

Abstract
The aim of this study was to functionally characterize the recombinant mouse P2X4 receptor and to compare its pharmacological properties with those of the human and rat orthologues.
Whole cell recordings were made from rafts of HEK-293 cells stably expressing recombinant mouse, rat or human P2X4 receptors, using Cs-aspartate containing electrodes (3–8 MΩ) in a HEPES-buffered extracellular medium.
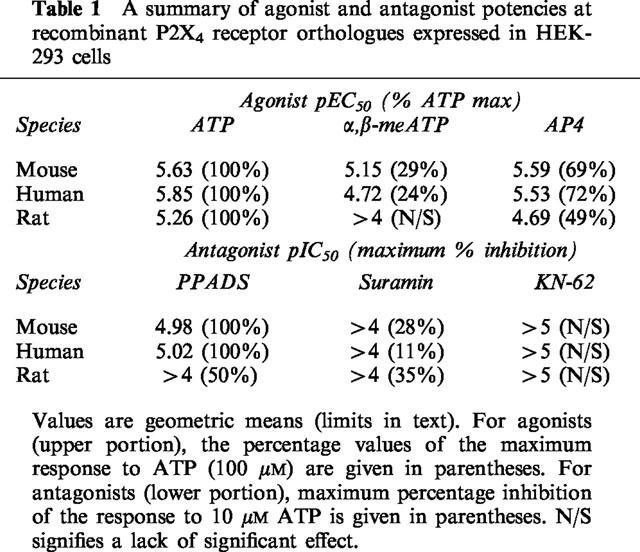
The agonist potency of ATP at the three species orthologues was similar, with mean EC50 values of 2.3 μM, 1.4 μM and 5.5 μM, respectively.
Adenosine-5′-tetraphosphate (AP4) acted as a partial agonist with respect to ATP at the mouse and human P2X4 receptors (EC50=2.6 and 3.0 μM), but was significantly less potent at the rat orthologue (EC50=20.0 μM). α,β-methylene adenosine-5′-triphosphate (α,β-meATP) also acted as a partial agonist, producing 29% of the maximum response at the mouse P2X4 and 24% at the human P2X4 receptor.
In contrast to the other species orthologues, α,β-meATP failed to elicit a significant agonist response at rat P2X4 receptors, and was found to act as an antagonist, with an IC50 of 4.6 μM, against 10 μM ATP.
Mouse P2X4 receptors were found to be sensitive to the antagonist, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) (IC50=10.5 μM), as were human P2X4 receptors (IC50=9.6 μM). The rat receptor however, showed a low sensitivity to PPADS (IC50>100 μM).
All three orthologues were relatively suramin-insensitive (IC50>100 μM) and insensitive to 1-[N,O-Bis(5-isoquinoline sulphonyl)benzyl]-2-(4-phenylpiperazine)ethyl]-5-isoquinoline sulphonamide (KN-62; IC50>3 μM).
Our results suggest that the pharmacological properties of the mouse receptor are most similar to the human P2X4 receptor, and differ markedly from the rat receptor.
Keywords: Mouse P2X4 receptor, P2X receptor, rundown, AP4, ATP
Introduction
P2X receptors form a family of ionotropic cation channels, activated by extracellular ATP. To date, cDNAs for seven subunits have been identified (Valera et al., 1994; Brake et al., 1994; Chen et al., 1995; Buell et al., 1996; Bo et al., 1995; Collo et al., 1996; Surprenant et al., 1996) and when expressed in heterologous systems, these produce functional homomeric receptors of unknown stoichiometry. Phenotypic characteristics of purinergic responses in vivo, in some cases, can be explained only by the existence of heteromeric receptors exhibiting novel pharmacological and kinetic phenotypes. Thus, hetero-oligomeric assembly of P2X2/3 (Lewis et al., 1995), P2X1/5 (Torres et al., 1998) and P2X4/6 (Lê et al., 1998) subunits have been described in vitro, and the novel pharmacological profiles exhibited by the heteromeric receptors have been shown to be similar to those seen in certain native tissues (e.g. Lewis et al., 1995). Notwithstanding this, the functional phenotypic features of certain homomeric P2X receptor assemblies do resemble phenotypes found in vivo, and thus the study of the operational features of homomeric P2X receptor subunit assemblies remains crucial for our understanding of the role of these channels in physiological function.
P2X4 receptors were originally cloned from rat brain (Bo et al., 1995) and were described as being relatively insensitive to the purinergic antagonists, suramin and pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS). The agonist, α,β-methylene-ATP (α,β-meATP) was shown to have little effect, and currents elicited by ATP were slowly desensitizing (Buell et al., 1996; Bo et al., 1995). Subsequently, both human (Garcia-Guzman et al., 1997) and murine orthologues of the P2X4 receptor have been isolated (Townsend-Nicholson et al., 1999; Simon et al., 1999), and the limited comparison of the three species orthologues so far performed have revealed operational differences.
To date there are no reports describing comparison of the properties of these orthologues in the same study, and even reports characterizing a single orthologue (i.e. the rat) describe some discrepancies. Thus, the response of homomeric rat P2X4 receptors to ATP desensitized slowly (Buell et al., 1996; Collo et al., 1996), although when expressed in oocytes, rat P2X4 receptors required 10–15 min to fully recover from desensitization (Bo et al., 1995). All studies to date show that homomeric rat P2X4 receptors are insensitive to α,β-meATP at concentrations up to 100 μM, when expressed in HEK-293 cells (Buell et al., 1996; Collo et al., 1996), although responses have been obtained from the rat orthologue when expressed in Xenopus oocytes (Lê et al., 1998). Bo et al. (1995) originally reported insensitivity to the antagonists, PPADS and suramin; findings supported by a study by Collo et al. (1996). However, studies by Séguéla et al. (1996) described modest inhibition by PPADS, and complete blockade by suramin, albeit at high concentrations, while Soto et al. (1996) described modest inhibition by suramin. It should be noted that high concentrations of suramin have been shown to have non-specific effects, and as such cannot be considered as being highly selective for P2 receptors (Balcar, 1995).
In contrast to the rat P2X4 receptor, recombinant human P2X4 channels have been shown to be sensitive to PPADS, and slightly sensitive to suramin (Garcia-Guzman et al., 1997; Dhulipala et al., 1998). Allosteric modulation of the human P2X4 receptor by Zn2+ (Soto et al., 1996) and cibacron blue (Miller et al., 1998) has also been demonstrated. The mouse P2X4 receptor is the most recently isolated P2X4 orthologue (Townsend-Nicholson et al., 1999; Simon et al., 1999), has 87 and 94% amino acid identity with the human and rat receptors, respectively, and has been characterized only when expressed in oocytes. Mouse P2X4 subunits produced rapid inward currents in response to ATP, which were potentiated, rather than blocked, by cumulative applications of low concentrations of PPADS or suramin (Townsend-Nicholson et al., 1999).
In this study, the whole cell configuration of the patch clamp technique was used to characterize recombinant P2X4 receptors. Each orthologue has been expressed in the same parental cell line, and this represents the first study where a full comparison between species orthologues of P2X4 receptor has been performed under the same experimental conditions. Full characterization of murine P2X4 receptors expressed in a mammalian system has yet to be described, and thus in this study we give particular emphasis to this orthologue. A preliminary account of these studies has been presented to the British Pharmacological Society (Jones et al., 1999).
Methods
Cell culture
Wild type human embryonic kidney (HEK-293) cells (1×106), devoid of endogenous P2X receptors (Chessell et al., 1998), were transfected with 4 μg of the mouse P2X4 pcDNA 3.1(−) supercoiled plasmid by electroporation (Easy-ject, Equibio, Kent, U.K.). The transfected cells were selected in DMEM nutrient mix supplemented with 10% FBS and 0.6 mg ml−1 geneticin sulphate (G418) for stable expression of the mouse P2X4 receptor (Simon et al., 1999). HEK-293 cells stably expressing mouse, rat or human P2X4 receptors were maintained in DMEM nutrient mix supplemented with FBS (10%) and 0.6 mg ml−1 G418. Cell lines were incubated in a water saturated atmosphere of 95% O2/5% CO2 at 37°C in 75 cm2 flasks (Costar, Bucks, U.K.) and were passaged by trypsinization (trypsin-EDTA 1× solution) when confluent. When required for study, cells were attached to glass coverslips (13 mm; Chance Propper Ltd, West Midlands, U.K.) and used for electrophysiological recording not less than 14 h after plating. All coverslips were used within 3 days.
Electrophysiological recording
For each experiment, coverslips were transferred to a perfused recording chamber (volume approximately 400 μl, flow rate 2 ml min−1) mounted on the stage of an inverted microscope (Nikon Diaphot, Nikon U.K., Kingston upon Thames, U.K.). ATP-evoked ionic currents were recorded using the whole cell configuration of the patch clamp technique (Hamill et al., 1981) from groups of four or more electrically coupled cells (cell rafts) unless otherwise stated. Cells were continuously perfused with external solution containing (in mM): NaCl 145, KCl 2, MgCl2 1, CaCl2 2, HEPES 10, D-Glucose 10 (pH 7.3, osmolarity 300 mOsm). Patch electrodes, with resistances of 3–8 MΩ, were pulled from 1.2 mm borosilicate glass (GC120F-10, Clarke Electromedical Supplies, Pangbourne, U.K.). Electrodes were firepolished and backfilled with internal solution (in mM): Cs aspartate 145, EGTA 11, HEPES 5, NaCl 2 (pH 7.3, osmolarity 290 mOsm).
Tight seal (>10 GΩ) currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, U.S.A.). Currents were filtered with a corner frequency of 1–5 kHz (8-pole Bessel filter), digitized at 2–10 KHz using a Digidata 1200A (Axon Instruments) interface, and stored on computer. Only cells with a residual series resistance of less than 20 MΩ were recorded from, and series resistance was uncompensated. In all experiments, cells were voltage clamped at −90 mV. For all experiments except the determination of current-voltage relationships, the junction potential was uncorrected.
Experimental procedures
In all experiments, agonists were applied using a computer-controlled fast-flow U-tube application system (Fenwick et al., 1982), modified to include an extra solenoid valve, which allowed both rapid application and removal of applied drugs (onset and offset latencies of approximately 90 and 50 ms, respectively; Chessell et al., 1997). Agonists applications of 1 s, with a 5 min wash period between subsequent doses, were used in the determination of concentration-effect curves.
Agonist concentration-effect curves for ATP, AP4 and α,β-meATP were determined. A single application of ATP (100 μM) was followed by sequentially increasing concentrations of the agonist under test (0.3–100 μM), separated by 5 min wash periods. A final control concentration of ATP (100 μM) was then applied, and the mean of both ATP responses was taken as 100% response. In this study, final responses to 100 μM ATP were not significantly different to the initial responses to 100 μM ATP.
Studies were carried out to investigate the effect of P2 antagonists pyridoxalphosphate-6-azophenyl-2′4′disulphonic acid (PPADS), suramin and 1-[N,O-Bis(5-isoquinoline sulphonyl)benzyl]-2-4-phenylpiperazine)ethyl]-5-isoquinoline sulphonamide (KN-62) on the current elicited by 10 μM ATP. In each case, two test applications of ATP (10 μM) were applied initially, followed by repeated applications of ATP (10 μM) in the presence of increasing concentrations of antagonists. Coverslips were preincubated for 5 min with each antagonist concentration prior to ATP exposure. Finally, ATP (10 μM) was applied twice, in the absence of any antagonist. Compounds being studied were included in the superfusate and U-tube drug application system at the appropriate concentration. A new coverslip was used for each experiment.
Data analysis
Data are expressed as a percentage of the maximum current elicited by ATP, or as a percentage of the first response observed in a particular experimental paradigm. Where appropriate, concentration-effect curves were fitted using a three parameter logistic equation (GraphPad Prism, San Diego, CA, U.S.A.). EC50 and IC50 values are expressed as the geometric mean±95% confidence intervals from 6–18 experiments, while individual responses are expressed as mean±s.e.mean. Statistical comparisons were made using the Student's paired or unpaired t-test, where appropriate, and the null hypothesis rejected when P<0.05.
Materials
Dulbecco's modified Eagle's medium/NutMix F12 (DMEM), foetal bovine serum (FBS), Geneticin (G418) sulphate and trypsin-EDTA were obtained from GibcoBRL (Paisley, U.K.). Adenosine-5′-triphosphate (ATP), α,β-methylene adenosine-5′-triphosphate (α,β-meATP), adenosine-5′-tetraphosphate (AP4), pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS), 1-[N,O-Bis(5-isoquinoline sulphonyl)benzyl]-2-(4-phenylpiperazine)ethyl]-5-isoquinoline sulphonamide (KN-62) were obtained from Sigma (Poole, U.K.) and Suramin was from Bayer. All other chemicals were purchased from Sigma or BDH Laboratory supplies (Leicester, U.K.). All stable cell lines were generated within Glaxo Wellcome laboratories.
Results
Effects of agonists
In a total of 81 experiments, ATP induced inward currents in all cells expressing recombinant mouse P2X4 receptors. In single cells, mean current amplitudes were 3.6±0.4 nA and mean rise times were 132±9.9 ms (n=9) in response to 10 μM ATP. In comparison, the maximum inward currents obtained from single cells in response to 10 μM ATP at the human and rat receptors were: 825±178 pA (n=11) and 82±21 pA (n=6) with mean rise times of 238±18.6 ms and 468±36.5 ms, respectively.
ATP acted as an agonist at the mouse, human and rat P2X4 receptors (Figure 1A), with EC50 values of 2.3 [1.3–4.3] μM (n=16), 1.4 [0.8–2.6] μM (n=6) and 5.5 [2.6–11.6] μM (n=7), respectively. Both α,β-meATP and AP4 acted as partial agonists relative to ATP at the mouse P2X4 receptor (maximum inward currents significantly (P<0.05) smaller than those observed using ATP), producing maximum responses of 29 and 69%, respectively, of that achieved with 100 μM ATP (Figure 1B,C). The EC50 values for AP4 and α,β-meATP at mouse P2X4 were not significantly different from that of ATP (2.6 [1.9–3.7] μM, n=6 and 7.0 [2.3–21.6] μM, n=9, respectively). Similar results were observed at the human P2X4 receptor, with AP4 having an EC50 value of 3.0 [1.4–6.0] μM (n=6) and reaching a maximum response of 72%, while α,β-meATP elicited a maximum response of 24% (EC50=19.2 [11.4–32.4] μM, n=9). AP4 was less potent at the rat receptor (P<0.05) (EC50=20.0 [4.6–87.4] μM, n=6) than at the mouse and human orthologues, and at 100 μM, elicited 49% of the maximum current. α,β-meATP failed to elicit a significant response at rat P2X4 receptors, at concentrations up to 100 μM. These results are summarized in Table 1.
Figure 1.
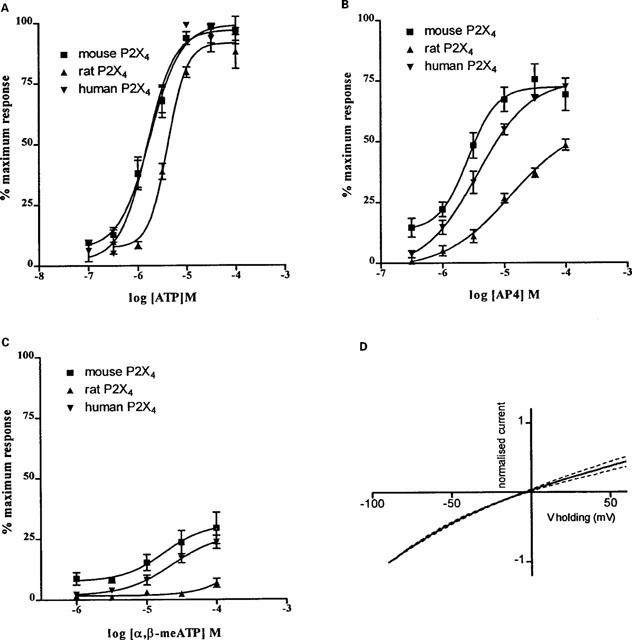
Concentration-effect curves for ATP (A), AP4 (B), and α,β-meATP (C) acting at mouse, human and rat P2X4 receptors expressed in HEK-293 cells. Serially increasing concentrations of ATP were applied for 1 s at 5 min intervals to rafts of electrically coupled cells. Symbols represent the mean±s.e.mean (n=6–18). (D) Current-voltage relationship of the mouse P2X4 receptor (10 μM ATP, normalized to the current at −90 mV, n=5).
Table 1.
A summary of agonist and antagonist potencies at recombinant P2X4 receptor orthologues expressed in HEK-293 cells
Effects of antagonists
Mouse P2X4 receptors were found to be sensitive to the P2 receptor antagonist PPADS (IC50=10.5 [7.5–14.8] μM (n=11), as were human P2X4 receptors (IC50=9.6 [4.8–19.0] μM, n=6) (Figure 2A; Table 1). The rat P2X4 receptor however, showed a low sensitivity to PPADS: the maximum concentration (100 μM) of antagonist tested caused a 50%±3 (n=8) inhibition in the response to ATP (10 μM). All three orthologues were relatively insensitive to suramin: the maximum concentration of antagonist tested (100 μM) caused a 28%±7, 11%±7 and a 35%±7 (n=6) reduction in ATP-induced response (at the mouse, human and rat P2X4 receptors, respectively).
Figure 2.
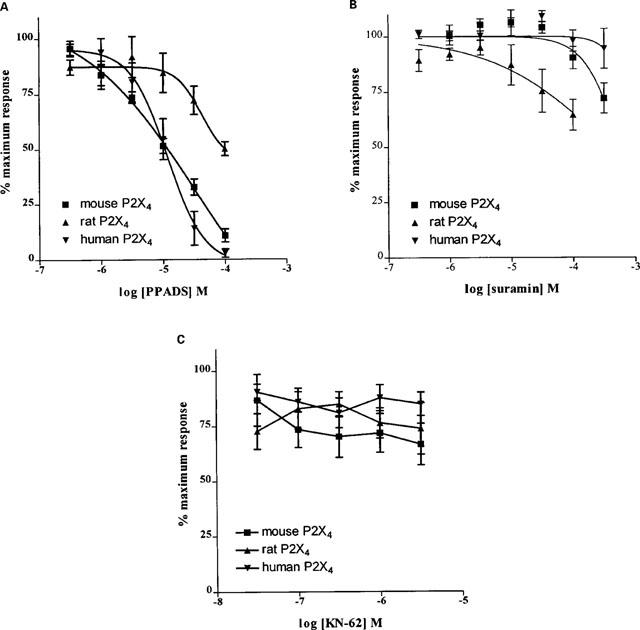
Effect of the purinergic antagonists, PPADS (A), suramin (B) and KN-62 (C) on ATP-evoked responses in mouse, human and rat P2X4 receptors expressed in HEK-293 cells. In all cases, whole cell currents were induced by 10 μM ATP, before and in the presence of serially increasing concentrations of antagonist. The inter-application time was 5 min, antagonists were included at the appropriate concentration in the superfusate and U-tube drug application system. Symbols are mean±s.e.mean (n=6–11).
Experiments were carried out to test the sensitivity of the P2X4 orthologues to 1-[N,O-Bis(5-isoquinoline sulphonyl)benzyl]-2-(4-phenylpiperazine)ethyl]-5-isoquinoline sulphonamide (KN-62), a potent antagonist at the human P2X7 receptor. No concentration-dependent inhibition was seen at any of the receptors (Figure 2C; Table 1), at concentrations of KN-62 up to 3 μM.
Antagonist effect of α,β-meATP
α,β-meATP acted as an agonist at the mouse and human P2X4 receptors, but failed to induce a significant response in cells expressing the rat orthologue. Studies were therefore carried out to test the antagonistic effect of α,β-meATP at the rat P2X4 receptor. α,β-meATP was found to act as an antagonist at the rat receptor, with an IC50 value of 4.6 [2.4–8.6] μM (n=9) causing a 91±3% inhibition of response to 10 μM ATP at 100 μM (Figure 3).
Figure 3.
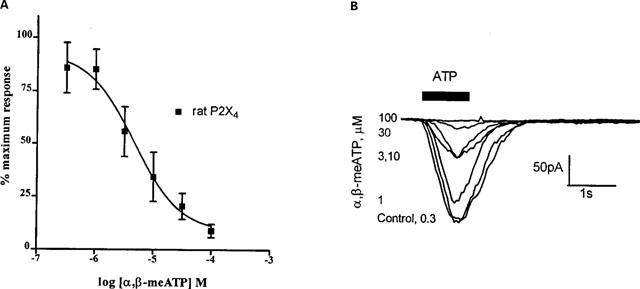
Effect of α,β-meATP on ATP-evoked responses in HEK-293 cells expressing recombinant rat P2X4 receptors. Whole cell currents were induced by 10 μM ATP, before and in the presence of serially increasing concentrations of α,β-meATP. The inter-application time was 5 min, α,β-meATP was included at the appropriate concentration in the superfusate and U-tube drug application system. Symbols are mean±s.e.mean (n=9). A representative trace of currents elicited by 10 μM ATP following preincubation of α,β-meATP is also shown (B).
Discussion
In this study we have characterized the recently isolated mouse P2X4 receptor (Simon et al., 1999) stably expressed in mammalian cells, and made a full comparison of the mouse, human and rat orthologues under identical experimental conditions. Both the mouse and human P2X4 receptors were found to be sensitive to the agonist, α,β-meATP and the antagonist, PPADS; while the rat orthologue was insensitive to both compounds. Uniquely, α,β-meATP acted as an antagonist at the rat P2X4 receptor.
In the present study, rafts of four or more electrically coupled cells were patched, as rundown of response has been reported to be a problem when recording from single cells expressing rat P2X4 receptors, using the whole cell configuration of the patch clamp technique (Miller et al., 1998). Similar rundown was seen at the mouse P2X4 receptor. Rundown of response is thought to be due to dialysis of the cytosolic components with the internal solution in the patching electrode, hence the reproducible responses gained from cell rafts. Clearly the use of cell rafts precludes detailed kinetic analyses, partly because of space-clamp considerations, and partly due to inadequate control of the concentration-clamp for agonists. However, these considerations do not preclude their use for pharmacological comparisons.
ATP induced rapid inward currents in 100% cells expressing each of the three species orthologues of P2X4. Differences in the mean amplitude of response are thought to reflect differences in receptor density, but this remains to be confirmed. Preliminary binding studies have demonstrated that it is possible to directly label P2X4 receptors in HEK-293 cells expressing the mouse orthologue, while no such binding can be detected in cells expressing the human or rat P2X4 receptors (A.D. Michel, unpublished observations), which suggests that the mouse receptor is expressed at a higher density than the human and rat orthologues.
The stable analogue of ATP, α,β-meATP has been commonly used in the characterization of P2X receptors. P2X4 receptors were typically considered to be insensitive to α,β-meATP, based on initial studies on the rat orthologue (Collo et al., 1996), but the subsequent cloning and characterization of the human P2X4 receptor (Garcia-Guzman et al., 1997) revealed modest sensitivity. In the present study, the mouse and human P2X4 receptors were found to be sensitive to α,β-meATP, where it acted as a partial agonist with respect to ATP. The action of this analogue of ATP has not previously been described at the mouse orthologue, but our results for the human P2X4 receptor are in agreement with the study by Garcia-Guzman et al. (1997). As expected, no response to α,β-meATP was observed at the rat P2X4 receptor (Collo et al., 1996; Séguéla et al., 1996), although a slight effect at 300 μM has been reported by Lê et al. (1998). Indeed, in the present study this compound acted as an antagonist at the rat receptor (see below).
Studies to date have tended to rely on the use of synthetic analogues of ATP such as 2-methylthio ATP, L-β,γ-methylene adenosine-5′-triphosphate, as well as α,β-meATP in the pharmacological characterization of P2X receptor subunits. We have also examined the action of an endogenous purinergic agonist, adenosine tetraphosphate (AP4), which has been described as acting with a potency several orders of magnitude higher than ATP itself (Small & Cooper, 1966). Cleavage of diadenosine pentaphosphate by asymetrical phosphodiesterases and diadenosine polyphosphate hydrolases yields AP4 (Zimmerman, 1996). This nucleotide also appears to be more resistant to ectoenzymic hydrolysis than ATP (Gualix et al., 1996; Mateo et al., 1998), a characteristic that may underlie its increased potency, and which may have physiological importance. All three P2X4 orthologues were found to be sensitive to AP4. The mouse and human receptors showed similar pharmacological profiles with AP4 acting as a partial agonist with EC50 values not significantly different to those of ATP. AP4 also acted as a partial agonist at the rat P2X4 receptor, but with a significantly lower maximum response than ATP.
Significant species differences have also been reported in the sensitivity of P2X4 orthologues to the P2 receptor antagonist, PPADS. We found the mouse and human P2X4 receptors to be PPADS-sensitive, while the rat receptor was relatively insensitive. Only one previous study has examined the effect of PPADS at the mouse orthologue expressed in oocytes (Townsend-Nicholson et al., 1999) and reported potentiation of ATP-induced currents at low concentrations of the antagonist. Although inhibition occurred at higher concentrations of PPADS, full inhibition was not achieved at 100 μM. Differences in expression systems might explain the conflicting results between our own study and that of Townsend-Nicholson et al. (1999), but differences in the protocols of agonist and antagonist applications are probably the major contributing factor. In the present study, HEK-293 cells expressing P2X4 receptors were pre-incubated with each antagonist concentration for 5 min before ATP was co-administered for 1 s, while Townsend-Nicholson et al. (1999) co-applied ATP and PPADS to oocytes expressing P2X4 orthologues for a prolonged period (60–180 s). It has been demonstrated that increased preincubation with antagonists can significantly increase the inhibition of ATP-induced currents seen at the rat P2X4 receptor (Lê et al., 1998) and it seems likely that the same is true at the mouse P2X4 receptor.
The human P2X4 receptor was found to be PPADS-sensitive, in accordance with previous studies (Garcia-Guzman et al., 1997), with complete inhibition of currents induced by 10 μM ATP being achieved at 100 μM PPADS. Our results from the rat P2X4 receptor also agree with the consensus from previous reports that the rat orthologue is significantly less sensitive to PPADS than the human or mouse receptors. The rat receptor has been described as ‘relatively insensitive' to PPADS because a number of studies report 10–20% inhibition of ATP-induced responses at high concentrations (>100 μM) of the antagonist (Soto et al., 1996; Buell et al., 1996) or with increased preincubation of antagonist concentrations (Lê et al., 1998). Only one study to date has reported near-complete inhibition of ATP-induced responses by 100 μM PPADS at the rat P2X4 receptor (Séguéla et al., 1996), and a 90 s preincubation period was required to inhibit currents induced by 1 μM ATP. Potentiation of ATP-induced currents at rat P2X4 receptors expressed in oocytes has also been reported at low PPADS concentrations (Bo et al., 1995), but no such effects were observed in the present study.
Low sensitivity to the antagonist suramin was observed at the mouse, human and rat P2X4 receptors. A slight antagonism of ATP-induced responses by high concentrations of suramin at the rat receptor has previously been reported by a number of groups (Soto et al., 1996; Buell et al., 1996) with longer antagonist preincubation times increasing the levels of inhibition seen (Lê et al., 1998). Garcia-Guzman et al. (1997) also reported low suramin-sensitivity at the human P2X4 receptor. It has been found that low concentrations of suramin potentiated ATP-induced currents at the mouse P2X4 receptor (Townsend-Nicholson et al., 1999), but, as discussed earlier, differences in protocol make comparisons with the present study difficult. Lack of specificity of suramin has also been reported (Balcar et al., 1995) and the synthesis of more selective P2 receptor antagonists would greatly facilitate the functional characterization of P2X receptors.
The isoquinoline derivative, KN-62, did not have an inhibitory effect on currents elicited by 10 μM ATP at any of the P2X4 orthologues tested. This compound has previously been reported to act as an antagonist at the human and mouse P2X7 receptors (Chessell et al., 1998; Gargett & Wiley., 1997), but had not previously been tested at the P2X4 subunits.
α,β-meATP was also tested as an antagonist at the rat P2X4 receptor, as this stable analogue of ATP did not induce inward currents at the rat orthologue. The finding that α,β-meATP was indeed capable of completely inhibiting ATP-induced currents at 100 μM was surprising given previous reports of a slight agonist effect at the same concentration (Lê et al., 1998). However, the same group previously described insensitivity of the rat P2X4 receptor to α,β-meATP (Séguéla et al., 1996) and the reasons for this discrepancy are unclear. Although α,β-meATP was originally described as a desensitizing agonist, antagonism by desensitization seems unlikely as α,β-meATP mediated inward currents were insignificant at the rat P2X4 receptor. It therefore seems that while α,β-meATP acts as a partial agonist at the mouse and human P2X4 receptors, it has low intrinsic activity at the rat orthologue, but is able to bind to the receptor and acts as an antagonist.
Identification of species differences in P2X4 receptors are important in the process of drug discovery. From the results gained in this study, it is clear that the human receptor is pharmacologically similar to the mouse P2X4 receptor, while the rat receptor shows marked phenotypic differences (Garcia-Guzman et al., 1997; Townsend-Nicholson et al., 1999). These phenotypic differences between species are themselves of considerable interest, and must be given critical importance when considering P2X4 receptors as drug targets in animal models of disease mechanisms.
Abbreviations
- α,β-meATP
α,β-methylene-ATP
- AP4
adenosine-5′-teteraphosphate
- ATP
adenosine-5′-triphosphate
- DMEM
Dulbecco's modified Eagle's medium
- FBS
foetal bovine serum
- KN-62
1-[N,O-Bis(5-isoquinoline sulphonyl)benzyl]-2-(4-phenylpiperazine)ethyl]-5-isoquinoline sulphonamide
- PPADS
pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid
References
- BALCAR V.J., DIAS L.S., LI Y., BENNETT M.R. Inhibition of [3H] CGP 39653 binding to NMDA receptors by a P2 antagonist, suramin. Neuroreport. 1995;7:69–72. [PubMed] [Google Scholar]
- BO X., ZHANG Y., NASSAR M., BURNSTOCK G., SCHOEPFER R. A P2X purinoceptor cDNA conferring a novel pharmacological profile. FEBS Lett. 1995;375:129–133. doi: 10.1016/0014-5793(95)01203-q. [DOI] [PubMed] [Google Scholar]
- BRAKE A.J., WAGENBACH M.J., JULIUS D. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature. 1994;371:519–523. doi: 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- BUELL G., LEWIS C., COLLO G., NORTH R.A., SURPRENANT A. An antagonist-insensitive P2X receptor expressed in epithelia and brain. EMBO J. 1996;15:55–62. [PMC free article] [PubMed] [Google Scholar]
- CHEN C.C., AKOPIAN A.N., SIVILOTTI L., COLQUHOUN D. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- CHESSELL I.P. , MICHEL A.D., HUMPHREY P.P.A. Properties of the pore-forming P2X7 purinoceptor in mouse NTW8 microglial cells. Br. J. Pharmacol. 1997;121:1429–1437. doi: 10.1038/sj.bjp.0701278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHESSELL I.P., MICHEL A.D., HUMPHREY P.P.A. Effects of antagonists at the human recombinant P2X7 receptor. Br. J. Pharmacol. 1998;124:1314–1320. doi: 10.1038/sj.bjp.0701958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLO G., NORTH R.A., KAWASHIMA E., MERLO-PICH E., NEIDHART S., SURPRENANT A. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHULIPALA P.D., WANG Y.X., KOTLIKOFF M.I. The human P2X4 receptor gene is alternatively spliced. Gene. 1998;207:259–266. doi: 10.1016/s0378-1119(97)00647-1. [DOI] [PubMed] [Google Scholar]
- FENWICK E., MARTY A., NEHER E. A patch clamp study of bovine chromaffin cells and of their sensitivity to acetylchloine. J. Physiol. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCIA-GUZMAN M., SOTO F., GOMEZ-HERNANDEZ Characterization of recombinant human P2X4 receptor reveals pharmacological differences to the rat homologue. Mol. Pharmacol. 1997;51:109–118. doi: 10.1124/mol.51.1.109. [DOI] [PubMed] [Google Scholar]
- GARGETT C.E., WILEY J.S. The isoquinoline derivative KN-62 is a potent antagonist of the P2Z-receptor of human lymphocytes. Br. J. Pharmacol. 1997;120:911–917. doi: 10.1038/sj.bjp.0701081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUALIX J., ABAL M., PINTOR J., MIRAS-PORTUGAL M.T. Presence of ε-adenosine tetraphosphate in chromaffin granules after transport of ε-ATP. FEBS Lett. 1996;391:195–198. doi: 10.1016/0014-5793(96)00732-6. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- JONES C.A., CHESSELL I.P., SIMON J., BARNARD E.A., MICHEL A.D., HUMPHREY P.P.A. Operational properties of mouse P2X4 receptors – a species comparison. Br. J. Pharmacol. 1999;128:38P. [Google Scholar]
- LÊ K.T., BABINSKI K., SEGUELA P. Central P2X4 and P2X6 channel subunits coassemble into a novel heteromeric ATP receptor. J. Neurosci. 1998;18:7152–7159. doi: 10.1523/JNEUROSCI.18-18-07152.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS C., NEIDHART S., HOLY C., NORTH R.A., BUELL G., SURPRENANT A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- MATEO J., GARCIA-LECEA M., MIRAS-PORTUGAL M.T., CASTRO E. Ca2+ signals mediated by P2X-type purinoceptors in cultured cerebellar purkinje cells. J. Neurosci. 1998;18:1704–1712. doi: 10.1523/JNEUROSCI.18-05-01704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER K.J., MICHEL A.D., CHESSELL I.P., HUMPHREY P.P. Cibacron blue allosterically modulates the rat P2X4 receptor. Neuropharmacol. 1998;37:1579–1586. doi: 10.1016/s0028-3908(98)00153-1. [DOI] [PubMed] [Google Scholar]
- SÉGUÉLA P., HAGHIGHI A., SOGHOMONIAN J.J., COOPER E. A novel neuronal P2X ATP receptor ion channel with widespread distribution in the brain. J. Neurosci. 1996;16:448–455. doi: 10.1523/JNEUROSCI.16-02-00448.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMON J., CHESSELL I.P., JONES C.A., MICHEL A.D., BARNARD E.A., HUMPHREY P.P. Molecular cloning and characterisation of splice variants of the mouse P2X4 receptor. Br. J. Pharmacol. 1999;126:20P. [Google Scholar]
- SMALL G.D., COOPER C. Studies on the occurrence and biosynthesis of adenosine tetraphosphate. Biochem. 1966;5:26–33. doi: 10.1021/bi00865a004. [DOI] [PubMed] [Google Scholar]
- SOTO F., GARCIA-GUZMAN M., GOMEZ-HERNANDEZ J.M., HOLLMANN M., KARSCHIN C. P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc. Natl. Acad. Sci. U.S.A. 1996;93:3684–3688. doi: 10.1073/pnas.93.8.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SURPRENANT A., RASSENDREN F., KAWASHIMA E., NORTH R.A. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- TORRES G.E., HAINES W.R., EGAN T.M., VOIGT M.M. Co-expression of P2X1 and P2X5 receptor subunits reveals a novel ATP-gated ion channel. Mol. Pharmacol. 1998;54:989–993. doi: 10.1124/mol.54.6.989. [DOI] [PubMed] [Google Scholar]
- TOWNSEND-NICHOLSON A., KING B.F., WILDMAN S.S., BURNSTOCK G. Molecular cloning, functional characterization and possible cooperativity between the murine P2X4 and P2X4a receptors. Br. Res. 1999;64:246–254. doi: 10.1016/s0169-328x(98)00328-3. [DOI] [PubMed] [Google Scholar]
- VALERA S., HUSSY N., EVANS R.J., ADAMI N., NORTH R.A., SURPRENANT A., BUELL G. A new class of ligand-gated ion channel defined by P2X receptor for extracellular ATP. Nature. 1994;371:516–519. doi: 10.1038/371516a0. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN H. Extracellular purine metabolism. Drug Dev. Res. 1996;39:337–352. [Google Scholar]