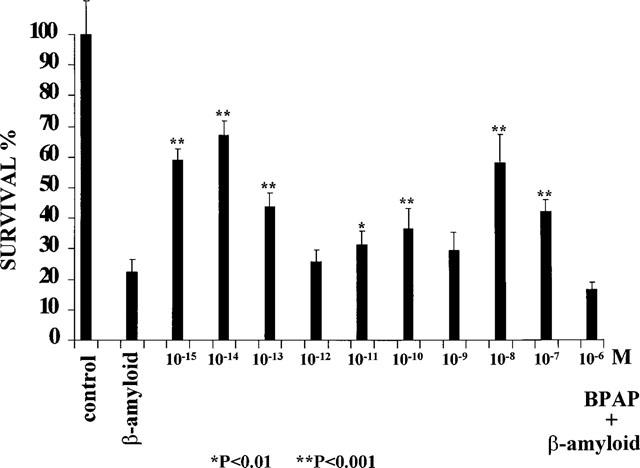
Survival of cultured rat hippocampal neurons in the absence (Control=100%) and presence of 20 μM β-amyloid (22.41±7.20%) and the protective effect of BPAP on β-amyloid induced neurotoxicity. Vertical lines show s.d. Statistics: Dunnett's t-test.
Effect of (−)BPAP on tyramine induced contractions in the pulmonary artery strip of the rabbit
The pulmonary artery strip of the rabbit is a highly sensitive preparation to detect the catecholamine releasing effect of an indirectly acting amine. Tyramine, by inducing the outflow of noradrenaline from the neuronal stores, elicits contraction of the artery strip in a dose-dependent manner. This effect of tyramine is blocked by uptake inhibitors and also by (−)deprenyl and (−)PPAP which interfere with the binding of tyramine to the plasma membrane amine transporter (for review, see Knoll, 1998). Similarly to (−)deprenyl and (−)PPAP, (−)BPAP (2 mg l−1) fully inhibited the tyramine induced release of noradrenaline from the pulmonary artery strip (N=6).
Effect of (−)BPAP on brain MAO activity
Tryptamine is the highly specific endogenous substrate to MAO-A in the brain. As a tryptamine derived substance, (−)BPAP proved to be a weak, selective inhibitor of MAO-A. The IC50 values in the mouse brain for (−)BPAP were found to be 4.0×10−7 M for MAO-A and 2.8×10−4 M for MAO-B and for clorgyline, the commonly used MAO-A inhibitor, 4.2×10−11 M for MAO-A and 2.6×10−4 M for MAO-B.
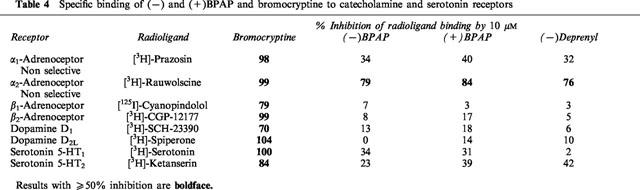
Specific binding of (−)BPAP to catecholamine and serotonin receptors
Drugs used in therapy as agonists or antagonists of one or another type of the pre- or postsynaptic catecholamine or serotonin receptors show substantial specific binding capacity to the whole group of these receptors. We measured the specific binding of (−)deprenyl, (−)BPAP and (+)BPAP to catecholamine and serotonin receptors and bromocryptine, the dopamine D2 receptor agonist, was selected as a reference substance, because (−)deprenyl is known to act primarily as a stimulant of the dopaminergic system in the brain. In contrast to (−)deprenyl and both enantiomers of BPAP, which showed in 10 μM concentration a specific binding capacity to the α2-adrenoceptors only, bromocryptine inhibited in the same concentration the radioligand binding to all catecholamine and serotonin receptors examined (Table 4).
Table 4.
Specific binding of (−) and (+)BPAP and bromocryptine to catecholamine and serotonin receptors
Discussion
Our first observation pointing to enhanced catecholaminergic activity during developmental longevity was the surprising finding that food deprived 2-month-old rats were significantly more active in an open field than their 4-month-old peers (Knoll, 1969; Knoll & Miklya, 1995). We proved thereafter that the basic activity of the catecholaminergic and serotoninergic neurons in the brain is significantly higher between weaning and the end of the 2nd month of age, than either before or after that period (Knoll & Miklya, 1995). The finding that the brain constituents, β-phenylethylamine (PEA) and its indol-analogue, tryptamine, enhance significantly the nerve stimulation induced release of [3H]-noradrenaline, [3H]-dopamine and [3H]-serotonin from the isolated rat brain stem was the first direct evidence for the operation of a previously unrevealed ‘catecholaminergic and serotoninergic activity enhancer (CAE/SAE) mechanism' in the central nervous system, which works on a higher activity level from weaning to sexual maturity, drops back thereafter rapidly to the preweaning level and is then subject to a continuous, slow age-related decline until death (Knoll, 1994; 1997).
(−)Deprenyl (Selegiline), for the time being the only CAE substance in clinical use free of the catecholamine releasing property, is metabolized to methamphetamine and amphetamine and is also a highly potent and selective MAO-B inhibitor. To get rid of these, from point of view of the CAE effect, disadvantageous properties of the compound, we synthetized in the early '90s deprenyl analogues not metabolized to amphetamines and free of the MAO inhibitory effect (Knoll et al., 1992).
Our selected reference compound, (−)BPAP, the benzofuran analogue of (−)PPAP, a much more potent enhancer of the impulse propagation mediated release of catecholamines and serotonin in the brain than either (−)deprenyl or (−)PPAP and a compound structurally unrelated to PEA and the amphetamines, seems to be an especially promising experimental tool for studying the nature and the physiological role of the CAE/SAE mechanism in the brain.
(−)BPAP is, with the exception of the α2-adrenoceptors, devoid of significant specific binding capacity to the catecholamine or serotonin receptors examined. The (−)BPAP induced enhancement of the impulse propagation mediated release of the transmitter from catecholaminergic neurons in the brain is however unrelated to an inhibition of α2-adrenoceptors, as yohimbine, the selective α2-adrenoceptor antagonist did not enhance the release of [3H]-noradrenaline from the isolated brain stem. Considering the low concentration of (−)BPAP needed to enhance the impulse propagation mediated release of noradrenaline, dopamine and serotonin from the neurons in the brain, the data in Table 4 clearly prove that the CAE/SAE effect of (−)BPAP is unrelated to stimulation of the catecholamine and serotonin receptors examined.
(−)BPAP does not induce the release of catecholamines from their storage sites in the nerve terminals and does not inhibit the plasma membrane amine transporter, it interferes however, like (−)deprenyl and (−)PPAP, with the uptake of the indirectly acting amines.
(−)BPAP, obviously because of its close structural similarity to tryptamine, is a weak, selective inhibitor of MAO-A, but this effect is from pharmacological point of view not significant. The effect of (−)BPAP in the shuttle box is due exclusively to its CAE effect, as this substance is 10 times more potent than clorgyline in antagonizing tetrabenazine induced depression (see shuttle box data), while 10,000 times less potent than clorgyline in inhibiting MAO-A activity (see data regarding MAO inhibition).
All in all, we may conclude that (−)BPAP activates the catecholaminergic and serotoninergic neurons in the brain as a highly selective and highly potent CAE/SAE substance. As the CAE/SAE effect of (−)BPAP is unrelated to the catecholamine and serotonin receptors examined, to the plasma membrane amine transporter or to MAO, it is reasonable to search for a specific macromolecular target for (−)BPAP in the brain. Ekblom et al. (1998) has investigated if brain tissue from MAO-B knockout mice contain such ‘non-MAO' binding sites for radiolabelled (−)deprenyl. Interestingly, no binding of (−)deprenyl was observed in this study, indicating that (−)deprenyl does not act directly via a macromolecular target.
Considering, as shown in Figure 1, the relation between chemical structure and pharmacological spectrum, we may summarize in retrospection the developments leading from amphetamine to (−)BPAP as follows. The endogenous amine PEA, the parent compound of the amphetamines, has two effects. It is primarily a CAE/SAE substance and in higher concentration a releaser of catecholamines and serotonin (Knoll, 1994; Knoll et al., 1996b). Metabolized by MAO-B, PEA is a short acting compound. The attachment of a methyl-group to the α-carbon in PEA (β-isopropylamine, amphetamine) converts the MAO-B substrate to a weak reversible inhibitor of both types of MAO. Accordingly, amphetamine is a long acting central nervous system stimulant. It is still believed that amphetamines exert their stimulating effects in the therapeutic dose-range exclusively by releasing catecholamines (in higher doses also serotonin) from their storage sites in nerve terminals (cf. Hoffman & Lefkovitz, 1996). The analysis of the peculiar behaviour of (−)deprenyl (Knoll et al., 1996a) threw, however, finally, light on the dual effect of PEA and the amphetamines. We realized that the introduction of the propargyl group, a relatively bulky substitution, to the nitrogen in methamphetamine (deprenyl) led to the loss of the catecholamine releasing effect while the CAE effect survived. It was with all probability the catecholamine releasing property of PEA, tyramine and the amphetamines which masked their CAE effect and hindered its detection for decades. The crucially important step forward was the discovery that tryptamine is like PEA an enhancer of the impulse propagation mediated release of catecholamines and serotonin in the brain. The tryptamine derived new compounds are now the first potent and selective enhancers of exocytosis of noradrenaline, dopamine and serotonin in the brain, which are structurally unrelated to amphetamines. (−)BPAP, the selected reference compound for further studies is a highly potent, selective CAE/SAE substance.
We demonstrated that PEA, tyramine and (−)methamphetamine (cf. Knoll et al, 1996b), as well as, (−)deprenyl and (−)PPAP (cf. Knoll et al., 1996a) enhanced the inward Ca2+ current in the sino-auricular fibres of the frog heart in a range of low concentration and were ineffective in higher concentration. Similarly, tryptamine in 0.25 mg l−1 concentration enhanced the nerve stimulation induced release of serotonin from the rat brain stem and was ineffective in 2.5 mg l−1 concentration. This phenomenon is detectable also in the experiments shown in Tables 2 and 3, as well as in the series of experiments performed on cultured hippocampal neurons shown in Figure 5. Further studies are needed to explain the peculiar dose-response relationship regarding the CAE/SAE effect of (−)BPAP.
(−)Deprenyl is a unique drug. Life-long (−)deprenyl medication which, due to its CAE effect, maintains the nigrostriatal dopaminergic neurons on a higher activity level, slows the age-related decline of sexual and learning performances of rats and prolongs their life (for review see Knoll, 1998; Kitani et al., 1998). The unique effect of (−)deprenyl on longevity described by us (Knoll, 1988; Knoll et al., 1989; 1994) was unequivocally confirmed in studies performed with rats (Milgram et al., 1990; Kitani et al., 1993), hamsters (Stoll et al., 1997) and dogs (Ruehl et al., 1997). Conflicting data were published regarding mice (Ingram et al., 1993; Archer & Harrison, 1996).
(−)Deprenyl slows the progression of Parkinson's disease (Tetrud & Langston, 1989; Parkinson Study Group, 1989) and Alzheimer's disease (Sano et al., 1997), and on the other hand, it exerts a peculiar, so-called ‘neuroprotective' effect (Tatton & Greenwood, 1991). These effects can reasonably be related to the CAE effect of the drug (Knoll, 1998). Based on the data presented in this paper, we expect that (−)BPAP will be substantially more potent than (−)deprenyl in slowing the progression of Parkinson's disease and Alzheimer's disease. We also anticipate that this substance will be a much more effective antidepressant in the clinic than (−)deprenyl, which was shown to have this effect (cf. Varga & Tringer, 1967; Mann & Gershon, 1980) but was never registered for this purpose.
BPAP inhibited significantly the β-amyloid induced neurotoxicity in the cultured hippocampal neurons in two distinct ranges of concentration, one with a peak of 10−14 M and one with a peak of 10−8 M (Figure 5). The mode of effect of BPAP on the hippocampal neurons is surprisingly identical with the mode of effect of (−)BPAP on the noradrenergic neurons (see the (−)BPAP induced enhancement of the release of noradrenaline from the isolated locus coeruleus in Table 3) indicating the essential identity of the BPAP-sensitive mechanism in the noradrenergic and hippocampal neurons.
As a matter of fact there is a conspicuous similarity between the BPAP induced effect on the cultured rat hippocampal neurons and the one induced by (−)deprenyl in rats treated with the drug for years during their post-developmental phase of life. In the (−)deprenyl experiment we picked out of a population of 1600 rats the animals with the lowest and the highest sexual performance and demonstrated, on the one hand, that the ‘high performing' rats lived significantly longer than their ‘low performing' peers, and on the other hand, that (−)deprenyl treatment transformed the low performing rats into significantly higher performing ones, which lived then as long as their saline treated high performing peers (Knoll et al., 1994). We assume that high performing, longer living rats possess a more efficient catecholaminergic brain machinery than their low performing peers and the treatment with (−)deprenyl, a CAE substance, acts accordingly. Whatever performance we measure in a random population, we always find a huge variation in efficiency, ranging from very low to very high performing individuals. This is true also regarding the performance of cells in a population of cultured neurons. We may look at the 20% of the cultured hippocampal neurons which survived in the presence of β-amyloid, as ‘high performing' cells, those possessing the most efficient BPAP-sensitive activation mechanism. Adding to the cultured neurons BPAP in the optimum concentration (10−14 M), the highly potent and selective enhancer of this regulation made each neuron higher performing and the surviving rate increased from 20% to 70%.
BPAP has obviously the same effect on the noradrenergic, dopaminergic, serotoninergic and hippocampal neurons. It may stimulate endogenous substances which enhance the activity of the neurons according to their physiological need. The high potency of BPAP in stimulating this regulation makes search in the brain after much more potent endogenous ‘enhancer' substances than PEA and tryptamine, reasonable.
Abbreviations
- BPAP
1-(benzofuran-2-yl)-2-propylaminopentane.HCl
- CAE
catecholaminergic activity enhancer
- MAO-B
B-type monoamine oxidase
- PEA
β-phenylethylamine.HCl
- PPAP
1-phenyl-2-propylaminopentane.HCl
- SAE
serotoninergic activity enhancer
References
- ARCHER J.R., HARRISON D.E. L-Deprenyl treatment in aged mice slightly increases life spans, and greatly reduces fecundity by aged males. J. Gerontol. Biol. Sci. 1996;31A:B448–B453. doi: 10.1093/gerona/51a.6.b448. [DOI] [PubMed] [Google Scholar]
- EKBLOM J., ORELAND L., CHEN K., SHIH J.C. Is there a ‘non-MAO' macromolecular target for L-deprenyl?: Studies on MAOB mutant mice. Life Sci. 1998;63:181–186. doi: 10.1016/s0024-3205(98)00370-1. [DOI] [PubMed] [Google Scholar]
- HOFFMAN B.B., LEFKOWITZ R.J.Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists Goodman and Gilman's The Pharmacological Basis of Therapeutics, Ninth edition 1996New York: McGraw-Hill; 199–248.Hardman, J.G. & Limbird, L.E. (eds) [Google Scholar]
- INGRAM D.K., WIENER H.L., CHACHICH M.E., LONGO J.M., HENGEMIHLE J., GUPTA M. Chronic treatment of aged mice with L-deprenyl produced marked MAO-B inhibition but no beneficial effects on survival, motor performance, or nigral lipofuscin accumulation. Neurobiol. Aging. 1993;14:431–440. doi: 10.1016/0197-4580(93)90101-g. [DOI] [PubMed] [Google Scholar]
- KITANI K., KANAI S., IVY G.O., CARRILLO M.C. Assessing the effects of deprenyl on longevity and antioxidant defenses in different animal models. Ann. N.Y. Acad. Sci. 1998;854:291–306. doi: 10.1111/j.1749-6632.1998.tb09910.x. [DOI] [PubMed] [Google Scholar]
- KITANI K., KANAI S., SATO Y., OHDA M., IVY G.O., CARRILLO M.C. Chronic treatment of (−)deprenyl prolongs the life span of male Fischer 344 rats: Further evidence. Life Sci. 1993;52:281–288. doi: 10.1016/0024-3205(93)90219-s. [DOI] [PubMed] [Google Scholar]
- KNOLL J. New York: Hafner Publishing Company; 1969. The theory of active reflexes. p. 131. Budapest: Publishing House of the Hungarian Academy of Sciences. [Google Scholar]
- KNOLL J. The striatal dopamine dependency of lifespan in male rats. Longevity study with (−)deprenyl. Mech. Ageing Dev. 1988;46:237–262. doi: 10.1016/0047-6374(88)90128-5. [DOI] [PubMed] [Google Scholar]
- KNOLL J. Memories of my 45 years in research. Pharmacol. Toxicol. 1994;75:65–72. doi: 10.1111/j.1600-0773.1994.tb00326.x. [DOI] [PubMed] [Google Scholar]
- KNOLL J. Sexual performance and longevity. Exp. Gerontol. 1997;32:539–552. doi: 10.1016/s0531-5565(96)00157-x. [DOI] [PubMed] [Google Scholar]
- KNOLL J. (−)Deprenyl (Selegiline) a catecholaminergic activity enhancer (CAE) substance acting in the brain. Pharm. Toxicol. 1998;82:57–66. doi: 10.1111/j.1600-0773.1998.tb01399.x. [DOI] [PubMed] [Google Scholar]
- KNOLL J., DALLO J., YEN T.T. Striatal dopamine, sexual activity and lifespan. Longevity of rats treated with (−)deprenyl. Life Sci. 1989;45:525–531. doi: 10.1016/0024-3205(89)90103-3. [DOI] [PubMed] [Google Scholar]
- KNOLL J., ECSERI Z., KELEMEN K., NIEVEL J., KNOLL B. Phenylisopropylmethylpropinylamine (E-250) a new psychic energizer. Arch. Int. Pharmacodyn. Thér. 1965;155:154–164. [PubMed] [Google Scholar]
- KNOLL J., KNOLL B., TÖRÖK Z., TIMÁR J., YASAR S. The pharmacology of 1-phenyl-2-propylaminopentane (PPAP), a deprenyl-derived new spectrum psychostimulant. Arch. Int. Pharmacodyn. Thér. 1992;316:5–29. [PubMed] [Google Scholar]
- KNOLL J., MAGYAR K. Some puzzling effects of monoamine oxidase inhibitors. Adv. Bioch. Psychopharmacol. 1972;5:393–408. [PubMed] [Google Scholar]
- KNOLL J., MIKLYA I. Multiple, small dose administration of (−)deprenyl enhances catecholaminergic activity and diminishes serotoninergic activity in the brain and these effects are unrelated to MAO-B inhibition. Arch. Int. Pharmacodyn. Thér. 1994;328:1–15. [PubMed] [Google Scholar]
- KNOLL J., MIKLYA I. Enhanced catecholaminergic and serotoninergic activity in rat brain from weaning to sexual maturity. Rationale for prophylactic (−)deprenyl (selegiline) medication. Life Sci. 1995;56:611–620. doi: 10.1016/0024-3205(94)00494-d. [DOI] [PubMed] [Google Scholar]
- KNOLL J., MIKLYA I., KNOLL B., MARKÓ R., KELEMEN K. (−)Deprenyl and (−)1-phenyl-2-propylaminopentane [(−)PPAP], act primarily as potent stimulants of action potential–transmitter release coupling in the catecholaminergic neurons. Life Sci. 1996a;58:817–827. doi: 10.1016/0024-3205(96)00014-8. [DOI] [PubMed] [Google Scholar]
- KNOLL J., MIKLYA I., KNOLL B., MARKÓ R., RÁCZ D. Phenylethylamine and tyramine are mixed-acting sympathomimetic amines in the brain. Life Sci. 1996b;58:2101–2114. doi: 10.1016/0024-3205(96)00204-4. [DOI] [PubMed] [Google Scholar]
- KNOLL J., YEN T.T., MIKLYA I. Sexually low performing male rats die earlier than their high performing peers and (−)deprenyl treatment eliminates this difference. Life Sci. 1994;54:1047–1057. doi: 10.1016/0024-3205(94)00415-3. [DOI] [PubMed] [Google Scholar]
- MANN J.J., GERSHON S. A selective monoamine oxidase-B inhibitor in endogenous depression. Life Sci. 1980;26:877–882. doi: 10.1016/0024-3205(80)90350-1. [DOI] [PubMed] [Google Scholar]
- MILGRAM N.W., RACINE R.K., NELLIS P., MENDONCA A., IVY G.O. Maintenance on L-deprenyl prolongs life in aged male rats. Life Sci. 1990;47:415–420. doi: 10.1016/0024-3205(90)90299-7. [DOI] [PubMed] [Google Scholar]
- PARKINSON STUDY GROUP Effect of (−)deprenyl on the progression disability in early Parkinson's disease. New Engl. J. Med. 1989;321:1364–1371. doi: 10.1056/NEJM198911163212004. [DOI] [PubMed] [Google Scholar]
- RUEHL W.W., ENTRIKEN T.L., MUGGENBURG B.A., BRUYETTE D.S., GRIFFITH W.G., HAHN F.F. Treatment with L-deprenyl prolongs life in elderly dogs. Life Sci. 1997;61:1037–1044. doi: 10.1016/s0024-3205(97)00611-5. [DOI] [PubMed] [Google Scholar]
- SANO M., ERNESTO C., THOMAS R.G., KLAUBER M.R., SCHAFER K., GRUNDMAN M., WOODBURY P., GROWDON J., COTMAN C.W., PFEIFFER E., SCHNEIDER L.S., THAL L.J. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. New Engl. J. Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- STOLL S., HAFNER U., KRANZLIN B., MULLER W.E. Chronic treatment of Syrian hamsters with low-dose selegiline increases life span in females but not males. Neurobiol. Aging. 1997;18:205–211. doi: 10.1016/s0197-4580(97)00009-2. [DOI] [PubMed] [Google Scholar]
- TATTON W.G., GREENWOOD C.E. Rescue of dying neurons: a new action for deprenyl in MPTP parkinsonism. J. Neurosci. Res. 1991;30:666–672. doi: 10.1002/jnr.490300410. [DOI] [PubMed] [Google Scholar]
- TETRUD J.W., LANGSTON J.W. The effect of (−)deprenyl (selegiline) on the natural history of Parkinson's disease. Science. 1989;245:519–522. doi: 10.1126/science.2502843. [DOI] [PubMed] [Google Scholar]
- TIPTON K.F. Determination of monoamine oxidase. Meth. Find. Exptl. Clin. Pharmacol. 1985;7:361–367. [PubMed] [Google Scholar]
- VARGA E., TRINGER L. Clinical trial of a new type of promptly acting psychoenergetic agent (phenyl-isopropylmethyl-propinylamine HCl, E-250) Acta Med. Acad. Sci. Hung. 1967;23:289–295. [PubMed] [Google Scholar]
- WATT J.A., PIKE C.J., WALENCEWICZ-WASSERMAN A.J., COTMAN C.W. Ultrastructural analysis of β-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Res. 1994;661:147–156. doi: 10.1016/0006-8993(94)91191-6. [DOI] [PubMed] [Google Scholar]