

Abstract
To examine the possible cardiotoxicity of the antimalarial drug mefloquine, increasing doses (0.3–30 mg kg−1) were given i.v. to anaesthetized guinea-pigs. Mefloquine did not alter ECG intervals significantly but gradually increased systolic blood pressure (at 3 mg kg−1) then had a depressor effect (at 10 mg kg−1). Death due to profound hypotension, probably resulting from cardiac contractile failure or AV block, occurred after either 10 mg kg−1 (2/6) or 30 mg kg−1 (4/6) mefloquine.
In isolated cardiac preparations mefloquine (3–100 μM) did not alter the effective refractory period but at the higher concentrations resting tension increased. Developed tension was reduced by 100 μM mefloquine in left atria (from 5.8±1.7 to 2.2±0.4 mN) whereas in papillary muscles although 30 μM mefloquine reduced developed tension (from 2.6±0.5 to 1.1±0.1 mN) subsequent addition of 100 μM caused a marked, but not sustained, positive inotropic effect (from 1.2±0.1 to 3.8±0.8 mN).
In single ventricular myocytes, mefloquine (10 μM) shortened action potential duration (e.g. APD90 from 285±29 to 141±12 ms) and reduced the amplitude of the systolic Ca2+ transient. These effects were accompanied by a decrease in the L-type Ca2+ current.
These results indicate that the main adverse effect of mefloquine on the heart is a negative inotropic action. This action can be explained by blockade of L-type Ca2+ channels.
Keywords: Mefloquine, ECG intervals, AV block, cardiac contractility, hypotension, effective refractory period, action potential duration, Ca2+ (intracellular), Ca2+ current
Introduction
In recent years there has been growing concern about the cardiac toxicity of certain antimalarial drugs. Mefloquine is currently the drug of choice for the treatment of chloroquine-resistant malaria and is also recommended for prophylaxis (Winstanley, 1996). In 1992, Laothavorn et al. reported that mefloquine did not alter ECG intervals (PR, QRS and QTc) and concluded that cardiotoxicity from mefloquine was unlikely. The following year, however, it was suggested that mefloquine may potentiate halofantrine-induced QT prolongation and associated cardiac arrhythmias (Nosten et al., 1993). Mefloquine is structurally similar to other quinoline antimalarial drugs such as chloroquine and quinine which are cardiotoxic. Quinine, and its diasteroisomer quinidine, cause QT prolongation and associated arrhythmias such as torsade de pointes (Jaeger et al., 1987; Ben-David & Zipes, 1993; Napolitano et al., 1994) whereas chloroquine has a greater effect on the QRS interval (Riou et al., 1988). In experimental studies in anaesthetized rats and rabbits, chloroquine dose-dependently decreased cardiac contractility and prolonged the PR and QRS intervals (Hughes & Coker, 1994).
We have investigated the acute cardiotoxicity of halofantrine and reported QTc prolongation, and at high doses, atrioventricular block (AV) block in anaesthetized guinea-pigs (Batey et al., 1997). Currently, there is a lack of data on mefloquine from experimental studies, where a wide range of doses can be examined. Before designing experiments to investigate interactions between halofantrine and mefloquine, more information about the possible effects of mefloquine per se is required. The aim of this work was therefore to examine the effects of mefloquine on the heart in vivo and in vitro, and to gain some insight into the mechanisms underlying any observed effects. Some of the in vitro data have been presented to the British Pharmacological Society (Coker & Batey, 1996) and the Physiological Society (Díaz et al., 1998).
Methods
In vivo experiments
Animal preparation
Experiments were performed in male Dunkin-Hartley guinea-pigs (720–850 g, Halls, Burton-on-Trent, U.K.), anaesthetized with sodium pentobarbitone (30 mg kg−1 i.p.). The trachea was cannulated to allow ventilation (Bioscience pump) with room air at a rate of 54 strokes min−1, and a stroke volume of 15 ml kg−1 body weight. Blood gases were monitored using a Corning 158 pH/blood gas analyser. A carotid artery and a jugular vein were cannulated for recording of arterial blood pressure and i.v. administration of drugs, respectively. Blood pressure was measured with a Bell and Howell type 4-422 transducer connected to a Grass 7P1/7DA amplifier, and a limb lead ECG was monitored from subcutaneous needle electrodes using a Grass 7P4 amplifier. Either lead I or lead II was recorded, depending on which gave the better separation of P waves from the T wave of the preceding complex. The amplifiers were connected to a Po-Ne-Mah computerized data acquisition system (Linton, Diss, Norfolk, U.K.), and signals were recorded at a sampling rate of 1000 Hz. Subsequently, the data was replayed and the PR, QRS and QT intervals were measured by manual positioning of on screen markers. At least four complexes were measured and averaged at each time point. The values for QT interval were corrected for heart rate to give QTc using Bazett's formula expressed in ms as recommended by Molnar et al. (1995). Anaesthesia was maintained as necessary, by administration of further doses of sodium pentobarbitone (1.5–3 mg kg−1 i.v.).
Experimental protocol
After a 10 min stabilisation period, initial recordings of blood pressure, heart rate and ECG were obtained and then the lowest dose of mefloquine (0.3 mg kg−1) was administered. Subsequent doses of 1, 3, 10, and 30 mg kg−1 mefloquine were given at 25 min intervals. To prevent the mefloquine from precipitating out in the cannula, heparinized glucose solution (5% w w−1 D-glucose containing 10 U ml−1 heparin), was used to flush the drug through into the circulation. Mefloquine was administered in a volume of 1 ml kg−1. Data were recorded continuously, and subsequently retrieved from the computer at 5, 10, 15 and 20 min after each dose of drug.
In vitro cardiac muscle preparations
Male Dunkin-Hartley guinea-pigs (380–530 g) were killed by a blow to the head followed by exsanguination. The thorax was opened and the heart removed rapidly and placed in a Krebs solution of the following composition (mM): NaCl 119, KCl 3.8, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, D-glucose 10, CaCl2 1.9. The left atrium and at least one right ventricular strip and one left papillary muscle were dissected out rapidly and a thread attached to one end of the tissue. The opposite end of each preparation was impaled on one pole of a bipolar platinum electrode, which was then placed in Krebs solution in a 30 ml organ bath maintained at 37°C and gassed with 95% O2/5% CO2. Preparations were suspended under a resting tension of 10 mN and paced at 1 Hz with square wave pulses of 5 ms duration at twice threshold voltage, using Grass S48 or S88 stimulators. Developed tension was measured using Lectromed UF1 isometric force transducers (sensitivity range, up to 570 mN) connected to Lectromed 5240 pre-amplifiers and a Lectromed MT6 recorder. Preparations were allowed to equilibrate for 1 h. During this time the preparations were washed three to four times and the resting tension reset if necessary. At the end of the equilibration period any preparations which showed spontaneous activity or required more than 10 V stimulation voltage were discarded.
The effective refractory period was measured using a modification of the extra stimuli method of Scholtysik (1980). At 30 s intervals a series of five extra stimuli were applied. These extra stimuli were delivered at the same pulse width, voltage and frequency as the 1 Hz pacing stimulus, but at a known delay after each normal pacing stimulus. The delay between normal and extra stimuli was increased gradually until an increase in developed tension was noted in response to the extra stimuli. The lowest delay at which this occurred was assumed to be the effective refractory period. Care was taken to check the threshold voltage at regular intervals, and to adjust the stimulation voltage if necessary, to ensure that all measurements of effective refractory period were made at twice threshold stimulation voltage.
Experimental protocol
Three measurements for the control value of effective refractory period were obtained, after which each preparation was washed. Ten minutes later the effective refractory period was checked again and then the lowest concentration of mefloquine (3 μM) was added to the organ bath. The effective refractory period was measured at 5, 10, 20, 30, 40 and 50 min after addition of mefloquine. After the last effective refractory period measurement the preparation was washed and the effective refractory period checked 10 min later, immediately prior to the addition of the next concentration of mefloquine. This sequence was repeated in the presence of 10, 30 and 100 μM mefloquine. Resting and developed tensions were measured at all of the above time points. The effects of equivalent volumes of vehicle were tested in separate tissues taken from different hearts, using exactly the same protocol.
Experiments in single myocytes
Cell isolation
Single ventricular myocytes were isolated from guinea-pig hearts using a collagenase and protease protocol as described previously (Eisner et al., 1989). Guinea-pigs (either sex) of about 200 g were killed by stunning and cervical dislocation. The thoracic cavity was opened and the heart excised rapidly and perfused retrogradely through the aorta with a nominally Ca2+ free solution for 6 min at 37°C. The Ca2+ free solution contained (mM): NaCl, 134, D-glucose 11, HEPES 10, KCl 4, MgSO4 1.2, Na2HPO4 1.2, pH 7.2 with NaOH. Collagenase (Sigma Type IV) and Protease (Sigma Type XIV) were added to a final concentration of 14.9 and 0.16 units ml−1 respectively and perfused for a further 9 min until the heart was flaccid. The perfusate was then changed to a taurine containing solution for 10 min. The taurine solution contained (mM): NaCl 108, taurine 50, D-glucose 11, HEPES 10, KCl 4, MgSO4 1.2, Na2HPO4 1.2, pH 7.2 with NaOH. The left ventricle was then dissected from the heart and cut into small pieces, suspended in the taurine containing solution and gently triturated to release single cells. The isolated cells were stored at room temperature in the taurine containing solution until use.
Measurement of intracellular Ca2+ ([Ca2+]i.)
Cells were loaded with the cell permeant acetoxymethyl ester form of the [Ca2+]i sensitive fluorescent indicator Indo-1 (2.5 μM for 5 min, followed by at least 30 min deesterification) (Molecular Probes. OR, U.S.A.) and placed in the superfusion chamber on the stage of an inverted microscope adapted for epifluorescence (Nikon Diaphot 300, Nikon, Japan). Indo-1 fluorescence was excited at 340 nm and measured at 400 and 500 nm (O'Neill et al., 1990).
Electrophysiological measurements
These were achieved by using the perforated patch technique (Horn & Marty, 1988) with amphotericin-B. Low resistance pipettes (1–3 MΩ) were filled with the following solution: (in mM) CsCH3O3S 125, CsCl 20, NaCl 12, HEPES 10, MgCl2 5, titrated to pH 7.2 with CsOH and a final amphotericin concentration of 240 μg.ml−1. An Axoclamp 2A (Axon Instruments, Foster City CA, U.S.A.) was used. Action potentials were elicited by injection of depolarizing current pulses (2 nA of 4 ms duration). In voltage clamp experiments the switch clamp was used at a chopping rate of 2–4 kHz. Cells were bathed in a control solution of the following composition (in mM): NaCl 135, KCl 4, HEPES 10, D-glucose 11, CaCl2 1.8, MgCl2 1, titrated to pH 7.4 with NaOH.
All experiments were carried out at 23°C. Cells were paced at 0.33 Hz and the action potentials and fluorescence signals were digitized at 2 kHz and stored on a 586 computer using Axoscope 1.1 software (Axon Instruments Inc.) for later analysis.
Drugs/reagents
(±)-Mefloquine hydrochloride, a gift from F. Hoffman-La Roche (Basel, Switzerland), was dissolved in propylene glycol and diluted such that the final concentration of propylene glycol was always⩽1% for in vitro experiments. For in vivo experiments mefloquine was dissolved in dimethylacetamide (40%) / propylene glycol (60%) v v−1 to give a stock solution of 50 mg ml−1 which was diluted with 5% w v−1 D-glucose solution to give appropriate concentrations for the i.v. administration of each dose of mefloquine. All salts for Krebs solutions were of AnalaR grade or higher and obtained from BDH (Poole, U.K.), or Fisons (Loughborough, U.K.). DMSO was purchased from Fisons (Loughborough, U.K.) Collagenase, protease, Tween 80, N,N-dimethylacetamide and propylene glycol were obtained from Sigma (Poole, U.K.). Heparin sodium (mucous) injection, was obtained from CP Pharmaceuticals (Wrexham, U.K.).
Statistics
Where appropriate, values are expressed as the mean±s.e.mean of n experiments. Shapiro-Wilk tests revealed that some data might not be distributed normally. Comparisons within group, between two values were therefore made using two-tailed Wilcoxon's signed ranks tests whereas Friedman tests were used for within group comparisons of three or more values. Paired t-tests were used for comparisons between two normally distributed values. A probability of P<0.05 was considered to be significant.
Results
Effects of mefloquine in vivo
Administration of doses of mefloquine up to 3 mg kg−1, caused transient dose-dependent reductions in arterial blood pressure which returned to pre-injection values within 1 min. These transient depressor effects were accompanied by a gradual rise in systolic pressure, which reached statistical significance 20 min after injection of 3 mg kg−1 (Figure 1). The pulse pressure also increased significantly from a baseline value of 16±1 to 19±1 mmHg, 20 min after administration of 1 and 3 mg kg−1 mefloquine. Higher doses of mefloquine caused more pronounced biphasic decreases in blood pressure. Within the first minute after administration of 10 mg kg−1 mefloquine, blood pressure decreased by 50–60%, then recovered within the next minute, sometimes to values in excess of those recorded before drug administration. This initial response was followed by a secondary decline in pressure which varied in magnitude among individuals. Two out of the six guinea-pigs died as a consequence of this secondary decline in pressure after 10 mg kg−1 mefloquine. The remaining animals died after administration of 30 mg kg−1 mefloquine, three of them within one minute, and the fourth within 6 min. Heart rate tended to decrease after administration of 10 mg kg−1 mefloquine (Figure 1), partly as a consequence of atrioventricular (AV) block in some animals, but none of the apparent changes in heart rate reached statistical significance.
Figure 1.
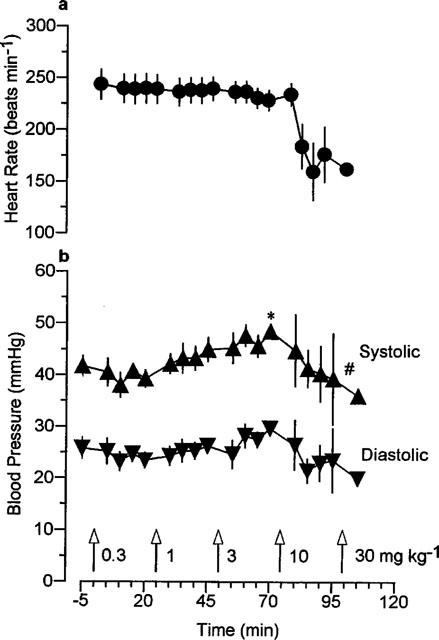
The effects of increasing doses of mefloquine on (a) heart rate and (b) systolic and diastolic arterial blood pressure in anaesthetized guinea-pigs. Values are mean with vertical bars indicating s.e.mean, n=6. *P<0.01 compared with time=−5 min, #P<0.01 compared with time=70 min, Friedman test.
ECG intervals were measured from normal complexes at intervals throughout the experiments. There were no significant alterations in PR, QRS or QTc intervals in response to mefloquine but there was a marked increase in variation, particularly of QTc intervals after 10 mg kg−1 mefloquine (Figure 2). This variation coincided with the apparent reductions in heart rate mentioned above (see Figure 1). Although the ECG intervals were not altered significantly some changes in ECG morphology were noted, particularly an enlargement of T waves after injection of the higher doses of mefloquine. The lack of effect of the vehicle on haemodynamics and the ECG has been reported previously (Batey et al., 1997).
Figure 2.
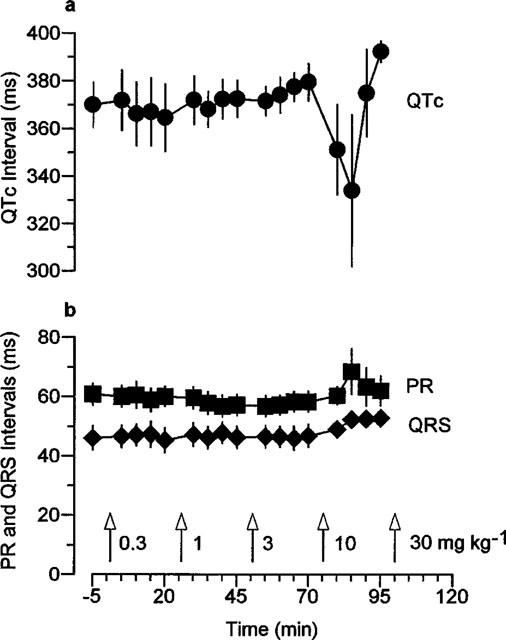
The effects of increasing doses of mefloquine on (a) the QTc interval and (b) the PR and QRS intervals in anaesthetized guinea-pigs. Values are mean with vertical bars indicating s.e.mean, n=6.
Effects of mefloquine on the effective refractory period and contractile function of isolated cardiac muscle preparations
Mefloquine did not alter the effective refractory period of left atrial preparations significantly (Figure 3). In left papillary muscles, the effective refractory period tended to decline with addition of increasing concentrations of mefloquine, but the apparent reduction in effective refractory period with 100 μM mefloquine just failed to reach statistical significance (Figure 4). The vehicle did not alter effective refractory period in either atrial or ventricular preparations (Figures 3 and 4).
Figure 3.
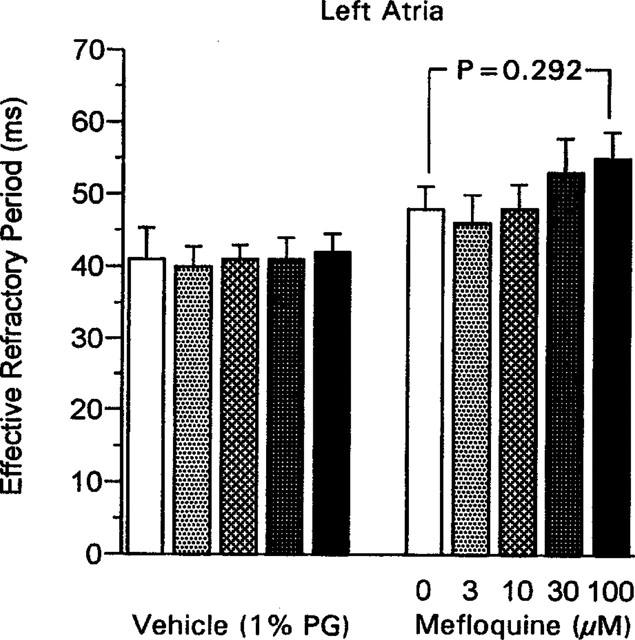
The effective refractory period in guinea-pig isolated left atria measured 50 min after addition of each concentration of mefloquine, or equivalent volumes of propylene glycol (PG; 1% final concentration) in the vehicle group. Values are mean with vertical bars indicating s.e.mean; vehicle group n=5, mefloquine group n=6. Statistical comparison, Wilcoxon test.
Figure 4.

The effective refractory period in guinea-pig isolated left papillary muscles measured 50 min after addition of each concentration of mefloquine, or equivalent volumes of propylene glycol (PG; 1% final concentration) in the vehicle group. Values are mean with vertical bars indicating s.e.mean; vehicle group n=4, mefloquine group n=7. Statistical comparison, Wilcoxon test.
The effects of mefloquine on resting and peak developed tensions in left atria and left papillary muscles measured throughout these experiments are shown in Figures 5 and 6 respectively. As time progressed, in the presence of the two higher concentrations of mefloquine, the resting tension of the left atria increased. This is clearly an effect of the drug since it was not seen in a separate group of tissues which received vehicle (Figure 5b). Resting tension also increased with time in the presence of 30 and 100 μM mefloquine in the left papillary muscles but not in the vehicle-treated tissues (Figure 6). The effects of mefloquine on the developed tension varied between the atrial and ventricular preparations. In the left atria only a negative inotropic effect was seen with 100 μM mefloquine (Figure 7), whereas in the ventricular preparations both negative and positive inotropic effects were seen. Initially, in the left papillary muscles a negative inotropic effect was seen with 30 μM mefloquine, then on addition of 100 μM mefloquine there was a marked increase in the force of contraction which peaked at 5 min then declined (Figure 7). The results obtained in right ventricular strips for the effects of mefloquine on resting and developed tensions were qualitatively and quantitatively similar to those in left papillary muscles.
Figure 5.
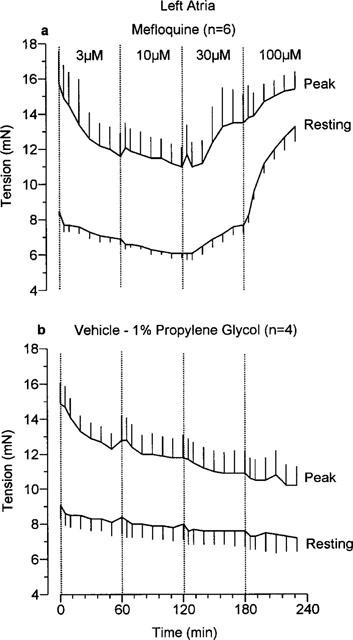
The effects of (a) mefloquine and (b) vehicle (propylene glycol, 1% final concentration) on resting tension and peak tension (resting+developed tension) in guinea-pig isolated left atria. Values are mean with vertical bars indicating s.e.mean.
Figure 6.
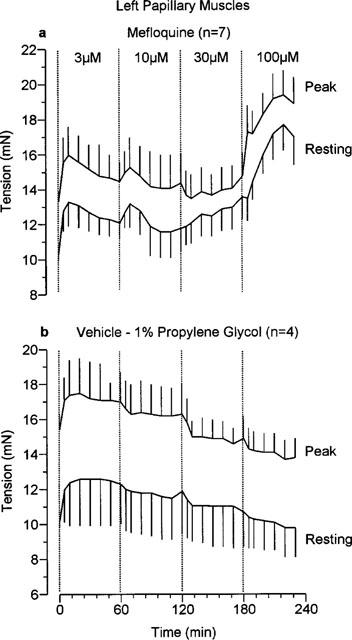
The effects of (a) mefloquine and (b) vehicle (propylene glycol, 1% final concentration) on resting tension and peak tension (resting+developed tension) in guinea-pig isolated left papillary muscles. Values are mean with vertical bars indicating s.e.mean.
Figure 7.
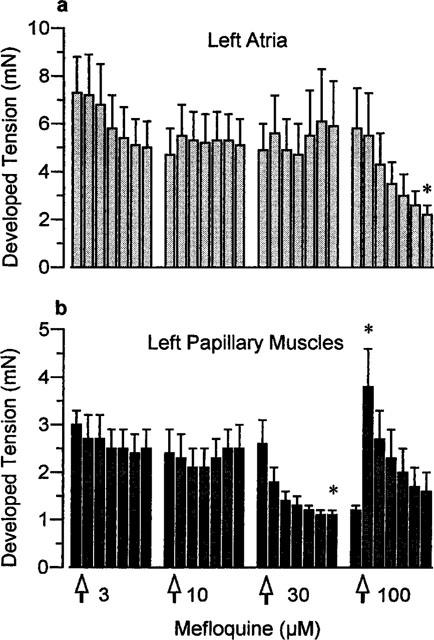
The effects of mefloquine on the developed tension (peak-resting tension) in (a) left atria (n=6) and (b) left papillary muscles (n=7). Values are mean with vertical bars indicating s.e.mean. *P<0.05 compared with value immediately before addition of that concentration of mefloquine, Wilcoxon test.
Effects of mefloquine on single ventricular myocytes
Superfusion of single ventricular myocytes with mefloquine (10 μM) shortened the action potential duration (mainly during the plateau phase) and reduced the amplitude of the systolic Ca2+ transient (Figure 8). Superfusion with the vehicle alone had no effect on either the action potential duration (Figure 8b) or the systolic Ca2+ transient (not shown). Higher concentrations of mefloquine (25 μM) caused irreversible changes in [Ca2+]i and the cells became completely inexcitable. Figure 8b summarizes data from eight cells. It is clear that there is a significant reduction in the action potential duration in the presence of mefloquine. The effects of mefloquine on both the action potential duration and the amplitude of the systolic Ca2+ transient could arise from an effect of the drug on the L-type Ca2+ channel. Therefore, in subsequent experiments we examined the effects of mefloquine in voltage-clamped myocytes. Figure 9 shows a typical example of such experiments. Again mefloquine produced inhibitory effects. There was a marked reduction in both the size of the Ca2+ current and the systolic Ca2+ transient. As well as decreasing the magnitude of the systolic Ca2+ transient, mefloquine decreased resting [Ca2+]i in six out of eight cells. One possible explanation of this is that, at least at the holding potential of −40 mV used in these experiments, the L-type Ca2+ channel has a finite opening probability and thereby contributes to the resting Ca2+ influx into the cell. Inhibition of this current by mefloquine would therefore be expected to decrease resting Ca2+.
Figure 8.
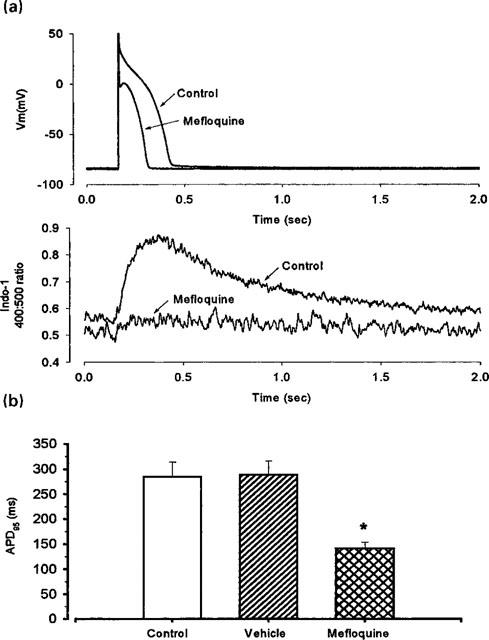
Effects of 10 μM mefloquine on the action potential and on the Ca2+ transient. (a) Typical records of action potential (upper traces) and Ca2+ transient (lower traces) during control conditions and after superfusion with 10 μM mefloquine. (b) Mean data from eight cells (from left to right as specified for each bar) showing APD95 under control conditions, during superfusion with vehicle (0.1 % propylene glycol) and with 10 μM mefloquine. Vertical bars indicate s.e.mean. *P<0.05, compared with control, paired t-test.
Figure 9.

Mefloquine inhibits the L-type Ca current. The data show current records (top) and changes in [Ca2+]i (as monitored by Indo-1) in a single myocyte before and after perfusing with 10 μM mefloquine. The membrane potential was held at −40 mv and 100 ms duration depolarizing pulses applied to 0 mV at 0.33 Hz. Under these conditions superfusion of the cell with 0.1% propylene glycol did not produce any alterations in the L-type Ca2+ current (not shown).
Discussion
These results are the first report of the effects of the antimalarial drug mefloquine on cardiac electrical activity and contractility in vitro and in vivo in experimental animals. They demonstrate clearly that mefloquine can reduce cardiac contractility and that this action can be explained by blockade of L-type Ca2+ current. However, even at doses that caused profound hypotension, mefloquine did not alter ECG intervals significantly in vivo.
ECG intervals
Previous clinical studies (Laothavorn et al., 1992) suggested that mefloquine has little if any effect on ECG intervals, but other ECG abnormalities have been reported. Mefloquine-induced sinus bradycardia occurred often in patients with malaria (Laothavorn et al., 1992) and a case of aberrant atrioventricular conduction has been reported (Richter et al., 1997). In the present in vivo experiments, mefloquine did not alter heart rate significantly, but AV block did occur in some animals after administration of the higher doses. The lack of effects of mefloquine on ECG intervals reported by Laothavorn et al., (1992) is contradicted by a subsequent claim that mefloquine prolongs the QTc interval in man (Davies et al., 1996). Although Davies et al. (1996) reported a 10 ms prolongation of the QTc interval which was statistically significant, the QTc interval was also prolonged by 10 ms in the placebo group but this did not reach statistical significance because of greater variance in the baseline values. It is unlikely that this apparent effect of mefloquine reported by Davies et al. (1996) is of biological significance, and this view is reinforced by the present animal studies where a wide range of doses of mefloquine, including those with other adverse cardiac effects, did not alter ECG intervals.
Cardiac contractility
In all the in vivo experiments, administration of the higher doses of mefloquine was fatal because of marked hypotension, presumably resulting from cardiac contractile failure. Similar effects have also been observed in anaesthetized rabbits (Coker et al., 1998). With the lower doses of mefloquine, and before the onset of hypotension with the higher doses, an increase in pulse pressure was seen in vivo. This observation agrees with the findings in isolated ventricular preparations where a non-sustained positive inotropic effect occurred. In vitro, however, the positive inotropic effect was not sustained and in both atrial and ventricular preparations prolonged incubation with mefloquine revealed a negative inotropic action. There are no other reports of the effects of mefloquine on isolated cardiac muscle, but similar effects have been observed with other structurally related antimalarial drugs such as quinine and chloroquine (Marshall & Ojewole, 1978). With chloroquine and quinine the initial positive inotropic effects were prevented by reserpine pretreatment, cocaine or propranolol and were concluded to be due to enhanced noradrenaline release. Further studies would be necessary to determine whether the positive inotropic effect of mefloquine occurred via similar mechanisms.
Sustained reductions in cardiac contractility occurred with chloroquine in guinea-pig atria (Marshall & Ojewole, 1978; Tona et al., 1990) and in perfused turtle (Ikhinmwin et al., 1981), rabbit (Essien & Ette, 1986) and guinea-pig (Tona et al., 1990) hearts. Tona et al., (1990) also reported that ventricular muscle was much more sensitive to the negative inotropic action of chloroquine than atrial muscle. A similar effect was seen here with mefloquine, where higher concentrations were required to reduce contractions in atria than in papillary muscles.
Studies on possible mechanisms underlying the negative inotropic activity of chloroquine and other antimalarials also suggest similarities with the action of mefloquine investigated here. Raising external Ca2+ concentrations reversed the cardiodepressant effects of chloroquine and quinine (Ikhinmwin et al., 1981; Tona et al., 1990). Decreased mitochondrial Ca2+ binding and accumulation was suggested to be related to the negative inotropic effect of chloroquine (Essien & Ette, 1986). Marshall & Ojewole (1978) also concluded that the cardiac effects of the antimalarial drugs could be explained by calcium antagonism. The present electrophysiological studies in single ventricular myocytes demonstrate clearly that 10 μM mefloquine shortened action potential duration, reduced the intracellular Ca2+ transient markedly and reduced L-type inward Ca2+ current. These actions can all be explained by blockade of L-type Ca2+ channels. Mefloquine has been reported to block L-type Ca2+ current in paramecia (Nori & Barry, 1997). In brain microsomes, mefloquine inhibited IP3-induced Ca2+ release with an IC50 of 42 μM (Lee & Go, 1996), whereas in skeletal muscle microsomes the predominant effect of mefloquine was inhibition of uptake of Ca2+ into microsomes, again with a similar IC50 of 43 μM (Go et al., 1995). Although the possibility of actions of mefloquine on cardiac intracellular calcium handling, e.g. at the level of the sarcoplasmic reticulum, or mitochondria, or on Na+/Ca2+ exchange cannot be excluded, simple blockade of L-type Ca2+ channels is sufficient to explain the negative inotropic action of mefloquine.
Relevance to clinical use
Substantial effects on action potential duration, [Ca2+]i and Ca2+ current were observed with 10 μM mefloquine in single ventricular myocytes. Higher concentrations of 30 and 100 μM were required in isolated ventricular and atrial preparations before any significant effects were seen. In the present in vivo experiments, blood samples were taken and attempts made to measure mefloquine concentrations. However, due to small sample volumes, reliable results could not be obtained. Maximum plasma concentrations of mefloquine in healthy volunteers ranged from 0.6 to 0.8 μM after a single 250 mg oral dose and 1.4 to 4.5 μM after 750 mg p.o., whereas peak plasma concentrations of 5.4 μM were seen in patients with malaria (see Karbwang & White, 1990 for review). In more recent studies, the plasma mefloquine concentrations measured after five consecutive weekly oral doses of 250 mg ranged from 0.94 to 5.37 μM (mean±s.d. 2.35±0.94 μM; Davies et al., 1996). Thus with concentrations only two to five times higher than those found routinely in clinical studies, mefloquine had substantial effects on single ventricular myocytes.
Although higher concentrations of mefloquine were required for effects in isolated tissue preparations (particularly papillary muscles) compared to single myocytes, this observation should be interpreted with caution. The atrial and ventricular preparations are not perfused, and the access of drugs to their site of action cannot be ensured. It is possible that it is only cells on the surfaces of the preparations that were exposed to the concentrations known to be added to the organ bath. Indeed, the report of much greater sensitivity to chloroquine of ventricular tissue compared to atrial tissue actually compared the effects of chloroquine on isolated atria in organ baths to those on Langendorff perfused hearts (Tona et al., 1990). A perfused preparation such as the Langendorff heart, or a superfused preparation such as single myocytes, receives drugs in a manner much closer to the in vivo situation.
Mefloquine is very insoluble in water and a parenteral formulation is not available for clinical use, thus absolute bioavailability cannot be determined accurately (Karbwang & White, 1990). The lipophilic nature of mefloquine is associated with extensive distribution into tissues and a very long elimination half-life. These factors add to the difficulty in trying to extrapolate from in vitro studies and in vivo studies in anaesthetized guinea-pigs with mefloquine given i.v., to clinical studies with orally administered mefloquine. The only adverse cardiovascular effects of mefloquine documented in clinical studies are effects on heart rate or rhythm, but it has been stated that mefloquine worsens the orthostatic hypotension associated with malaria (Karbwang & White, 1990, citing unpublished observations). The present results support this latter observation and provide a possible explanation.
Conclusions
These experiments indicate that mefloquine can have adverse effects on the cardiovascular system. Cardiac contractility is affected rather than the electrical activity of the heart and the effects of mefloquine can be explained by blockade of L-type Ca2+ channels. Although the effects on cardiac contractility are unlikely to have serious consequences at normal therapeutic concentrations of mefloquine, they could be influential in situations of overdose, interactions with other drugs, or conditions where cardiac contractility is already compromised.
Acknowledgments
We thank F. Hoffman-La Roche Ltd., Basel, Switzerland for providing mefloquine.
Abbreviations
- APD
action potential duration
- AV
atrioventricular
- ECG
electrocardiogram
References
- BATEY A.J., LIGHTBOWN I.D., LAMBERT J.P., EDWARDS G., COKER S.J. Comparison of the acute cardiotoxicity of the antimalarial drug halofantrine in vitro and in vivo in anaesthetized guinea pigs. Br. J. Pharmacol. 1997;122:563–569. doi: 10.1038/sj.bjp.0701402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEN-DAVID J., ZIPES D.P. Torsade de pointes and proarrhythmia. Lancet. 1993;341:1578–1582. doi: 10.1016/0140-6736(93)90708-o. [DOI] [PubMed] [Google Scholar]
- COKER S.J., BATEY A.J. Mefloquine, an antimalarial drug: effects on the contractile function and the effective refractory period of guinea pig isolated cardiac muscle preparations. Br. J. Pharmacol. 1996;119:95P. [Google Scholar]
- COKER S.J., LIGHTBOWN I.D., HUGHES D.A., LAMBERT J.P., EDWARDS G. Mefloquine potentiates halofantrine-induced QTc prolongation by altering the distribution of halofantrine. Br. J. Pharmacol. 1998;123:88P. doi: 10.1038/sj.bjp.0703823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES T.M.E., DEMBO L.G., KAYE-EDDIE S.A., HEWITT B.J., HISLOP R.G., BATTY K.T. Neurological, cardiovascular and metabolic effects of mefloquine in healthy volunteers: a double blind, placebo-controlled trial. Br. J. Clin. Pharmacol. 1996;42:415–421. doi: 10.1046/j.1365-2125.1996.04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAZ S.J., EISNER D.A., COKER S.J. Shortening of action potential duration by the antimalarial agent mefloquine in isolated guinea pig ventricular myocytes. J. Physiol. 1998;509P:149P–150P. [Google Scholar]
- EISNER D.A., NICHOLS C.G., O'NEILL S.C., SMITH G.L., VALDEOLMILLOS M. The effects of metabolic inhibition on intracellular calcium and pH in isolated rat ventricular cells. J. Physiol. 1989;411:393–418. doi: 10.1113/jphysiol.1989.sp017580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESSIEN E.E., ETTE E.I. Effects of chloroquine and didesethylchloroquine on rabbit myocardium and mitochondria. J. Pharm. Pharmacol. 1986;38:543–546. doi: 10.1111/j.2042-7158.1986.tb04635.x. [DOI] [PubMed] [Google Scholar]
- GO M.L., LEE H.S., PALADE P. Effects of mefloquine on Ca2+ uptake by crude microsomes of rabbit skeletal muscle. Arch. Int. Pharmacodyn. 1995;329:255–271. [PubMed] [Google Scholar]
- HORN R., MARTY A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES D.A., COKER S.J. Cardiovascular response to acute chloroquine poisoning. J. Mol. Cell. Cardiol. 1994;26:CCLIX. [Google Scholar]
- IKHINMWIN M.K., SOFOLA O.A., ELEBUTE O. The effects of calcium ions on the depression of cardiac contractility by chloroquine and quinine. Eur. J. Pharmacol. 1981;69:507–510. doi: 10.1016/0014-2999(81)90458-1. [DOI] [PubMed] [Google Scholar]
- JAEGER A., SAUDER P., KOPFERSCHMITT J., FLESCH F. Clinical features and management of poisoning due to antimalarial drugs. Medical Toxicology. 1987;2:242–273. doi: 10.1007/BF03259868. [DOI] [PubMed] [Google Scholar]
- KARBWANG J., WHITE N.J. Clinical pharmacokinetics of mefloquine. Clin. Pharmacokinet. 1990;19:264–279. doi: 10.2165/00003088-199019040-00002. [DOI] [PubMed] [Google Scholar]
- LAOTHAVORN P., KARBWANG J., NA BANGCHANG K., BUNNAG D., HARINASUTA T. Effect of mefloquine on electrocardiographic changes in uncomplicated falciparum malaria patients. Southeast Asean J. Trop. Med. Public Health. 1992;23:51–54. [PubMed] [Google Scholar]
- LEE H.S., GO M.L. Effects of mefloquine on Ca2+ uptake and release by dog brain microsomes. Arch. int. Pharmacodyn. 1996;331:221–231. [PubMed] [Google Scholar]
- MARSHALL R.J., OJEWOLE J.A.O. Comparative effects of some antimalarial drugs on isolated cardiac muscle of the guinea pig. Toxicol. Appl. Pharmacol. 1978;46:759–768. doi: 10.1016/0041-008x(78)90320-4. [DOI] [PubMed] [Google Scholar]
- MOLNAR J., WIESS J.S., ROSENTHAL J.E. The missing second: what is the correct unit for the Bazett corrected QT interval. Am. J. Cardiol. 1995;75:537–538. doi: 10.1016/s0002-9149(99)80603-1. [DOI] [PubMed] [Google Scholar]
- NAPOLITANO C., PRIORI S.G., SCHWARTZ P.J. Torsades de pointes. Mechanisms and management. Drugs. 1994;47:51–65. doi: 10.2165/00003495-199447010-00004. [DOI] [PubMed] [Google Scholar]
- NORI V.S., BARRY S.R. Toxic effects of antimalarial drugs in Paramecium: role of calcium channels. J. Comp. Physiol. A. 1997;180:473–480. doi: 10.1007/s003590050064. [DOI] [PubMed] [Google Scholar]
- NOSTEN F., TER KUILE F.O., LUXEMBERGER C., WOODROW C., KYLE D.E., CHONGSUPHAJAISIDDHI T., WHITE N.J. Cardiac effects of antimalarial treatment with halofantrine. Lancet. 1993;341:1054–1056. doi: 10.1016/0140-6736(93)92412-m. [DOI] [PubMed] [Google Scholar]
- O'NEILL S.C., DONOSO P., EISNER D.A. The role of [Ca2+]i and [Ca2+]i-sensitization in the caffeine contracture of rat myocytes: measurement of [Ca2+]i and [caffeine]i. J. Physiol. 1990;425:55–70. doi: 10.1113/jphysiol.1990.sp018092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICHTER J., BURBACH G., HELLGREN U., DENGLER A., BIENZLE U. Aberrant atrioventricular conduction triggered by antimalarial prophylaxis with mefloquine. Lancet. 1997;349:101–102. doi: 10.1016/S0140-6736(05)60885-9. [DOI] [PubMed] [Google Scholar]
- RIOU B., BARRIOT P., RIMAILHO A., BAUD F.J. Treatment of severe chloroquine poisoning. New Engl. J. Med. 1988;318:1–6. doi: 10.1056/NEJM198801073180101. [DOI] [PubMed] [Google Scholar]
- SCHOLTYSIK G.Retardation of aconitine-induced ECG-alterations in rats as an indication of membrane-stabilizing drug effects The rat electrocardiogram in pharmacology and toxicology 1980Oxford, Pergamon Press Ltd; 257–264.eds: Budden R. Detweiler D.K. & Zbinden G [Google Scholar]
- TONA L., NG Y.-C., AKERA T., BRODY T.M. Depressant effects of chloroquine on the isolated guinea-pig heart. Eur. J. Pharmacol. 1990;178:293–301. doi: 10.1016/0014-2999(90)90108-i. [DOI] [PubMed] [Google Scholar]
- WINSTANLEY P. Mefloquine: the benefits outweigh the risks. Br. J. Clin. Pharmacol. 1996;42:411–413. doi: 10.1046/j.1365-2125.1996.04754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]