

Abstract
It was postulated that swelling dependent chloride channels are involved in the proton secretion of parietal cells. Since omeprazole, lansoprazole and its acid activated sulphenamide form AG2000 are structurally related to phenol derivatives known to block swelling dependent chloride channels, we set out to test, whether these substances – which are known to block the H,K-ATPase – could also lead to an inhibition of swelling-dependent chloride channels. Swelling-dependent chloride channels – characterized in many different cell types – show highly conserved biophysical and pharmacological features, therefore we investigated the effect of omeprazole, lansoprazole and its acid activated sulphenamide form AG2000 on swelling-dependent chloride channels elicited in fibroblasts, after the reduction of the extracellular osmolarity.
Omeprazole, lansoprazole and its acid activated sulphenamide form AG2000 are able to block swelling-dependent chloride channels (IClswell).
Lansoprazole and its protonated metabolite AG2000 act on at least two different sites of the IClswell protein: on an extracellular site which seems to be in a functional proximity to the nucleotide binding site, and on an intracellular site which allows the formation of disulfide-bridges.
The inhibition of the proton pump and the simultaneous blocking of chloride channels by omeprazole, lansoprazole and its acid activated sulphenamide form AG2000, as described here could be an effective mode to restrict proton secretion in parietal cells.
Keywords: Chloride channels; lansoprazole; AG2000; omeprazole; cell volume regulation; gastric H,K-ATPase; IClswell
Introduction
The secretion of hydrochloric acid by gastric parietal cells is effected by the concerted action of several ion-conducting mechanisms. Beside the H,K-ATPase, an electroneutral H+/K+ antiporter (Hersey & Sachs, 1995; Sachs et al., 1995; Shull & Lingrel, 1986) which can be blocked by benzimidazoles (Barradell et al., 1992; McTavish et al., 1991; Ritter et al., 1998), a chloride extrusion mechanism on the apical side of parietal cells seems to be needed for the effective hydrochloric acid secretion (Asano et al., 1987; Cuppoletti & Sachs, 1984; Demarest et al., 1989). Cuppoletti and coworkers (Malinowska et al., 1995) proposed that a protein they had cloned, showing a 93% homology to ClC-2, is a molecular candidate for the chloride channel involved in gastric acid secretion. The ClC-2 chloride channel was originally cloned by Jentsch and coworkers (Gründer et al., 1992) and was shown to be associated with regulatory volume decrease (RVD). Therefore we set out to test whether volume-dependent chloride currents could be directly modulated by benzimidazoles.
Increase of the cytoplasmatic volume leads in a variety of different cells to the activation of volume-dependent chloride currents, which are outwardly rectifying, inactivating at holding potentials more positive than +40 mV and sensitive to chloride channel blockers like anthracene-9-COOH or nucleotides (Okada, 1997). We have extensively characterized these volume-dependent chloride currents in NIH3T3 fibroblasts in which they are paramount for effecting RVD (Gschwentner et al., 1996; 1995a,1995b).
Here we show that lansoprazole and its active metabolite AG2000, as well as omeprazole are able to block the swelling dependent chloride channel (IClswell) in NIH3T3 fibroblasts at micromolar concentrations.
Methods
Electrophysiology and cell culture
Fibroblasts (NIH3T3) were grown on glass cover slips in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal calf serum at 37°C, 5% CO2/95% air. Experiments were performed 24–48 h after splitting and re-culturing the cells. For whole-cell voltage-clamp experiments the method described by Marty & Neher (1983) was chosen to measure swelling-induced chloride currents (Gschwentner et al., 1996). These experiments were carried out at room temperature. Bath and pipette solutions were chosen to enable chloride current measurements, which were only accepted with series resistances of 2.5–12 MΩ. The isotonic extracellular solution used was composed of (in mM): NaCl 125, CaCl2 2.5, MgCl2 2.5, N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) 10, mannitol 50, pH 7.2 (adjusted with NaOH; measured osmolality 304±0.5 mmol kg−1). To reduce extracellular osmolarity, mannitol was omitted. One to two minutes after confirming whole-cell configuration, hypotonic conditions were established and the activated chloride current determined. To define the relative selectivity of SCN−,I− and gluconate for the induced chloride current, we exchanged NaCl for the corresponding sodium salt in the extracellular solution after 2–3 min of hypotonic stimulation. The current measurements were made by calculating the mean current over the interval of 400 ms for voltage steps from 0 mV to +40 mV. Fast exchange of the bath solution was obtained using a perfusion system with a flow rate of 10 ml min−1 and a bath volume of 200 μl. The filling solution of the patch pipette was (in mM): CsCl 125, MgCl2 5, ethyleneglycol-bis (β-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) 11, raffinose 50, HEPES 10, pH 7.2 (adjusted with CsOH; measured osmolality 314±0.5 mmol kg−1). In order to determine the dose-response curve for the different drugs, each concentration was tested individually on different cells. The formula used for the dose-response curves (% block) is 100−(Idrug /Imax·100). The IC50 values were obtained using Graphpad Prism 2.0 software (U.S.A.). For data acquisition and analysis an Axopatch 200A (Axon Instruments, U.S.A.) amplifier was used, controlled by an Apple computer, needed to run the amplifier and the program for analysis (HEKA, Germany). All current measurements were filtered at 1 kHz (analogue 4-pole BESSEL).
Chemicals
All chemicals used are of pro analysi grade. Omeprazole (5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl] sulfinyl]-1H-benzimidazole) was obtained from Astra (Austria). Takeda Pharmaceutical (Japan) kindly provided lansoprazole (2-[[[3-methyl-4 -(2,2,2 -trifluoroethoxy) -2-pyridyl]methyl]sulfinyl]-1H-benzimidazole) and AG2000 (4-methyl-3-(2,2,2-trifluoroethoxy)-5H-pyrido[1′,2′ : 4, 5][1, 2, 4]thiadiazino [2, 3-a]benzimidazole-13-ium tetrafluoroborate).
Statistical analysis
Where applicable, data are expressed as arithmetic mean±standard error of the mean (s.e.mean). Statistical analysis was made by t-test, where appropriate. Significant difference was assumed at P<0.05, indicated by one asterisk for 0.01<P<0.05 and two asterisks for P⩽0.01.
Results
Lansoprazole and its active metabolite AG2000 as well as omeprazole block swelling-dependent chloride channels
Under isotonic conditions in NIH3T3 fibroblasts a whole-cell chloride current density of 1.98±0.16 pA/pF (n=145) can be measured, using a voltage-step protocol from 0 mV (holding potential) to +40 mV. After a time interval of 180 s the reduction of the extracellular osmolarity by the omission of 50 mM mannitol leads to a significant increase in the chloride current density to 34.00±1.74 pA/pF (n=145) when using the same voltage-step procedure. In the presence of extracellular sodium chloride this swelling-induced current reverses at +1.15±0.3 mV (n=15) i.e. close to the calculated equilibrium potential for chloride of 0 mV (Figure 1).
Figure 1.
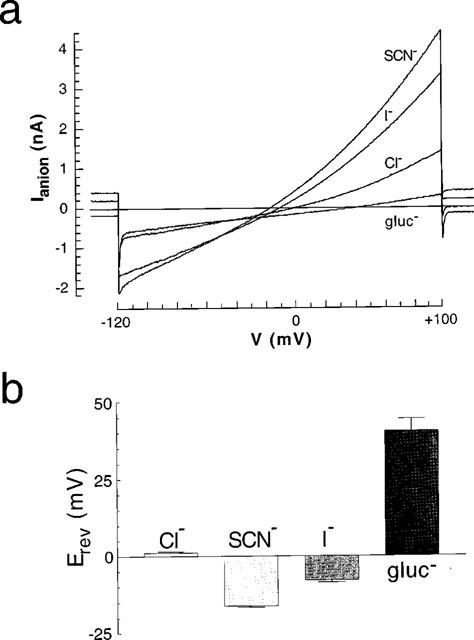
Relative permeabilities of swelling-dependent chloride currents for SCN−, I−, Cl− and gluconate (gluc−) anions. (a) The cells were held at 0 mV, and voltage ramps were made between −120 and +100 mV for 500 ms. (b) The summary of the reversal potentials (Erev) in the presence of the different anions is given.
Substituting chloride with SCN−,I− and gluconate shifts the reversal potential to the expected values of −16.18±0.4 mV (n=5), −7.89±0.8 mV (n=3) and +40.42±4.0 mV (n=7) respectively (Figure 1), indicating that the elicited current is mainly an anion selective current (Wöll et al., 1996). This swelling-induced chloride current (IClswell) is outwardly rectifying (Figures 1 and 2) and shows a voltage-dependent inactivation at potentials more positive than +40 mV, characteristics that are typical of IClswell in numerous cells. As shown in Figures 2 and 3, the ‘pro-drug' lansoprazole as well as its active metabolite AG2000 are able to block IClswell if added to the extracellular fluid. Original tracings are shown for lansoprazole in Figure 2a and for AG2000 in Figure 2c at increasing concentrations. In Figure 2b and d, the current density/voltage relation of IClswell is illustrated before and after the addition of the maximal concentration tested for each drug. After a time interval of 180 s, the drugs were omitted and the recovery of the current measured. The current elicited following the omission of lansoprazole (0.5 mM) was partially reversible (62.9±20.7%, n=7, of the current prior to the addition of the drug), whereas the current after the omission of AG2000 (100 μM) was further reduced and therefore not reversible (16.8±9.5%, s.d. n=2, of the current prior to the addition of the drug; Figure 2b and d). This finding is in accordance with earlier reports describing that AG2000 also produces an irreversible block on the H,K-ATPase (Hersey & Sachs, 1995; Sachs et al., 1995). At a concentration of 0.25 mM, the effect of lansoprazole proved to be completely reversible (101.7±13.5%, n=8, of the current prior to the addition of the drug).
Figure 2.
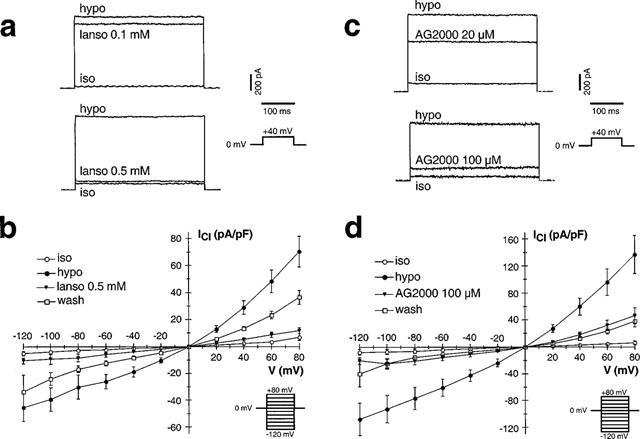
Lansoprazole and AG2000 are able to block IClswell in a dose-dependent manner. (a) At a concentration of 0.1 mM, lansoprazole led to a slight inhibition of IClswell in four out of eight experiments. In the presence of 0.5 mM, IClswell is blocked by 78.8±3.4% (n=7). Each concentration was tested individually, and therefore the original readings of two different cells are shown. The holding potential for both experiments was 0 mV. Repetitive voltage steps were made up to a potential of +40 mV for 400 ms. (b) The current-density/voltage relation is given for the current under isotonic (iso) conditions, after reduction of the extracellular osmolarity (hypo), under hypotonic conditions in the presence of 0.5 mM lansoprazole (lanso 0.5 mM) and 180 s after washing out the drug (wash). (c) The same experimental procedure as described in (a) is used for AG2000. At a concentration of 20 μM, AG2000 led to a moderate block of IClswell in five out of six experiments. At a concentration of 100 μM, AG2000 blocks IClswell by 70.4±9.5% (n=5). (d) According to (b), the current-density/voltage relation is given in the absence and presence of 100 μM AG2000. Note that the effect of AG2000 (100 μM) is not reversible, since 180 s after washing out the drug a further reduction of the current can be measured.
Figure 3.
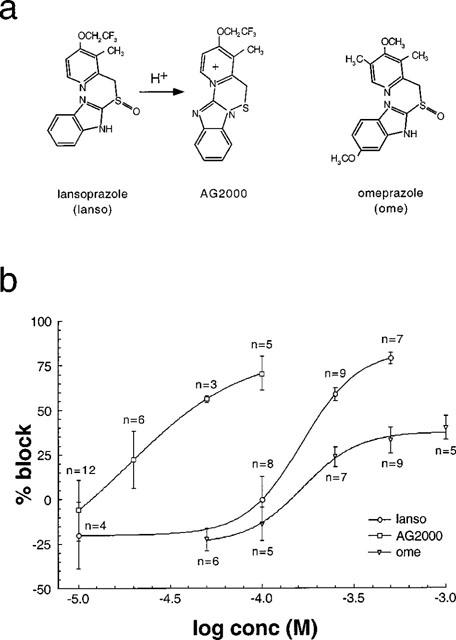
Lansoprazole and AG2000 as well as omeprazole block IClswell at different concentrations. (a) The molecular structures of omeprazole and lansoprazole are given. The substituted benzimidazole lansoprazole is rearranged after protonization to form a sulfenic acid that is further rearranged to a sulphenamide. The sulphenamide of lansoprazole is termed AG2000, and it interacts covalently with sulfhydryl groups of the membrane spanning H,K-ATPase. (b) The half-maximal concentration required to block IClswell (IC50) is ≈22 μM for AG2000 and ≈160 μM for lansoprazole (the holding potential is 0 mV and repetitive voltage steps to +40 mV are made). Omeprazole is significantly less active in blocking IClswell compared with lansoprazole.
The half-maximal concentration (IC50) that is required to block IClswell is ≈22 μM for AG2000 and ≈160 μM for lansoprazole (Figure 3). Additionally, we tested omeprazole, which proved to be significantly less active in blocking IClswell (Figure 3b) when compared with lansoprazole, to which it is structurally related (Figure 3a). At a concentration of 0.5 mM the blocking effected by lansoprazole was 78.8±3.4% (n=7), whereas the blocking effected by omeprazole at the same concentration was significantly lower, measuring only 32.9±7.1% (n=9).
If added to the extracellular solution, lansoprazole blocks IClswell faster than AG2000. As shown in Figure 4, the time constant for the block of IClswell was twice as fast for lansoprazole compared with its metabolite AG2000. The respective τ's are 57.9±8.6 s (n=7) for lansoprazole and 104.6±5.2 s (n=5) for AG2000. The addition of lansoprazole to the pipette solution at a concentration of 160 μM is not able to block IClswell, however, the addition of 22 μM AG2000 does lead to a significant reduction of IClswell (Figure 5).
Figure 4.
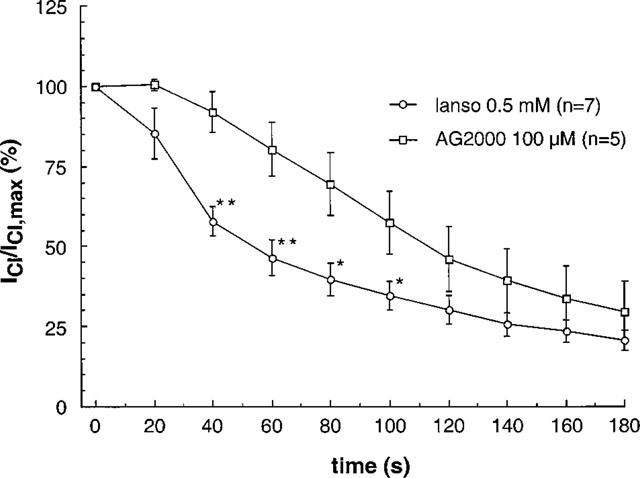
At the maximal concentration tested, lansoprazole and AG2000 block IClswell with different time constants. The block with lansoprazole is significantly faster (τ is 57.9±8.6 s; n=7) compared to AG2000 (τ is 104.6±5.2 s; n=5).
Figure 5.
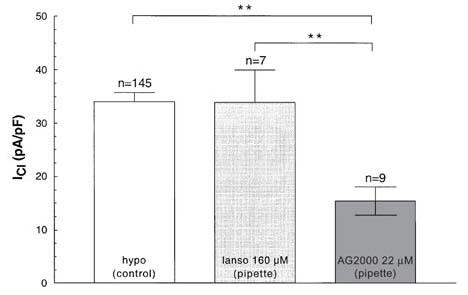
Intracellular (pipette) effect of lansoprazole (160 μM) and AG2000 (22 μM). The swelling-dependent chloride current (hypo (control)) can be blocked by intracellular AG2000, but not by lansoprazole (lanso).
Thymidine 5′-diphosphate (TDP) modifies the effect of lansoprazole, but has no effect on the blockage of IClswell by AG2000
We have shown that TDP is able to modify the effect of nucleoside analogues on IClswell (Gschwentner et al., 1995b). The inhibitory effect of AZT was abolished in the presence of 100 μM TDP (which per se is not able to reduce IClswell; Figure 6). We therefore concluded that TDP is competing with the binding site(s) for the nucleosides, thus leading to a competitive block of the IClswell inhibition. In order to test whether the putative extracellular binding site(s) for lansoprazole and/or AG2000 are similarly related to the TDP binding site(s), we tested the effect of both substances in the presence of 100 μM TDP. The effect of AG2000 (22 μM; IC50 calculated from Figure 3) can not be blocked by TDP (Figure 7b), whereas the effect of lansoprazole (160 μM; IC50 calculated from Figure 3) is annihilated when 100 μM TDP is present (Figure 7a). This compares with the effect produced by TDP in the presence of nucleoside analogues or phenol derivatives. Interestingly enough, following an increase of the lansoprazole concentration from 160 μM to 0.5 mM, the time constant τ for the block of IClswell is faster (33.3±5.2 s; n=7) compared to the time constant for the block of IClswell in the absence of TDP for the same drug (57.9±8.6 s; n=7).
Figure 6.
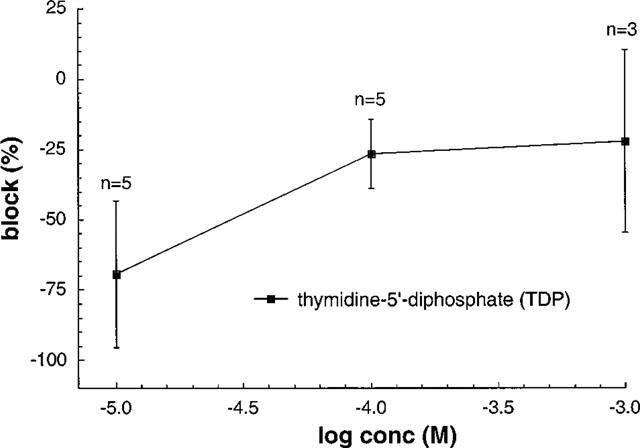
Dose-response curve of TDP on swelling-dependent chloride currents. No block can be observed at different concentrations tested.
Figure 7.
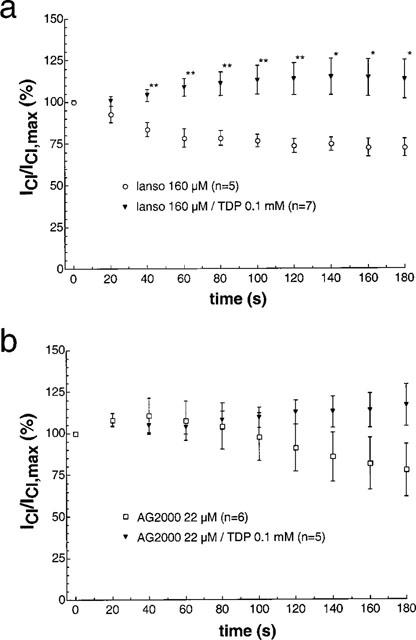
Effect of lansoprazole (lanso) and AG2000 in the presence of 0.1 mM TDP. (a) The block effected by lansoprazole (160 μM) is reduced in the presence of 0.1 mM TDP. (b) The block of AG2000 (22 μM) remains unaffected by the presence of 0.1 mM TDP.
Intracellular dithiothreitol modifies the AG2000 effect on IClswell
The sulphenamide AG2000 leads to an irreversible inhibition of the H,K-ATPase by interacting covalently with the sulfhydryl groups (Hersey & Sachs, 1995; Sachs et al., 1995). Dithiothreitol (DTT) is able to interfere with this reaction (Nagaya et al., 1990). At a concentration of 0.1 mM extracellular AG2000 is able to reduce IClswell by 70.5±9.5% (n=5; Figure 8a). The prior addition (>1 min) of 1 mM DTT to the extracellular solution does not alter this value significantly, however, the prior adding of the same concentration of DTT to the cytoplasm (pipette solution) significantly reduces the block of AG2000 from 70.5±9.5%, (n=5) to 45.3±5.5% (n=16) (Figure 8a). DTT alone does not have a significant effect on IClswell, if added on either side of the membrane (Figure 8b).
Figure 8.
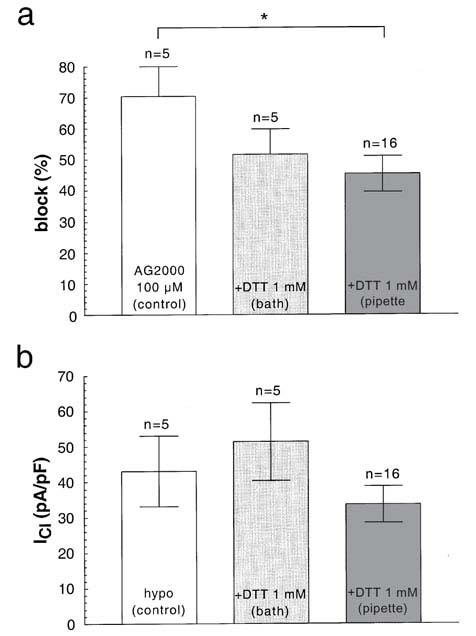
The effect of DTT in the presence and in the absence of AG2000. (a) The block effected by AG2000 (control) can be significantly reduced in the presence of 1 mM intracellular (pipette) DTT, but can not be reduced by adding DTT to the extracellular (bath) side (the cells were preincubated at least 1 min with DTT before adding AG2000). (b) DTT at a concentration of 1 mM is not able to block swelling-dependent chloride channels when added to either side.
Discussion
The H,K-ATPase and apical chloride channels in parietal cells are functionally linked (Asano et al., 1987; Cuppoletti & Sachs, 1984; Demarest et al., 1989; McGreevy et al., 1986; Reenstra et al., 1987; Starlinger et al., 1986). It has been suggested that the chloride channels in parietal cells involved in proton secretion are closely related to swelling-dependent chloride channels (Malinowska et al., 1995) since they resemble several features of those channels (Cuppoletti et al., 1993; Okada, 1997), e.g. sensitivity to anthracene-9-COOH (Cuppoletti & Sachs, 1984) and a permeability sequence of I−>Br−>Cl− (Saccomani et al., 1991). In addition, it has been shown for gastric glands isolated from rabbit stomach that hypotonicity is indeed able to stimulate acid secretion. This stimulation appears within the same time frame which is necessary for the stimulation of IClswell (Sernka, 1990). This, however, does not rule out the involvement of additional chloride channels in the ion conducting process of acid secretion.
Here we show that swelling-dependent chloride channels (IClswell) are sensitive to the benzimidazoles lansoprazole and, to a lesser extent, omeprazole. These substances have been described as direct inhibitors of the H,K-ATPase of parietal cells (Hersey & Sachs, 1995; Sachs et al., 1995). Both ‘pro-drugs' need to be protonated in an acidic environment in order to be able to restrict proton secretion (Sachs et al., 1995). In humans this protonization can only be substantially effected in the acid environment of the stomach (Sachs et al., 1995). Lansoprazole as well as AG2000 – the latter being the acid activated sulphenamide form of the ‘pro-drug' – are able to block IClswell. The IC50 value of the ‘pro-drug' is eight times higher compared with that of AG2000 when added to the extracellular solutions. The IC50 values for blocking IClswell in our experiments were tested at room temperature and within 2–5 min after the addition of the drugs. In order to estimate the therapeutic relevance of these IC50 values, the higher temperature and the much higher extent of availability in vivo of these drugs must be considered. In addition it must be kept in mind that we performed our experiments using fibroblasts, which are presumably not able to accumulate the activated sulphenamides a 1000 fold as it was shown for parietal cells. Due to these differences, the IC50 values for blocking IClswell in vivo in parietal cells are probably lower, and therefore close to the IC50 values measured for the blocking of acid secretion in these cells. It is important to mention that the block effected by lansoprlazole is much faster compared to the block by AG2000 (both drugs are added extracellularly). One explanation for these findings could be the existence of binding site(s) for lansoprazole on the extracellular side of the chloride channel and reaction site(s) for AG2000 at the cytoplasmic side of the channel protein. The intracellular site(s) appear to favour AG2000 over lansoprazole – since lansoprazole is ineffective if added to the cytoplasmic (pipette) side. The binding of AG2000 to the intracellular reaction site(s), which is followed by the blocking of IClswell, seems to involve the formation of disulfide-bridges – since the addition of DTT, which can interfere with the disulfide-bridge formation between sulfhydryl groups and the sulphenamide AG2000 is able to impair the effect of AG2000. Our experiments indicate that the site(s) for the disulfide-bridge formation between AG2000 and the IClswell protein is/are located intracellularly, whereas the disulfide-bridging between benzimidazoles and the H,K-ATPase was found to be on the extracytoplasmatic side (Hersey & Sachs, 1995).
The extracellular binding site(s) seem/s to be functionally related to the nucleotide-binding site(s), since the effect of lansoprazole is modified in the presence of TDP. Similarly, the effect of nucleoside analogues and phenol derivatives (Gschwentner et al., 1996; 1995b) on swelling dependent chloride channels in fibroblasts can be altered by the presence of TDP. A possible explanation for these observations might be that TDP is able to bind to nucleotide binding site(s) located at the outer mouth of swelling-dependent chloride channels without blocking the current while competing with other substances, which use the same binding site(s) (AZT or a closely related one (gossypol) which are both able to block IClswell). If the concentration of lansoprazole is increased from 160 μM to 0.5 mM in the presence of TDP, the time constant for the blockage becomes much faster. Our results therefore, suggest that there is a functional relationship between the nucleotide binding site(s) and the binding site(s) for lansoprazole on swelling dependent chloride channels. Furthermore, our experiments show that omeprazole, lansoprazole and AG2000 are able to block swelling-dependent chloride channels (IClswell) in fibroblasts. Similar channels could be involved in facilitating proton secretion, especially under metabolic stress of the gastric mucosa created by the restricted blood flow in the presence of high concentrations of catecholamines (Sanders, 1976; Semb, 1982). The metabolic stress leads to the swelling of the mucosal cells, which, in turn, is followed by the activation of IClswell (Lang et al., 1998). Additional experiments, however, are necessary in order to precisely define the role of IClswell in proton secretion.
Acknowledgments
We thank B. Oehl, C. Bazzini and M. Jakab for helpful discussion and/or critical reading of the manuscript. We greatfully acknowledge the support by B. Maier and H. Hasibeder from Takeda, Austria and M. Kuroda and N. Inatomi PhD from Takeda Pharmaceutical, Osaka, Japan, for supplying lansoprazole as well as AG2000. Expert technical assistance by E. Papp, A. Wimmer and G. Buemberger is gratefully acknowledged. This work was supported in part by grants from the Austrian Science Foundation grant #P10393-MED, P12337-MED and P13041-MED, the Austrian National Bank grant #6994/1, the EC BMH4-CT96-0602, the Gastein Foundation grant #FP46 and the ÖGLUT to M. Paulmichli.
Abbreviations
- AG2000
4-methyl-3-(2,2,2-trifluoroethoxy)-5H-pyrido[1′, 2′:4, 5][1, 2, 4] thiadiazino [2, 3-a] benzimidazole-13-ium tetrafluoroborate
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- EGTA
ethyleneglycol-bis (β-aminoethylether)-N,N,N′N′-tetraacetic acid
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- IClswell
swelling-dependent chloride channels
- lansoprazole
2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridyl]methyl]sulfinyl]-1H-benzimidazole
- omeprazole
5-methoxy-2-[[(4-methoxy-3, 5-dimethyl-2-pyridinyl)methyl] sulfinyl]-1H-benzimidazole
- RVD
regulatory volume decrease
References
- ASANO S., INOIE M., TAKEGUCHI N. The Cl-channel in Hog Gastric Vesicles is Part of the Function of H,K-ATPase. J. Biol. Chem. 1987;262:13263–13268. [PubMed] [Google Scholar]
- BARRADELL L.B., FAULDS D., MCTAVISH D. Lansoprazole. A review of its pharmacodynamic and pharmacokinetic properties and its therapeutic efficacy in acid related disorders. Drugs. 1992;44:225–250. doi: 10.2165/00003495-199244020-00007. [DOI] [PubMed] [Google Scholar]
- CUPPOLETTI J., BAKER A.M., MALINOWSKA D.H. Cl-channels of the gastric parietal cell that are active at low pH. Am. J. Physiol. 1993;264:C1609–C1618. doi: 10.1152/ajpcell.1993.264.6.C1609. [DOI] [PubMed] [Google Scholar]
- CUPPOLETTI J., SACHS G. Regulation of Gastric Acid Secretion via Modulation of a Chloride Conductance. J. Biol. Chem. 1984;259:14952–14959. [PubMed] [Google Scholar]
- DEMAREST J.R., LOO D.F., SACHS G. Activation of Apical Chloride Channels in the Gastric Oxyntic Cell. Science. 1989;245:402–404. doi: 10.1126/science.2474200. [DOI] [PubMed] [Google Scholar]
- GRÜNDER S., THIEMANN A., PUSCH M., JENTSCH T.J. Regions involved in the opening of ClC-2 chloride channel by voltage and cell volume. Nature. 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- GSCHWENTNER M., JUNGWIRTH A., HOFER S., WÖLL E., RITTER M., SUSANNA A., SCHMARDA A., REIBNEGGER G., PINGGERA G.M., LEITINGER M., FRICK J., DEETJEN P., PAULMICHL M. Blockade of swelling-induced chloride channels by phenol derivatives. Brit. J. Pharmacol. 1996;118:41–48. doi: 10.1111/j.1476-5381.1996.tb15364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GSCHWENTNER M., NAGL O.U., WÖLL E., SCHMARDA A., RITTER M., PAULMICHL M. Antisense oligonucleotides suppress cell-volume-induced activation of chloride channels. Pflügers Arch. 1995a;430:464–470. doi: 10.1007/BF00373882. [DOI] [PubMed] [Google Scholar]
- GSCHWENTNER M., SUSANNA A., WÖLL E., RITTER M., NAGL U.O., SCHMARDA A., LAICH A., PINGGERA G.M., ELLEMUNTER H., HUEMER H., DEETJEN P., PAULMICHL M. Antiviral drugs from the nucleoside analog family block volume-activated chloride channels. Mol. Med. 1995b;1:407–417. [PMC free article] [PubMed] [Google Scholar]
- HERSEY S.J., SACHS G. Gastric Acid Secretion. Physiol. Rev. 1995;75:155–189. doi: 10.1152/physrev.1995.75.1.155. [DOI] [PubMed] [Google Scholar]
- LANG F., BUSH G.L., RITTER M., VÖLKL H., WALDEGGER S., GULBINS E., HÄUSINGER D. Functional Significance of Cell Volume Regulatory Mechanisms. Physiol. Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- MALINOWSKA D.H., KUPERT E.Y., BAHINSKI A., SHERRY A.M., CUPPOLETTI J. Cloning, functional expression, and characterization of a PKA-activated gastric Cl-channel. Am. J. Physiol. 1995;268:C191–C200. doi: 10.1152/ajpcell.1995.268.1.C191. [DOI] [PubMed] [Google Scholar]
- MARTY A., NEHER E.Tight-seal whole-cell recording Single Channel Recording 1983New York; 107–122.In: Sakmann, B. & Neher, E. (eds) [Google Scholar]
- MCGREEVY J.M., BARTON R.G., HOUSINGER T. Chloride transport in bullfrog gastric mucosa. J. Surg. Res. 1986;40:462–466. doi: 10.1016/0022-4804(86)90216-7. [DOI] [PubMed] [Google Scholar]
- MCTAVISH D., BUCKLEY M.M., HEEL R.C. Omeprazole. An updated review of its pharmacology and therapeutic use in acid-related disorders. Drugs. 1991;42:138–170. doi: 10.2165/00003495-199142010-00008. [DOI] [PubMed] [Google Scholar]
- NAGAYA H., SATOH H., MAKI Y. Possible Mechanism for the Inhibition of Acid Formation by the Proton Pump Inhibitor AG-1749 in Isolated Canine Parietal Cells. J. Pharm. Exp. Therap. 1990;252:1289–1295. [PubMed] [Google Scholar]
- OKADA Y. Volume expansion-sensing outward-rectifying Cl− channel: fresh start to the molecular identify and volume sensor. Am. J. Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- REENSTRA W.W., BETTENCOURT J.D., FORTE J.G. Mechanisms of active Cl− secretion by frog gastric mucosa. Am. J. Physiol. 1987;252:G543–G547. doi: 10.1152/ajpgi.1987.252.4.G543. [DOI] [PubMed] [Google Scholar]
- RITTER M., SCHRATZBERGER P., ROSSMANN H., WÖLL E., SEILER K., SEIDLER U., REINISCH N., KÄHLER C.M., ZWIERZINA H., LANG H.J., LANG F., PAULMICHL M., WIEDERMANN C.J. Effect of inhibitors of Na+/H+-exchange and gastric H+/K+ ATPase on cell volume, intracellular pH and migration of human polymorphonuclear leucocytes. Br. J. Pharmacol. 1998;124:627–638. doi: 10.1038/sj.bjp.0701864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SACCOMANI G., PSARRAS C., SMITH P.R., KIRK K.L., SHOEMAKER R.L. Histamine-induced chloride channels in apical membrane of isolated rabbit parietal cells. Am. J. Physiol. 1991;260:C1000–C1011. doi: 10.1152/ajpcell.1991.260.5.C1000. [DOI] [PubMed] [Google Scholar]
- SACHS G., SHIN J.M., BRIVING C., WALLMARK B., HERSEY S. The Pharmacology of the Gastric Acid Pump: The H+,K+ ATPase. Annu. Rev. Pharmacol. Toxicol. 1995;35:277–305. doi: 10.1146/annurev.pa.35.040195.001425. [DOI] [PubMed] [Google Scholar]
- SANDERS D.J. A review: the agents and actions of sympathetic nerve and catecholamine inhibition of gastric mucosal function. Agents Actions. 1976;6:385–388. doi: 10.1007/BF01973207. [DOI] [PubMed] [Google Scholar]
- SEMB B.K. The effect of catecholamines on gastric mucosal flow. Scand. J. Gastroenterol. 1982;17:663–670. doi: 10.3109/00365528209181076. [DOI] [PubMed] [Google Scholar]
- SERNKA T.J. Direct hyposmotic stimulation of gastric acid secretion. Membr. Biochem. 1990;9:1–7. doi: 10.3109/09687689009026819. [DOI] [PubMed] [Google Scholar]
- SHULL G.E., LINGREL J.B. Molecular cloning of the rat Stomach (H+K+)-ATPase. J. Biol. Chem. 1986;261:16788–16791. [PubMed] [Google Scholar]
- STARLINGER M.J., HOLLANDS M.J., ROWE P.H., MATTHEWS J.B., SILEN W. Chloride transport of frog gastric fundus: effects of omeprazole. Am. J. Physiol. 1986;250:G118–G126. doi: 10.1152/ajpgi.1986.250.1.G118. [DOI] [PubMed] [Google Scholar]
- WÖLL E., GSCHWENTNER M., FÜRST J., HOFER S., BUEMBERGER G., JUNGWIRTH A., FRICK J., DEETJEN P., PAULMICHL M. Fluorescence-optical measurements of chloride movements in cells using the membrane-permeable dye diH-MEQ. Pflügers Arch. 1996;432:486–493. doi: 10.1007/s004240050160. [DOI] [PubMed] [Google Scholar]