

Abstract
cis-9,10-octadecenoamide (‘oleamide') accumulates in CSF on sleep deprivation. It induces sleep in animals (the trans form is inactive) but its cellular actions are poorly characterized. We have used electrophysiology in cultures from embryonic rat cortex and biochemical studies in mouse nerve preparations to address these issues.
Twenty μM cis-oleamide (but not trans) reversibly enhanced GABAA currents and depressed the frequency of spontaneous excitatory and inhibitory synaptic activity in cultured networks.
cis-oleamide stereoselectively blocked veratridine-induced (but not K+-induced) depolarisation of mouse synaptoneurosomes (IC50, 13.9 μM).
The cis isomer stereoselectively blocked veratridine-induced (but not K+-induced) [3H]-GABA release from mouse synaptosomes (IC50, 4.6 μM).
At 20 μM cis-oleamide, but not trans, produced a marked inhibition of Na+ channel-dependent rises in intrasynaptosomal Ca2+.
The physiological significance of these observations was examined by isolating Na+ spikes in cultured pyramidal neurones. Sixty-four μM cis-oleamide did not significantly alter the amplitude, rate of rise or duration of unitary action potentials (1 Hz).
cis-Oleamide stereoselectively suppressed sustained repetitive firing (SRF) in these cells with an EC50 of 4.1 μM suggesting a frequency- or state-dependent block of voltage-gated Na+ channels.
Oleamide is a stereoselective modulator of both postsynaptic GABAA receptors and presynaptic or somatic voltage-gated Na+ channels which are crucial for synaptic inhibition and conduction. The modulatory actions are strikingly similar to those displayed by sedative or anticonvulsant barbiturates and a variety of general anaesthetics.
Oleamide may represent an endogenous modulator for drug receptors and an important regulator of arousal.
Keywords: cis-9,10-octadecenoamide (oleamide); GABAA receptor; voltage-gated Na+ channel; sustained repetitive firing; cultured rat cortical neurons; mouse synaptoneurosomes/synaptosomes; transmitter release; intracellular Ca2+; hypnotic/anaesthetic/anticonvulsant drugs; electrophysiology/patch clamp; endogenous sleep regulator
Introduction
cis-9,10-Octadecenoamide (cOA or ‘oleamide') was originally isolated from sleep-deprived cats and an enzyme specific for its hydrolysis is present in rat brain (Cravatt et al., 1995). Synthetic cOA induced physiological sleep when injected into rats but little is known about its cellular mode of action. High-affinity interactions with recombinant 5HT2 receptors have been reported (Huidobro-Toro & Harris, 1996), but others conclude that the hypnogenic effects are probably not secondary to activation of G-proteins (Boring et al., 1996). More recent studies demonstrate that oleamide may bind to cannabinoid sites (IC50 of 10 μM) in rodent CNS, that it induces hypolocomotion in vivo which is sensitive to the CB1 antagonist SR141716A but that the brain lipid cannot elicit the same second messenger responses as acknowledged CB1 cannabinoid agonists (Cheer et al., 1999). At high μM concentrations cOA can deconvolute gap junctions in glia (Guan et al., 1997). To date, none of these putative receptor interactions have been shown to contribute to a net depressant effect for oleamide at a systems level.
Experiments in laboratory animals confirm that oleamide induces sleep and biologically active concentrations of oleamide in CSF have been determined (Basile et al., 1999). Recently we demonstrated that rat GABAA receptors and inhibitory synaptic currents were sensitive to modulation by oleamide (but not equally hydrophobic congeners like oleic acid). Recombinant human GABAA receptor isoforms expressed in oocytes were modulated by oleamide and diazepam only if a γ subunit was co-expressed with αβ (but the fatty acid amide response was insensitive to high concentrations of the specific benzodiazepine receptor antagonist flumazenil) (Lees et al., 1998a). Synthetic depressant drugs are acknowledged as stereoselective allosteric modulators of ion channel targets like the GABAA receptor (a crucial/widespread inhibitory channel) and the voltage-gated Na+ channel (important for generating action potentials in excitable membranes) (reviewed by Lees, 1998). Here we have examined the potential of oleamide isomers to modulate these key signalling molecules in an attempt to further delineate cellular mode of action and to identify targets contributing to the stereoselectivity of the oleamides in vivo, where the cis isomer is demonstrably more potent (Cravatt et al., 1995). We discuss the quantitative data and the intriguing hypothesis that oleamide represents an endogenous ligand for depressant drug recognition sites in the mammalian CNS.
Methods
Cell culture
Neuronal cultures were prepared from cerebral cortices of 17–18-day-old rat embryos (Lees & Leach, 1993). Cells were plated onto poly-D-lysine coated coverslips (200,000 cells ml−1) in Dulbecco's Modified Eagle Medium supplemented with 10% foetal calf serum and 100 units μg ml−1 penicillin/streptomycin. After 12–24 h the plating medium was replaced by a maintenance medium comprising neurobasal medium, with 2% B27 supplement, 1% glutamax (Gibco, Paisley, U.K.) and 100 units μg ml−1 penicillin/streptomycin. Cells were used in experiments after 14–28 days in vitro.
Chemical synthesis and suppliers
Details of the synthesis and purification of cOA have been published (Lees et al., 1998a). tOA: (E)-9-octadecenoic acid amide was synthesized as previously described (Roe at al., 1949). The white crystalline solid was recrystallized from ethanol and its melting point of 92–92.5°C was consistent with literature values. Unless otherwise stated, all chemicals and drugs were obtained from Sigma Aldrich Chemical Co. (Poole, Dorset, U.K.) or (Merck Ltd, Poole, Dorset, U.K.).
Electrophysiology
GABAA currents and spontaneous synaptic activity
Cultured neurones on polylysine coated coverslips were placed in a 5 mm perspex trench on the stage of an inverted microscope. The extracellular saline contained (mM): NaCl 142, KCl 5, CaCl2 2, MgCl2 2, HEPES 5, D-glucose 10, pH 7.4. Whole-cell pipette saline contained (mM): K-gluconate 142, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, pH 7.4. All potentials cited are those based on the preamplifier null potential and take no account of the liquid junction offset (circa+14 mV by direct measurement) inherent in the use of these asymmetrical solutions. Borosilicate patch pipettes (3–5 MΩ) were used. 60–80% series resistance compensation was applied at the List EP7 pre-amplifier. Neurons with input resistances of ⩾100 MΩ were studied. Whole cell currents were filtered at 1–3 KHz prior to digitization and monitored on a Gould chart recorder or analysed using WCP (John Dempster, Strathclyde University) or Spike 2 software (Cambridge Electronic Design, Cambridge, U.K.).
Isolation of Na+ spikes or SRF
Pyramidal cells were whole cell clamped in a physiological saline consisting of (mM): NaCl 142, KCl 2.5, CaCl2 1, MgCl2 2, CoCl2 2, HEPES 5, D-glucose 10, pH 7.4. The gluconate based pipette solution given above was used. Some data were obtained in amphotericin perforated mode (240 μg ml−1 in above pipette solution; 0.4% DMSO) which did not qualitatively alter modulatory effects. 3–10 MΩ pipettes were fabricated on a Mecanex BBCH puller. An Axoclamp 2B pre-amp was used in bridge mode to monitor cellular excitability and current pulses were delivered from a Grass S66 stimulator (see text for details of stimulus protocols). Cells were held at their resting membrane potential (only those with Em more negative than −50 mV were selected for study). Analogue data were filtered at 1 KHz and digitized (CED 1401 plus) for analysis using Win WCP software (courtesy of John Dempster, University of Strathclyde).
Pharmacology
cOA was dissolved in dimethylsulphoxide (DMSO) then diluted 1000× into saline: 0.1% DMSO produced no effect on the parameters reported here and was present in pre- and post-treatment phases of all experiments. To further facilitate dissolution of the modulator, all extracellular salines were supplemented with 0.1% bovine serum albumin (fraction V, Sigma) which was present during all phases of the reported experiments. cOA was formulated daily and perfused from glass reservoirs via teflon lines. GABA was rapidly (10–90% rise times <50 ms) and quantitatively delivered to cultured cells using the Y-tube technique (Murase et al., 1989) either alone or with oleamide isomers (previously equilibrated with the cell by constant superfusion through the recording chamber). Log concentration-response curves were fit to the Hill equation by non-linear regression (Graphpad Prism software, San Diego, CA, U.S.A.). Logarithmic values of EC50 and associated standard error were used for statistical comparisons. Student's t-tests (two-tailed throughout, and paired where appropriate) or ANOVA were used as indicated in the text. Significant effects are indicated by P<0.05 throughout. Data are expressed as mean±standard error (s.e.) of the mean.
Synaptoneurosomes and transmembrane potentials
Synaptoneurosomes were routinely prepared from the whole brain of a single CD1 mouse, using established procedures (Harris & Allan, 1985). Fifty-seven μl aliquots were held on ice in saline consisting of (mM): NaCl 125, KCl 5, CaCl2 1, MgSO4 1.2, HEPES 20, glucose 10, pH 7.4, supplemented with 1 mg ml−1 bovine serum albumin (to facilitate dissolution of fatty acid amides). The membrane potential of synaptoneurosomes at 37°C in rapidly stirred suspensions was determined using the lipophilic cation rhodamine 6G (Auichi et al., 1982) at a final concentration of 0.4 μM with continuous fluorescence monitoring (λex=520 nm; λem=550 nm). Test compounds or solvent blanks, dissolved in 3 μl DMSO, were injected into a suspension of synaptoneurosomes (0.65 mg protein per 3 ml saline) in a quartz cuvette at t=0 min. Rhodamine 6G was added at t=12 min and when the fluorescence stabilized (t=15 min), veratridine (66 μM) or K+ (35 mM) were applied.
Measurements were taken as soon as the fluorescence change following the depolarizing pulse reached steady state. Fluorescence changes were recorded on a Perkin-Elmer LS-50 fluorimeter and calibrated according to the equilibrium potential for potassium ions using the reduced Goldman equation.
Synaptosomes, transmitter release and pre-synaptic intraterminal Ca2+
Pure synaptosomes were isolated from the brains of CD1 mice on discontinuous Percoll gradients (Dunkley et al., 1986), and pre-incubated in saline containing 10 μM aminooxyacetic acid at 37°C.
An aliquot (2.5 ml) of this suspension was transferred to a tube containing 12.5 μCi of [3H]-GABA at a final concentration of 1 μM and incubated for 10 min.
Loading was terminated by adding 10 ml ice cold saline then centrifugation at 20,100×g for 10 min at 2°C.
The washed pellet was dispersed in 1.1 ml saline then a 90 μl aliquot (400 μg of protein) was placed on a GF/B filter in each of ten superfusion units. After superfusion of synaptosomes with 25 ml saline a total of ten 2 ml samples were collected from each line (10 ml of saline to pretreat with modulators and 10 ml to challenge with activator veratridine (50 M) or 35 mM KCl). Modulators were present during depolarizing challenge and the flow rate was 1 ml min−1 throughout.
Calcium fluorimetry
Synaptosomes were loaded with the acetoxymethyl ester of the calcium indicator fura-2 (according to Komulainen & Bondy, 1987) except that saline was supplemented with 2.5 mg ml−1 BSA and had no added calcium. Loading procedures were conducted in darkness. To determine [Ca2+]i, aliquots (0.25 mg synaptosomal protein) were resuspended in saline (3 ml) containing 2 mM Ca2+ at 37°C in a continuously stirred quartz cuvette in a Perkin Elmer LS-50 fluorimeter. Test compounds were equilibrated with synaptosomes after addition in DMSO (vehicle dilution 1 in 1000). Analyses were conducted using rapidly alternating (1.8 s) excitation wavelengths of 340 and 380 nm and emission intensity was sampled at 510 nm. Calibrations were performed using alkaline EGTA followed by SDS to establish the Rmin ratio and at a saturating concentration of calcium to establish Rmax. [Ca2+]i was calculated/calibrated using established methods (Grynkiewicz et al., 1985), assuming a dissociation constant of the fura-2: calcium complex of 224 nM. The autofluorescence of control (DMSO-treated) synaptosomes was measured and subtracted from total fluorescence. In these assays, and those involving measurements of GABA release and membrane potential, KCl was applied at the maximum effect concentration, while veratridine concentrations were close to those resulting in maximal effect.
Results
Stereoselective modulation of GABAA receptors and reversible depression spontaneous synaptic activity in cultured cortical neurones
As we have previously reported, cOA (3.2–64 μM) has the capacity to positively modulate GABAA receptors in whole cell patch-clamped cultured rat neurones and human cloned GABAA isoforms (Lees et al., 1998a). Here, in rat neocortical pyramidal cells, 20 μM cOA reversibly enhanced the amplitude of current evoked by Y-tube application of 3.2 μM GABA, reaching peak steady state values between 8–14 min (Figure 1a). This elevation was significant (Figure 1c: paired student's t-test, n=5, P=0.002). In contrast, the trans isomer (tOA) did not significantly modulate GABA-evoked current over a period of 14 min (Figure 1b,c: n=5; P=0.41). Modulation of the evoked GABA current by 20 μM cOA was invariably accompanied by a progressive decrease in the frequency of both IPSCs and EPSCs (Figure 1a). The reduced incidence of post-synaptic currents was significant after a 10 min exposure (IPSCs, P=0.044, n=7; EPSCs, P=0.037, n=7; Figure 1d). However, tOA (Figure 1b,d) had no effect on the rates of incidence of either inhibitory or excitatory synaptic currents (P=0.22 and 0.3 respectively, n= 4).
Figure 1.
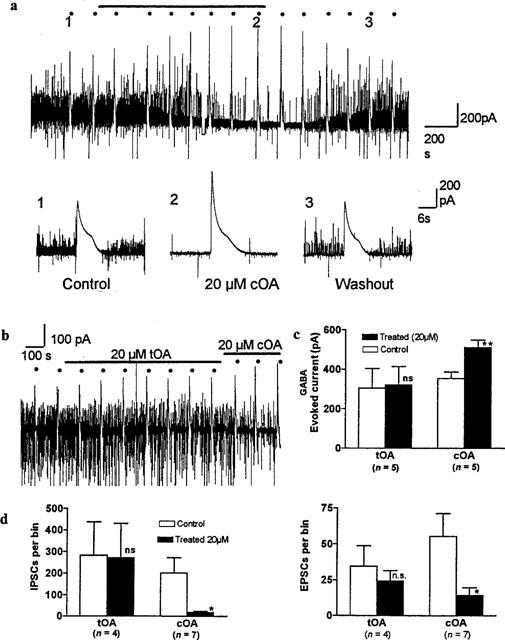
GABAA currents and spontaneous synaptic traffic are selectively modulated by the cis-isomer of oleamide. (a) Superfusion of 20 μM cOA (black bar) to a neurone reversibly enhanced the effects of 4 s pulses of exogenous GABA at 3.2 μM (indicated by black dots and shown at higher resolution in the traces below). Note the slow onset kinetics and the concurrent reduction in the incidence (and graded reduction in amplitude) of spontaneous synaptic currents. (b) Identical experiment with tOA giving only a marginal response in contrast to the initial phases of cOA superfusion. (c) Histogram (mean+s.e.mean) depicting compounded data from replicated experiments with exogenous GABA (ns, not significant; **P<0.01). (d) Histograms (mean+s.e.mean) depicting steady-state effects of oleamide isomers on the incidence of spontaneous synaptic currents (event detection by Spike 2 software with a bin width of 2 min). ns, not significant; *P<0.05.
Biochemical analysis of presynaptic effects of oleamide
We were intrigued by the selective depressant effects of the cis isomer on synaptic traffic which could be qualitatively mimicked by superfusing the cells with low concentrations of the Na+ channel blocker tetrodotoxin (Lees et al., 1998a), suggesting that the majority of the large synaptic events are evoked (secondary to spiking in presynaptic cells). Mouse brain synaptoneurosomes were incubated with the membrane potential dependent fluorescent probe rhodamine 6G, then depolarized, using the Na+ channel activator veratridine, or with a high concentration of K+. cOA, dose-dependently, blocked the veratridine-induced depolarization with an IC50 of 13.93 μM without affecting the K+-induced signal significantly at 20 μM (Figure 2a). The kinetics of this effect (approximately 10 min to equilibrium; t½=2.63 min) (Figure 2b) were comparable to those of both the GABA receptor modulation (Figure 3a) and the depressant effect on synaptic traffic observed in cultured cells (Figure 3b). The non-hypnotic tOA was unable to mimic this effect at 20 μM (the highest concentration we could formulate as a true solution). When we examined the release of [3H]-GABA from synaptosomes, a similar profile of activity was obtained (Figure 4a). Veratridine-induced secretion was inhibited by cOA (IC50: 4.58 μM) but 20 μM tOA proved inactive. Functional depression of phasic, evoked transmitter release by oleamide may underpin the progressive reduction in the amplitude of IPSCs noted in electrophysiological experiments (see Figure 1a). Neither of the isomers were able to significantly alter K+-induced secretion (compared to DMSO treatment blanks) (Figure 4b). Synaptosomal cytoplasmic free [Ca2+] concentrations were not affected by oleamides (20–40 μM) in the absence of a depolarizing challenge (Figure 5a). Again cOA at 20–40 μM blocked a very large proportion of the veratridine-induced rise in free Ca2+ compared to the K+-induced signal (although the latter was weakly depressed at high concentrations) (Figure 5b,c). In summary, cOA but not tOA was able to selectively block veratridine's effect on nerve terminals at concentrations approximating to (or below) those we have previously reported to modulate GABAA receptors in cultured cells suggesting that the voltage-gated Na+ channel may be a viable alternative target.
Figure 2.
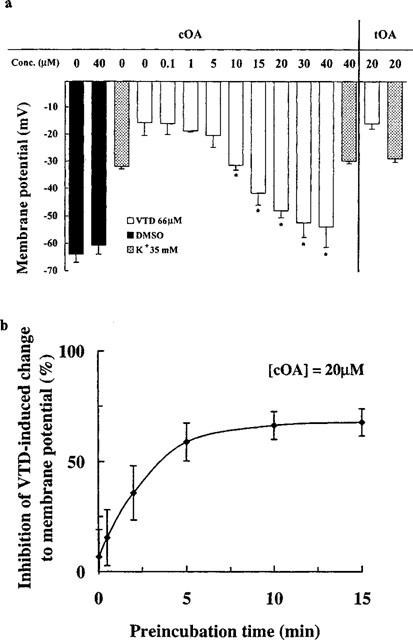
Biochemical data confirming the presynaptic effects of oleamide on mouse nerve terminal polarization and a selective interaction with the voltage-gated Na+ channel. (a) Effect of cOA and tOA on veratridine- (VTD) or K+-induced depolarization of mouse brain synaptoneurosomes. Apparent IC50 for cOA, by probit transformation, was 13.93 μM. Bars depict mean and s.e.mean of 3–4 independent determinations (*P<0.05). (b) Kinetic effects of 20 μM cOA on veratridine-induced (66 μM) depolarization of synaptoneurosomes. Each point represents mean (bars±s.e.mean) of three independent experiments. The data above were determined at equilibrium (15 min preincubation).
Figure 3.
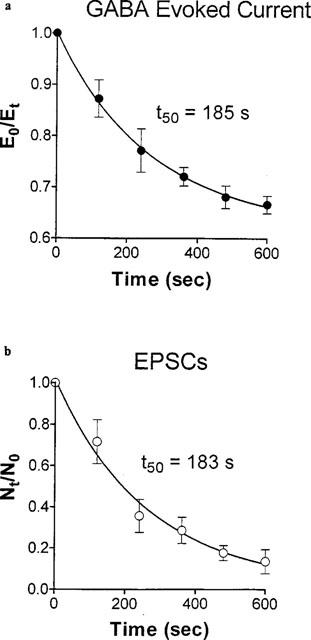
The modulatory actions of 20 μM cOA develop with similar, slow, kinetics. (a) Peak GABA evoked current immediately prior to cOA exposure (E0) were measured at the indicated time points (Et) and approach an asymptote at 10–12 min. (b) The number of excitatory events per 120 s bin pretreatment (N0) was measured after exposure to cOA (Nt) and declined with a very similar timecourse. Points represent mean±s.e.mean (n=4), throughout.
Figure 4.
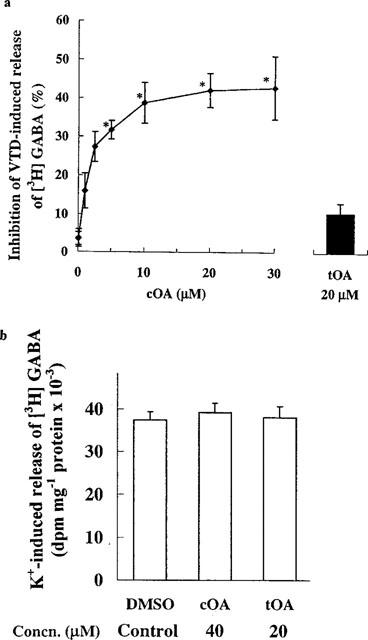
cOA stereoselectively blocked [3H]-GABA release from synaptosomes evoked by veratridine (VTD) but not by elevated K+. (a) Left: Inhibitory action of cOA on the release of [3H]-GABA from synaptosomes induced by 50 μM VTD. The IC50 by probit transformation was 4.58 μM. Right: Inability of 20 μM tOA to inhibit VTD-evoked release of [3H]-GABA from synaptosomes (P>0.05, n=3–10). (b) The oleamides were unable to significantly modulate K+-induced transmitter release (n=3; P>0.05).
Figure 5.
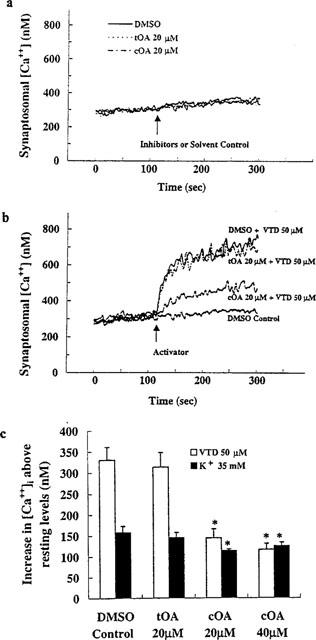
The effects of oleamides on [Ca2+]i in resting and depolarized nerve terminals. (a) Neither cOA or tOA can acutely alter synaptosomal [Ca2+]i at relatively high concentrations. (b) Continuous recording of synaptosomal [Ca2+]i showing the differential effects of trans- and cis-oleamide on veratridine (VTD)-dependent rises in [Ca2+]i. (c) Isomer dependence of blockade of veratridine- and K+-induced rises in intraterminal [Ca2+]i. Asterisks indicate a statistically significant difference from respective controls. (n=4–6; *P<0.05).
Selective depression of sustained repetitive firing
The functional significance of these biochemical data was examined in patch clamped cortical neurones in culture. Pyramidal cells were current-clamped at their resting membrane potential, and unitary action potentials were generated by the application of current pulses of 2 ms duration at a frequency of 1 Hz, and of sufficient amplitude to induce all-or-none (TTX-sensitive) events. For analysis, the waveforms of 50 consecutive action potentials were averaged and the parameters of amplitude, overshoot, rise-time, time to 50% repolarization and resting membrane potential were measured pre- and post-treatment. cOA had no significant effect on any of the parameters measured from the TTX-sensitive low frequency evoked spikes at either 20 μM (n=4) or 64 μM (n=7) (Figure 6a,b). In contrast, cOA strongly depressed sustained repetitive firing (SRF) elicited by protracted current passage (500 ms duration, 0.1 Hz) through the recording microelectrode (Figure 6c). At all concentrations tested, cOA had no effect on the initial action potential elicited by the depolarizing current pulse. However, SRF (defined here as the train of potentials occurring subsequent to this primary action potential) was inhibited in a concentration-dependent manner (Figure 6d). Sixty-four μM cOA completely and rapidly abolished SRF (4/4) (Figure 6c), with an approximate time for block to 50% of pre-treatment response (t50) of 39±3 s (n=4). Ten μM cOA was found to completely abolish SRF (all excepting the primary spike) in some cases (4/10) but this occurred over a longer time course (t50=174±49 s). The extent of the steady-state block at intermediate doses was reduced by hyperpolarizing the impaled cell (not shown) indicating a voltage-dependent supression of burst firing. Collation of data interpolated from dose responses (following exposure to test concentrations for at least 15 min) in individual replicates yielded an apparent EC50 of 4.39±0.47 μM (n=5). Data pooled from all experiments were fitted by non-linear regression to a sigmoid curve with an EC50 of 4.12±0.024 μM (mean±log s.e.mean) and a Hill slope of 2.9 (95% C.I. 2.1–3.6) (Figure 6d). Both oleic acid (32 μM) and tOA (20 μM) were very weak antagonists of SRF after 15 min exposure (Figure 6e).
Figure 6.
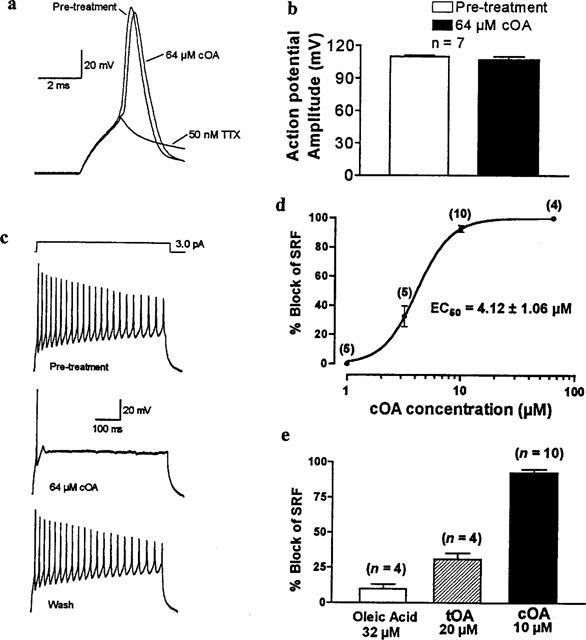
cis-Oleamide is a specific state-dependent antagonist of the voltage-gated Na+ channel. (A) Spikes evoked by current passage through the patch micro-electrode (at 1 Hz, averages of 50 consecutive sweeps) were not altered by 64 μM cOA but were fully blocked by 50 nM TTX. (b) Compounded data from seven replicated experiments on unitary action potentials. Bars represent mean+s.e.mean. (c) In contrast cOA (10–64 μM) markedly suppressed sustained repetitive firing (SRF) in salines containing Co2+ to suppress voltage-gated Ca2+ currents. (d) Block of SRF was concentration- and voltage-dependent (the blocking action could be partially reversed by hyperpolarization of the impaled cell, not shown). Each data point represents mean±s.e.mean for the indicated number of replicates. Note the very steep slope (Hill coefficient of approximately 3). (e) The high affinity block produced by cOA was not seen with high concentrations of hydrophobic congeners.
Discussion
Selectivity of oleamide for CNS receptors and channels
Our data suggest that oleamide is a stereoselective modulator of both the voltage-gated Na+ channel and the GABAA receptor complex. Only the hypnotic (cis) isomer was active at both model targets, equally hydrophobic congeners were inactive and the reciprocal modulatory effects were effected at concentrations overtly suppressing activity in cultured neural circuits. Rat cortical neurones express 5HT2 (e.g., LopezGimenez et al., 1997) receptors, at least in vivo, and these have been cited as putative high-affinity targets for oleamide (Huidobro-Toro & Harris, 1996). In this and a previous study (Lees et al., 1998a), functional antagonistic effects of oleamide on synaptic activity were detected only at concentrations in excess of 3–10 μM consistent with a functional role for the molecular targets studied here but not with a higher affinity interaction with the G-protein coupled serotonin receptors. The isomer dependence and lack of effect with oleic acid is consistent with a receptor-mediated response and our earlier studies (Lees et al., 1998a,1998b) on GABAA receptor isoforms of varying subunit composition (expressed in the invariant lipid membrane of amphibian oocytes) suggest that specific domains on channel proteins themselves may be targeted by the brain lipid. The slow-onset kinetics of cOA at both molecular targets may reflect the involvement of a second messenger or merely time for partitioning of the very hydrophobic molecule into target membranes (complete solution exchange is achieved at the cell within 20 s of selecting a novel saline for superfusion through the bath). The depression of spontaneous synaptic traffic was equally slow so it may result from enhanced polysynaptic inhibitory tone and or depressant effects on Na+ channels (as indicated by SRF suppression) leading to reduced transmitter release from nerve terminals.
Relevance of the concentrations used
The modulatory actions at both GABAA receptors and voltage-gated Na+ spikes are entirely consistent with limitation of neuronal excitability and sedation in the intact animal, but are the concentrations used physiologically relevant in vivo? Extracellular levels of synthetic intraperitoneal oleamide required to induce sleep (Cravatt et al., 1995) or dose-dependent psychomotor depression (Cheer et al., 1999) can be only crudely extrapolated (see Boring et al., 1996; Lees et al., 1998a) but initial concentrations in brain could lie in the range 6–60 μM. Recently, refinements in analytical methods suggest that resting physiological oleamide levels in rat CSF are approximately 170 nM (Hanus et al., 1999) and following sleep deprivation levels in CSF approximate to 450 nM (Basile et al., 1999). Both of these values are below the threshold for modulation of GABAA receptors and presumed Na+ channels (reported in this study) but would be sufficient to exert an increase in 5HT binding affinity at ketanserin labelled sites (see Cheer et al., 1999). However,the very high affinity modulatory effect previously reported on metabotropic 5HT receptors, at concentrations between 1–100 nM (Huidobro-Toro & Harris, 1996), would be saturated even at physiological levels (in the absence of sleep deprivation). Bearing in mind the extreme hydrophobicity (calculated log P>6.5) of oleamide and the selective, membrane associated, distribution of its degradative amidohydrolase enzyme (e.g., Thomas et al., 1997) it is conceivable that the lipid may represent a short range messenger which accumulates and exerts its hypnogenic effects within the cell membrane itself at discrete anatomical sites.
Analogies with depressant drugs
Complementary modulation of both ligand and voltage-gated ion channels was historically perceived as being unusual, with most therapeutic drugs and toxins showing a high degree of selectivity for either one or other target (e.g., Franks & Lieb, 1994; Catterall, 1992). Similarly, physiological regulation is thought to be channel selective or, as reported for protein kinase C, reduces both voltage-gated sodium currents and GABA-gated chloride conductances (Sigel & Baur, 1988). In recent years, however, the phenomenon of state-dependent modulation of voltage-gated channels by depressant drugs has been acknowledged as an important feature of their therapeutic effects and manageable clinical toxicity (Catterall, 1987). Molecules which have long been acknowledged as GABAA modulators, like barbiturates (Frenkel et al., 1993; Macdonald & McLean, 1986) and volatile anaesthetics (Rehberg et al., 1996), also have the capacity to block burst firing or depolarized/inactivated channels (rodent or human proteins are equally susceptible) preferentially. In the above studies, state-dependent Na+ channel modulation and GABAA modulation (Macdonald & McLean, 1986) were effected at, or below, concentrations achieved in the interstitial fluid of sedated, seizure-free or anaesthetized patients (see Franks & Lieb, 1994). We have reported that the homo-oligomeric GABAC receptor (insensitive to biculline but in structural terms unequivocally a member of the GABA-gated ionotropic receptor superfamily) which is composed of rho subunits is uniquely insensitive to the positive modulatory effects of both isoflurane and oleamide (Lees et al., 1998b). This rho subunit was originally cloned in retinal tissue and has a relatively restricted distribution in CNS so one would not expect it to play a pivotal role in anaesthesia. These analogies with inhalational anaesthetics have been examined only to a very limited extent in vivo. In rats intra peritoneal oleamide did not alter the minimal alveolar concentration (MAC) of desflurane suggesting that it does not synergize anaesthetics in vivo like other acknowledged GABAA modulators although pharmacokinetic factors may limit its bioavailability in such studies (Yost et al., 1998).
Anticonvulsants like phenytoin, carbamezepine and lamotrigine inhibit sustained repetitive firing (Cheung et al., 1992) and paroxysmal bursting by promoting Na+ channel inactivation (Lang et al., 1993; Leach et al., 1995) but are not noted for GABAA modulation at therapeutic concentrations (e.g., Macdonald & McLean, 1986; Lees & Leach, 1993). A potent anticonvulsant butyrolactone (a single congener from a class of molecules which are recognised GABAA modulators) can concurrently block Na+ currents in a state-dependent manner, reinforcing the importance of these actions as determinants of pharmacological profile (Hill et al., 1998). The fact that oleamide can modulate both ion channels in an analogous manner to phenobarbitone and selectively depress sustained repetitive firing is indicative of its anticonvulsant potential. Anaesthetic and anticonvulsant drug effects on voltage-gated Ca2+ channels are also thought to be important in their therapeutic actions and clinical utility (Beck et al., 1998; Study, 1994). The depression of K+-induced elevation of [Ca2+]i, albeit weakly in our experiments, by oleamide may be due to inhibition of a non-inactivating voltage-gated Ca2+ channel such as the L-type channel which is a member of the same structural superfamily as the Na+ channel α subunit but does not contribute strongly to phasic transmitter release transmitter at CNS synapses (Dunlap et al., 1995).
Oleamide as an endogenous hormone: implications for drug discovery
Overall, it is an intriguing possibility that oleamide may represent an endogenous ligand for depressant drug binding sites in mammalian CNS. This hypothesis is of more than academic interest. Our results suggest that oleamide has a similar molecular mode of action to therapeutically important and widely prescribed drugs used globally for the treatment of anxiety, sleep disorders, epilepsy and even to induce anaesthesia in the operating room (see also Lees, 1998; Lees et al., 1998a). These synthetic drugs are of undoubted therapeutic and commercial importance but none of them have ideal properties. Many epileptic patients are refractory to treatment, most of the drugs have a ‘marginal therapeutic ratio' and many are habit forming, produce hangovers, tolerance or retrograde amnesia (e.g., Costa & Guidotti, 1996). A more detailed understanding of the molecular pharmacology of endogenous depressant hormones like oleamide holds much promise for safe and effective treatments for a range of debilitating conditions in a vast pharmaceutical market.
Acknowledgments
Thanks to the Wellcome Trust (G. Lees)/NSERC of Canada (R.A. Nicholson) for financial support and to Helen Jackson for technical assistance with cell cultures.
Abbreviations
- BSA
bovine serum albumin
- cOA
cis-9,10-octadecenoamide (oleamide)
- DMSO
Dimethylsulphoxide
- tOA
(E)-9-octadecenoic acid
- SRF
sustained repetitive firing
- VTD
veratridine
References
- AUICHI T., DIAMATSU T., NAKAYA K., NAKAMURA Y. Fluorescence changes of rhodamine 6G associated with changes of membrane potential in synaptosomes. Biochim. Biophys. Acta. 1982;685:289–296. doi: 10.1016/0005-2736(82)90070-0. [DOI] [PubMed] [Google Scholar]
- BASILE A.S., HANUS L., MENDELSON W.B. Characterization of the hypnotic properties of oleamide. Neuroreport. 1999;10 5:947–951. doi: 10.1097/00001756-199904060-00010. [DOI] [PubMed] [Google Scholar]
- BECK H., STEFFENS R., ELGER C.E., HENMANN U. Voltage-dependent Ca++ currents in epilepsy. Epilepsy Res. 1998;32:321–332. doi: 10.1016/s0920-1211(98)00062-x. [DOI] [PubMed] [Google Scholar]
- BORING D.L., BERGLUND B.A., HOWLETT A.C. Cerebrodiene, arachidonyl-ethanolamide, and hybrid structures: potential for interaction with brain cannabinoid receptors. Prostagland. Leukot. Essent. Fatty-Acids. 1996;55 3:207–210. doi: 10.1016/s0952-3278(96)90100-3. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Common modes of drug action on sodium channels: local anesthetics, antiarrhythmics and anticonvulsants. TIPS. 1987;8:57–65. [Google Scholar]
- CATTERALL W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- CHEER J.F., CADOGAN A-K., MARSDEN C.A., FONE K.C.F., KENDALL D.A. Modification of 5HT2 receptor mediated behaviour in the rat by oleamide and the role of cannabinoid receptors. Neuropharmacology. 1999;38:533–541. doi: 10.1016/s0028-3908(98)00208-1. [DOI] [PubMed] [Google Scholar]
- CHEUNG H., KAMP D., HARRIS E. An in vitro investigation of the action of lamotrigine on neuronal voltage-activated sodium channels. Epilepsy Res. 1992;13:107–112. doi: 10.1016/0920-1211(92)90065-2. [DOI] [PubMed] [Google Scholar]
- COSTA E., GUIDOTTI A. Benzodiazepines on trial: a research strategy for their rehabilitation. TIPS. 1996;17:192–200. doi: 10.1016/0165-6147(96)10015-8. [DOI] [PubMed] [Google Scholar]
- CRAVATT B.F., PROSPERO-GARCIA O., SIUZDAK G., GILULA N.B., HENRIKSON S.J., BOGER D.L., LERNER R.A. Chemical characterisation of a family of brain lipids that induce sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- DUNKLEY P.R., JARVIE P.E., HEATH J.W., KIDD K.G.J., ROSTAS J.A.P. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986;372:115–129. doi: 10.1016/0006-8993(86)91464-2. [DOI] [PubMed] [Google Scholar]
- DUNLAP K., LUEBKE J.I., TURNER T.J. Exocytotic Ca++ channels in mammalian central neurons. TINS. 1995;18:89–98. [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- FRENKEL C., DUCH D.S., URBAN B.W. Effects of i.v. anaesthetics on human brain sodium channels. Br. J. Anaesthesia. 1993;71:15–24. doi: 10.1093/bja/71.1.15. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca++ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GUAN X.J., CRAVATT B.F., EHRING C.R., HALL J.E., BOGER D.L., LERNER R.A., GILULA N.B. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J. Cell Biol. 1997;139:1785–1792. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANUS L.O., FALES H.M., SPANDE T.F., BASILE A.S. A gas-chromatographic-mass spectral assay for the quantitation of oleamide in biological fluids. Anal. Biochem. 1999;270:159–166. doi: 10.1006/abio.1999.4083. [DOI] [PubMed] [Google Scholar]
- HARRIS R.A., ALLAN A.M. Functional coupling of γ-aminobutyric acid receptors to chloride channels in brain membranes. Science. 1985;228:1108–1110. doi: 10.1126/science.2581319. [DOI] [PubMed] [Google Scholar]
- HILL M.W., REDDY P.A., COVEY D.F., ROTHMAN S.M. Inhibition of voltage-dependent sodium channels by the anticonvulsant γ-aminobutyric acid type A receptor modulator, 3-benzyl-3-ethyl-2-piperidinone. J. Pharm. Exp. Ther. 1998;285:1303–1309. [PubMed] [Google Scholar]
- HUIDOBRO-TORO J.P., HARRIS R.A. Brain lipids that induce sleep are novel modulators of 5-hydroxytryptamine receptors. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8078–8082. doi: 10.1073/pnas.93.15.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMULAINEN H., BONDY S.C. Increased free intrasynaptosomal Ca2+ by neurotoxic organometals: distinctive mechanisms. Toxicol. Appl. Pharmacol. 1987;88:77–86. doi: 10.1016/0041-008x(87)90271-7. [DOI] [PubMed] [Google Scholar]
- LANG D.G., WANG C.M., COOPER B.R. Lamotrigine, phenytoin and carbamezepine interactions on the sodium current present in N4TG1 mouse neuroblastoma cells. J. Pharm. Exp. Ther. 1993;266:829–835. [PubMed] [Google Scholar]
- LEACH M.J., LEES G., RIDDALL D.R. Lamotrigine mechanisms of action Antiepileptic drugs 1995Raven Press Ltd: New York; 861–869.In: Levy, R.H. et al., eds4th edn [Google Scholar]
- LEES G. Molecular mechanisms of anaesthesia: light at the end of the channel. Br. J. Anaesthesia. 1998;81 4:491–493. doi: 10.1093/bja/81.4.491. [DOI] [PubMed] [Google Scholar]
- LEES G., EDWARDS M.D., HASSONI A.A., GANELLIN C.R., GALANAKIS D. Modulation of GABAA receptors and inhibitory synaptic currents by an endogenous CNS sleep regulator cis-9,10 octadecenoamide (cOA) Br.J. Pharmacol. 1998a;124:873–882. doi: 10.1038/sj.bjp.0701918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEES G., EDWARDS M.D., WILLOTT R. Effects of isoflurane and cis-9,10-octadecenoamide (cOA) on the ρ1 GABA receptor. J. Physiol. (London) 1998b;509:P190–P191. [Google Scholar]
- LEES G., LEACH M.J. Studies on the mechanism of action of the novel anticonvulsant lamotrigine (lamictal) using primary neuroglial cultures from rat cortex. Brain Res. 1993;612:190–199. doi: 10.1016/0006-8993(93)91660-k. [DOI] [PubMed] [Google Scholar]
- LOPEZGIMENEZ J.F., MENGOD G., PALACIOS J.M., VILARO M.T. Selective visualization of rat brain 5-HT2A receptors by autoradiography with [H-3]MDL 100,907. Naunyn-Schmiedebergs Arch. Pharmacol. 1997;356 4:446–454. doi: 10.1007/pl00005075. [DOI] [PubMed] [Google Scholar]
- MACDONALD R.L., MCLEAN M.J. Anticonvulsant drugs: mechanisms of action. Advances in Neurology, vol. 44 1986Raven Press, New York; 713–736.In: (Delgado-Escuetta A.V. et al. eds.) [PubMed] [Google Scholar]
- MURASE K., RYU P.D., RANDIC M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely dissociated dorsal horn neurons. Neurosci. Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- REHBERG B., XIAO Y.-H., DUCH D.S. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anaesthetics. Anesthesiology. 1996;84 5:1223–1233. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- ROE E.T., SCANLAN J.T., SWERN D. Fatty Acid Amides I. Preparation of amides of oleic and the 9,10-dihydroxy stearic acids. J. Amer. Chem. Soc. 1949;71:2215–2218. [Google Scholar]
- SIGEL E., BAUR R. Activation of protein kinase C differentially modulates neuronal Na+, Ca2+ and γ-aminobutyrate type A channels. Proc. Natl. Acad. Sci. U.S.A. 1988;85:6192–6196. doi: 10.1073/pnas.85.16.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUDY R.E. Isoflurane inhibits multiple voltage-gated calcium currents in hippocampal pyramidal neurons. Anesthesiology. 1994;81:104–116. doi: 10.1097/00000542-199407000-00016. [DOI] [PubMed] [Google Scholar]
- THOMAS E.A., CRAVATT B.F., EHRING C.R., HALL J.E., BOGER D.L., LERNER R.A., GILULA N.B. Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution in neurons within the rat central nervous system. J. Neurosci. Res. 1997;50 6:1047–1052. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- YOST C.S., HAMSON A.J., LEONOUDAKIS D., KOBLIN D.D., BORNHEIM L.M., GRAY A.T. Oleamide potentiates benzodiazepine-sensitive gamma-aminobutyric acid receptor activity but does not alter minimum alveolar anesthetic concentration. Anesthes. Analges. 1998;86 6:1294–1300. doi: 10.1097/00000539-199806000-00031. [DOI] [PubMed] [Google Scholar]