

Abstract
Endothelium-derived hyperpolarizing factor (EDHF) has recently been identified as potassium released from endothelial cells into the myo-endothelial space. The present study was designed to test this hypothesis.
In rat small mesenteric arteries, mounted in a wire myograph, relaxation to acetylcholine or potassium was not significantly changed following incubation with oxadiazolo-quinoxalin-1-one (ODQ, 4 μM) and indomethacin (10 μM, n=9).
Maximal relaxations to acetylcholine occurred in all arteries, were maintained and were significantly greater (P<0.01, n=9) than the transient relaxations to potassium, which only occurred in 30–40% of vessels.
Removal of the vascular endothelium abolished relaxant responses both to potassium and acetylcholine (P<0.005, n=9).
Compared with responses in 5.5 mM potassium PSS, relaxation responses to added potassium in arteries maintained in 1.5 mM potassium PSS were more marked and were not dependent on the presence of an intact endothelium (n=8).
Incubation with BaCl2 (50 μM) significantly inhibited the maximal relaxant response to potassium in the presence of an intact endothelium in 5.5 mM potassium PSS (P<0.05, n=4), but had no effect on relaxation of de-endothelialized preparations in 1.5 mM potassium PSS (n=5).
Treatment with ouabain (0.1 mM) abolished the relaxant response to potassium in 1.5 mM potassium PSS (P<0.001, n=9), but only partly inhibited the maximal relaxant response to acetylcholine in 5.5 mM potassium PSS (P<0.01, n=5).
These data show that at physiological concentrations of potassium an intact endothelium is necessary for potassium-induced relaxation in rat mesenteric arteries. Furthermore, the response to potassium is clearly different to that from acetylcholine, indicating that potassium does not mimic EDHF released by acetylcholine in these arteries.
Keywords: Potassium, acetylcholine, EDHF, rat mesenteric artery, endothelium dependent relaxation
Introduction
The vascular endothelium mediates agonist stimulated, endothelium-dependent vasodilatation through the release of substances whose primary site of action is vascular smooth muscle cells. These substances include nitric oxide (Furchgott & Zawadzki, 1980) and vasodilator prostanoids. A third component of endothelium-dependent vasodilatation, termed endothelium-derived hyperpolarizing factor (EDHF), acts in a nitric oxide-independent, prostanoid-independent manner. EDHF is a unique endothelial factor in that vascular relaxation appears to be entirely dependent upon smooth muscle hyperpolarization. The chemical identity of EDHF is not clear but candidates include the cytochrome P450-derived epoxyeicosatrienoic acids (Adeagbo, 1997; Hecker et al., 1994) and endocannabinoids (Randall et al., 1996). It has recently been proposed that EDHF is potassium released from charybdotoxin and apamin sensitive potassium channels on the vascular endothelium which then acts on inwardly-rectifying potassium channels and Na+/K+ ATPases on the smooth muscle cells to induce hyperpolarization (Edwards et al., 1998). The present study investigated whether exogenously added potassium can mimic EDHF in rat mesenteric small arteries.
Methods
Arterial mounting and experimental design
Adult Wistar Rats of either sex were killed by stunning followed by cervical dislocation, mesenteries collected and segments of third order arteries (200–300 μm internal diameter) dissected and mounted in a Mulvany-Halpern wire myograph (Danish myotechnology, Denmark). Arterial segments were incubated at 37°C either in PSS (composition mM: NaCl 118, NaHCO3 25, KCl 4.5, KH2PO4 1, CaCl2 2.4, MgSO4 1, glucose 6, pH 7.4) or in HEPES-buffered PSS (HPSS) (composition mM: NaCl 118, NaHCO3 25, KCl 4.7, KH2PO4 1.2, CaCl2 1.8, MgSO4 1.2, HEPES 5, glucose 11, pH 7.4) under normalized tension. Arterial segments were constricted with noradrenaline or phenylephrine (10 μM) and cumulative relaxation-response curves to acetylcholine (10−10–10−4 M) and then potassium (0.5–15 mM, added concentration) constructed. The above protocol was then repeated in low potassium PSS or HPSS, in which potassium was reduced to 3.2, 1.5 or 1.0 mM by isotonic replacement with NaCl. Relaxation response curves to potassium were repeated after incubation with ouabain (0.1 mM) for 30 min in low potassium solution. In order to isolate the relaxant component due to endothelium-derived hyperpolarizing factor the above solutions were supplemented with indomethacin (10 μM) and either [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 4 μM), or Nω-nitro-L-arginine methyl ester (L-NAME, 400 μM) under the same conditions. There was no difference in the behaviour of the arteries in these differing solutions. Cumulative relaxation response curves were finally carried out to sodium nitroprusside (SNP, 10−9–10−4 M) in the absence and presence of ODQ and indomethacin. In addition, cumulative relaxation response curves were performed, as described above, on a parallel group of arteries from which the endothelium had been removed by rubbing with a hair.
A sub group of arteries, which showed significant relaxation responses to potassium, were incubated in the presence of BaCl2 (50 μM) for 30 min. Concentration-effect curves to potassium or acetylcholine were then repeated. Arteries were then washed with PSS for 30 min and concentration-effect curves performed again. These studies were all carried out in the continued presence of indomethacin and ODQ. A further group of mesenteric arteries were incubated with 0.1 mM ouabain or 0.1 mM ouabain plus 50 μM BaCl2 for 30 min, or with 0.1 mM charybdotoxin plus 0.1 μM apamin for 10 min. Cumulative concentration-effect curves to acetylcholine were then carried out in the additional presence of 4 μM ODQ and 10 μM indomethacin.
In a separate study, mesenteric arteries mounted in PSS were incubated with 4 μM ODQ and 10 μM indomethacin and cumulative relaxation response curves to acetylcholine were performed. Arteries were then incubated with Nω-nitro-L-arginine (L-NOARG, 100 μM) for 60 min and concentration-effect curves to acetylcholine repeated in the combined presence of ODQ, indomethacin and L-NOARG.
In order to minimize any effects due to gender differences, female rats were used throughout, except where stated in the text or figure legends.
Drugs
Acetylcholine, Indomethacin, Nω-nitro-L-arginine (L-NOARG), Nω-nitro-L-arginine methyl ester (L-NAME), [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), phenylephrine and Sodium Nitroprusside were all obtained from the Sigma Chemical Company (Poole, Dorset, U.K.). Noradrenaline (DL- Arterenol) was obtained from ICN Biomedicals Inc, (Aurora, OH, U.S.A.). All other chemicals were analytical grade, obtained from standard suppliers. Chemicals were dissolved in water except for ODQ, which was dissolved in dimethyl sulphoxide.
Analysis and statistics
Relaxation responses are expressed as a percentage decline of the maximal contractile response. In the text and figure legends data represents mean±s.e.mean and n represents the number of animals in each group. At least two arterial segments were taken from each animal used. Data from individual concentration-response curves were transformed as median-effect plots (Chou & Talalay, 1984) and EC50 values obtained using a concentration-effect analysis computer program (Biosoft). Maximal changes in tension for whole concentration-effect curves (Emax) and EC50 values were compared using paired or unpaired Student's two-tailed t-test with P values <0.05 taken as significant. When s.e.mean values differed, comparisons were made using Mann-Whitney U-tests.
Results
Mesenteric small arteries with an intact endothelium showed concentration-dependent relaxation in response to acetylcholine (Figure 1: EC50 0.08±0.012 μM, Emax 90±5.2%, n=9). Relaxation was sustained, even at higher concentrations of acetylcholine. The incremental addition of potassium to the same group of mesenteric arteries resulted in either no response (60–70% of vessels) or an initial reduction in tension followed by a contraction at potassium concentrations exceeding 10 mM (30–40%) (Figure 2: EC50 2.57±0.13 mM, Emax 34±7.3%, n=9). Examples of representative relaxation responses to acetylcholine and to potassium are shown in Figure 3. When relaxation response curves were performed on a parallel group of de-endothelialized arteries, relaxations were abolished to both acetylcholine (Figure 1: EC50 0.52±0.19 μM P<0.05, Emax 12.2±2.9%, P<0.001, n=9) and potassium (Figure 2: EC50 2.64±0.4 mM P>0.05, Emax 4.5±1.4% P<0.005, n=9). By contrast, the addition of potassium to arteries maintained in low potassium PSS in the presence of indomethacin and ODQ, resulted in concentration-dependent relaxation responses in both intact and de-endothelialized vessels (Figure 4: EC50 1.91±0.27 mM, Emax 94.4±1.74% plus endothelium, EC50 1.71±0.12 mM, Emax 93±0.97% minus endothelium, n=8). This relaxation in response to added potassium was abolished following incubation with 0.1 mM ouabain.
Figure 1.
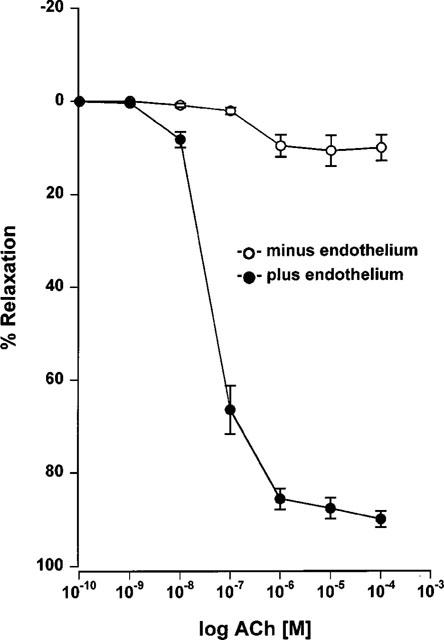
Cumulative concentration-response curves to acetylcholine of endothelium denuded and non-denuded Wistar mesenteric small arteries preconstricted with 10 μM noradrenaline in 5.5 mM potassium PSS (n=9).
Figure 2.
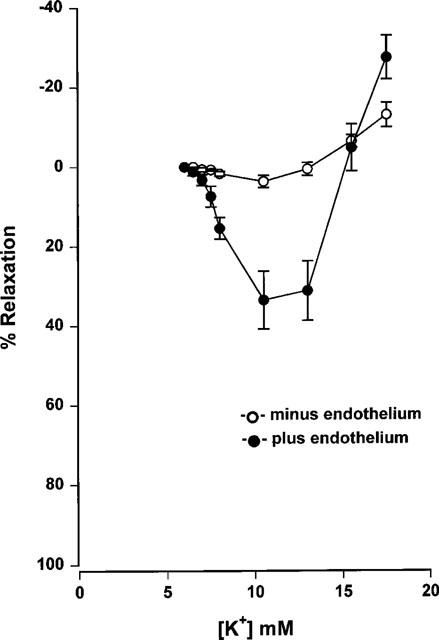
Cumulative concentration-response curves to potassium of endothelium denuded and non-denuded Wistar mesenteric small arteries preconstricted with 10 μM noradrenaline in 5.5 mM potassium PSS (n=9).
Figure 3.
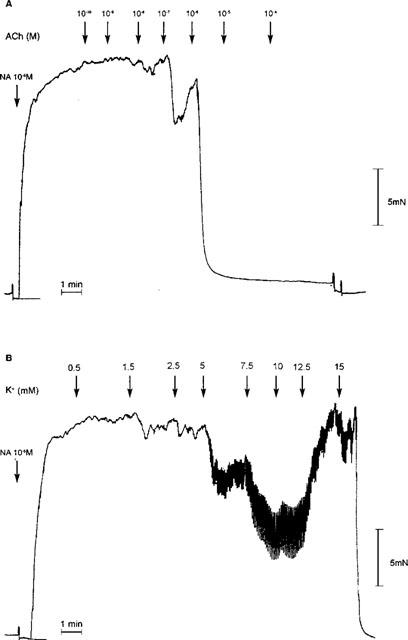
Representative tracings showing isometric tension (mN) with time for relaxation responses to (A) acetylcholine and (B) potassium of an endothelium intact Wistar mesenteric small artery equilibrated in 5.5 mM potassium PSS. The artery was preconstricted with 10 μM noradrenaline in the presence of 4 μM ODQ and 10 μM indomethacin and exposed to cumulative increasing acetylcholine or potassium concentrations as indicated by the arrows.
Figure 4.
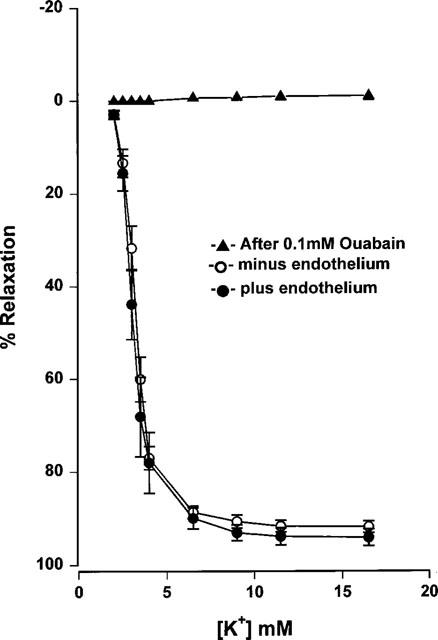
Cumulative concentration-response curves to potassium of endothelial denuded and non-denuded Wistar mesenteric small arteries incubated in 1.5 mM potassium PSS. Arteries were preconstricted with 10 μM noradrenaline in the presence of indomethacin and ODQ (n=8). Concentration-response curves were repeated following incubation with ouabain (0.1 mM) for 30 min.
Incubation of endothelium intact arteries with ODQ and indomethacin in PSS had little effect on the relaxation response to 1 μM acetylcholine (Figure 5). By contrast, relaxation responses to SNP were significantly reduced (Figure 5: EC50 1.38±0.4 μM, Emax 67.7±4.2% before ODQ plus indomethacin, EC50 11.7±1.7 μM P<0.001, Emax 18.5±2.5% P<0.001 after ODQ plus indomethacin, n=8). This suggests that the major part of the relaxation response to acetylcholine is mediated by a mechanism unaffected by prostacyclin or nitric oxide, typical of an EDHF response. The responses to acetylcholine and potassium of a series of arteries were investigated following treatment with ODQ and indomethacin. The magnitude of the relaxation evoked by 1 μM acetylcholine was always greater than that induced by 5 mM potassium (P<0.01, n=9). Similarly, a comparison of whole concentration-effect curves following treatment with indomethacin and ODQ showed that maximal relaxation to acetylcholine (EC50 0.104±0.018 μM, Emax 85.4±7.5%, n=9) was significantly greater than to potassium (EC50 2.99±0.31 mM, Emax 38.9±11.3%, P<0.01, n=9). In common with responses to acetylcholine, relaxation responses to potassium were unaffected by treatment with ODQ and indomethacin (n=9).
Figure 5.
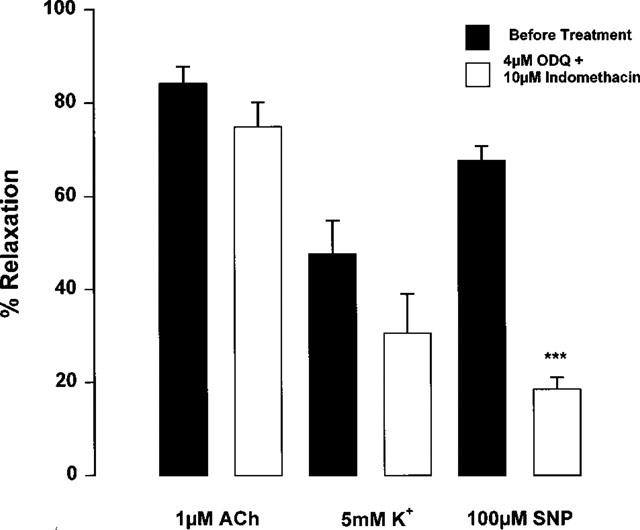
Effect of treatment with 4 μM ODQ and 10 μM indomethacin on relaxation to 1 μM acetylcholine, 5 mM potassium or 100 μM SNP of Wistar mesenteric small arteries preconstricted with 10 μM noradrenaline and maintained in 5.5 mM potassium PSS. ***P<0.0001 No treatment vs after indomethacin and ODQ, Student's t-test (n=8).
Maximal relaxation responses to acetylcholine in the presence of ODQ and indomethacin were not altered in the additional presence of L-NOARG (100 μM) (Figure 6: EC50 0.13±0.02 μM, Emax 83±6.9% before L-NOARG, EC50 0.35±0.04 μM P<0.001, Emax 80.7±5.9% P>0.05 after L-NOARG, n=5).
Figure 6.
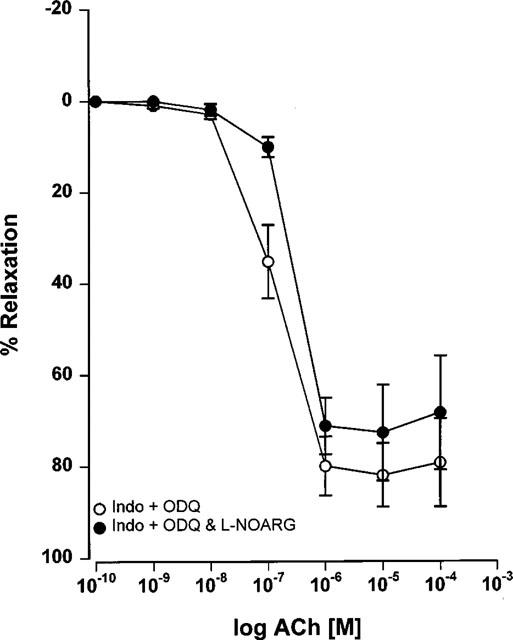
Effect of the additional presence of 100 μM L-NOARG on concentration-response curves to acetylcholine for Wistar mesenteric small arteries. Arteries were preconstricted with 10 μM noradrenaline and incubated in the presence of 4 μM ODQ and 10 μM indomethacin in 5.5 mM potassium PSS (n=5).
Incubation with BaCl2 (50 μM) partly inhibited relaxation responses both to acetylcholine (EC50 0.09±0.02 μM, Emax 83.5±2.6% before BaCl2 EC50 0.43±0.12 μM P<0.05, Emax 59.3±3.7% P<0.001 after BaCl2, n=4) and potassium (EC50 2.96±0.22 mM, Emax 69.6±10.4% before BaCl2, EC50 2.7±0.24 mM P>0.05, Emax 30.48±9.9% P<0.05 after BaCl2, n=4) in endothelium-intact mesenteric arteries in the presence of ODQ and indomethacin. BaCl2 (50 μM) had no effect on the extent of potassium-evoked relaxation of de-endothelialized arteries in low potassium solutions (Figure 7: EC50 1.28±0.04 mM, Emax 90.3±2.5% before BaCl2 EC50 2.57±0.36 mM P<0.05, Emax 88.14±4.24% P>0.05 after BaCl2, n=5). By contrast to the complete inhibition of relaxation to potassium for arteries maintained in low potassium PSS, incubation with ouabain (0.1 mM) in 5.5 mM potassium PSS only partly inhibited relaxation responses to acetylcholine in the continued presence of ODQ and indomethacin (Figure 8: EC50 0.11±0.04 μM, Emax 81.2±3.1% before ouabain, EC50 0.57±0.1 μM P<0.01, Emax 63.6±3.6% P<0.01 after ouabain, n=5). In the presence of the combination of ouabain and BaCl2, relaxation to acetylcholine was more markedly inhibited (Figure 8: EC50 0.19±0.04 μM, Emax 76.9±5.5% before BaCl2 plus ouabain, EC50 0.82±0.15 μM P<0.01, Emax 26.6±4.2% P<0.01 after BaCl2 plus ouabain, n=4). By contrast, relaxation to acetylcholine was abolished in the combined presence of apamin (0.1 μM) plus charybdotoxin (0.1 μM, n=7).
Figure 7.
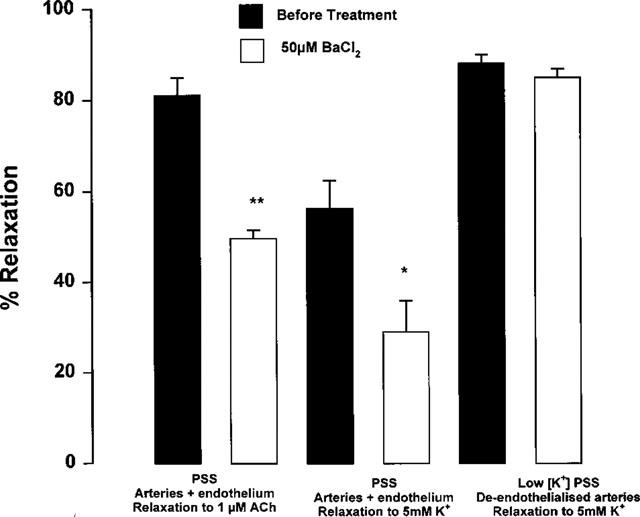
Effect of treatment with 50 μM BaCl2 on relaxation responses to 1 μM acetylcholine or 5 mM potassium of Wistar mesenteric small arteries incubated in 5.5 mM potassium PSS or 1.5 mM potassium PSS. Relaxation responses were all carried out on noradrenaline (10 μM) preconstricted arteries in the presence of 4 μM ODQ and 10 μM indomethacin. *P<0.05 before treatment vs after BaCl2. **P<0.01 before treatment vs after BaCl2 (n=4).
Figure 8.
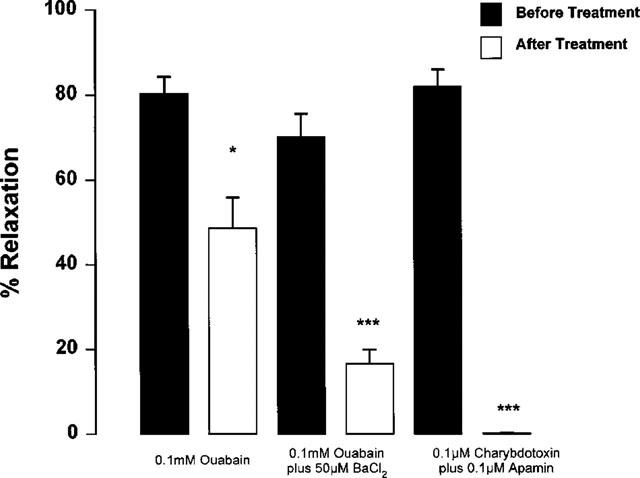
Effect of treatment with 0.1 mM ouabain, 0.1 mM ouabain plus 50 μM BaCl2 or 0.1 μM charybdotoxin plus 0.1 μM apamin on relaxation to 1 μM acetylcholine for Wistar mesenteric small arteries incubated in 5.5 mM potassium PSS. Relaxation responses were all carried out on noradrenaline (10 μM) preconstricted arteries in the presence of 4 μM ODQ and 10 μM indomethacin. *P<0.05, ***P<0.0001 before treatment vs after treatment (n=4).
Relaxation responses to potassium in mesenteric arteries showed a marked dependence on bathing solution potassium concentrations (Figure 9). Decreasing bathing potassium concentrations resulted in an increase in the potency of, and maximum response to added potassium (EC50 2.9±1.1 mM, Emax 97.3±1.0% in 1 mM potassium; EC50 3.8±5.2 mM, Emax 42.2±8.0% in 3.2 mM potassium; EC50 could not be calculated, Emax 4.2±2.0% in 5.9 mM potassium, n=7). By comparison, maximal relaxation to 1 μM acetylcholine was not affected by changing the PSS potassium concentration (n=7, Figure 10).
Figure 9.
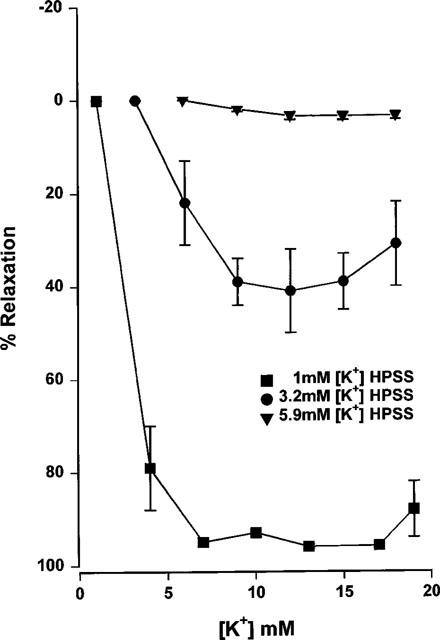
Cumulative concentration-response curves to potassium, carried out in 1, 3.2 or 5.9 mM potassium HPSS of endothelium intact mesenteric small arteries from male Wistar rats. Arteries were preconstricted with 10 μM phenylephrine and relaxation responses were all carried out in the presence of 10 μM indomethacin and 400 μM L-NAME (n=7).
Figure 10.
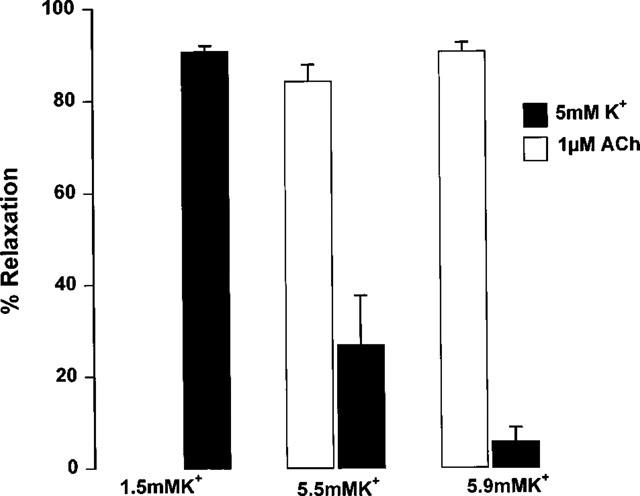
Relaxation response to 1 μM acetylcholine or 5 mM potassium carried out in 1.5, 5.5 or 5.9 mM potassium HPSS, of endothelium intact mesenteric small arteries from male Wistar rats. Arteries were preconstricted with 10 μM phenylephrine and relaxation responses carried out in the presence of 10 μM indomethacin and 400 μM L-NAME (n=7).
Discussion
In the present study the pattern and characteristics of relaxation of small mesenteric arteries to externally added potassium clearly differ from those to acetylcholine. Acetylcholine induced a large sustained relaxation, whilst potassium-induced relaxations were significantly smaller and associated with a subsequent re-contraction. There was also a greater variability in the response to potassium for endothelium intact arteries, with the majority showing little or no response to the addition of potassium while showing consistent relaxation to acetylcholine in the same arteries. Furthermore, relaxation responses to potassium were clearly potentiated when the initial potassium concentration was decreased, whilst those to acetylcholine showed little change. These observations are in agreement with a recent study by Quignard et al. (1999). These workers clearly showed a more extensive hyperpolarization of vascular smooth muscle cells to acetylcholine in endothelium intact guinea-pig carotid and porcine coronary arteries than was seen following addition of 5 mM potassium. This suggests that certain arteries show different physiological responses to acetylcholine and potassium, acetylcholine inducing a sustained hyperpolarization that was absent following potassium stimulation. Vascular smooth muscle cell hyperpolarization is the likely explanation for the extensive relaxation seen to acetylcholine in our study, whilst relaxation to potassium appears to be mediated by a different mechanism.
The observation that the majority of the relaxation response to acetylcholine was maintained following incubation with ODQ and indomethacin, whilst that to SNP was attenuated suggests that the major part of the response to acetylcholine is mediated by a non-nitric oxide, non-prostanoid mechanism. This activity has been correlated with smooth muscle hyperpolarization in other studies (Parkington et al., 1995) and is mediated by the putative EDHF. The observation that relaxation to acetylcholine in the presence of ODQ and indomethacin was abolished following treatment with the combination of charybdotoxin and apamin further supports the idea that this activity represents EDHF (Corriu et al., 1996; Chataigneau et al., 1998; White & Hiley, 1997; Zygmunt & Hogestatt, 1996). Direct comparisons of relaxation responses to potassium and to acetylcholine in our study clearly revealed different characteristics, which were well maintained after incubation with indomethacin and ODQ. This observation suggests that potassium itself cannot mimic acetylcholine-, and hence EDHF-induced, relaxation.
In normal potassium solution, relaxation responses to both acetylcholine and potassium were clearly dependent upon an intact endothelium. The observation that an intact endothelium is required for potassium-induced relaxation is clearly inconsistent with the idea that potassium is EDHF. By contrast, relaxation responses to potassium for arteries incubated in low potassium concentrations occurred in all vessels tested, and in an endothelium-independent manner, indicating that in low potassium solution, potassium re-addition causes direct relaxation. Potassium can also be seen to induce a larger and more potent relaxant response than that observed in arteries maintained in normal potassium solution.
Potassium has been shown to induce a direct hyperpolarization and relaxation of vascular smooth muscle cells by two mechanisms. Following depletion, subsequent re-addition of potassium stimulates the ouabain-sensitive Na+/K+ ATPase causing hyperpolarization and relaxation in an endothelium independent manner (McCarron & Halpern, 1990). In our study, relaxation to externally added potassium in arteries incubated in low potassium solutions, clearly showed properties consistent with activation of the Na+/K+ ATPase. In solutions containing physiological concentrations of potassium the addition of extra potassium in the range 6–16 mM stimulates hyperpolarization and relaxation in certain arteries by stimulation of the activity of inward rectifying potassium channels (McCarron & Halpern, 1990; Knot et al., 1996; Johnson et al., 1998). Indeed, stimulation of KIR by potassium released from the endothelium is a mechanism through which non-nitric oxide, non-prostanoid (EDHF) mediated relaxation has been proposed to act (Edwards et al., 1998). However, it should be noted that there is no good evidence for the presence of KIR channels on mesenteric arteries (for review see Quayle et al., 1997).
The characteristics of potassium-induced relaxation in our study differ from those that would be expected if KIR channels were responsible. Activation of KIR in cerebral and coronary arteries has been shown to mediate an endothelium-independent dilatation that is abolished by treatment with barium (Knot et al., 1996). These differences suggest that relaxation to externally added potassium in our study is not dependent on KIR channel activation. This conclusion is supported by McCarron et al. (1991) who reported that mesenteric arteries from WKY rats failed to show a barium sensitive dilatation in response to externally added potassium, whilst such an activity was present in small cerebral arteries. In contrast, Edwards et al. (1998) reported an extensive, endothelium-independent relaxation to potassium in hepatic arteries from Sprague Dawley rats, similar in extent to that seen to acetylcholine. The differences between these studies might reflect differences in KIR density between different arterial beds, different strains of rat, or with differing arterial size (Quayle et al., 1996).
The characteristics of potassium-induced relaxations in our studies are clearly different from those reported in the investigations of Edwards et al. (1998). A reversal of relaxation at potassium concentrations in excess of 15 mM was seen in the present study, whilst in the study of Edwards', relaxation in similar arteries continued in the range 20–25 mM. It is difficult to rationalize how these differences arise, but differences in the strain and age of rat used and the nature and concentration of the pre-contracting agent may play a part. Relaxation responses to potassium for arteries incubated in 5.5 mM potassium solution were seen to be somewhat variable and this might have contributed to the differences in the profile of potassium-mediated relaxations seen between the studies. In agreement with the study of Edwards et al. (1998) we observed inhibition of the EDHF response by ouabain and the combination of BaCl2 plus ouabain. However, the extent of this inhibition was not as great as that seen to the combination of charybdotoxin and apamin, neither was it as great as the effect of ouabain on potassium induced relaxation in low potassium PSS. This suggests that under the conditions of this study, blockade of the sodium potassium ATPase and KIR are unable to block the EDHF response.
The mechanism of endothelial dependency in the response to potassium in our study is not clear. Rubanyi & Vanhoutte (1988) described an endothelial dependent relaxation response to the addition of 15 mM potassium in canine femoral arteries. Extra-luminal addition of potassium mediated a partial relaxation, smaller in extent than that seen with acetylcholine and associated with subsequent re-contraction. This activity was suggested to result from stimulation of the vascular endothelium by substances released following potassium stimulation of perivascular nerves. Such a mechanism could underlie the potassium responses seen in our study. Alternatively, it might reflect stimulation of endothelial KIR by added potassium leading to endothelial cell hyperpolarization. This hyperpolarization could potentially be communicated directly to the smooth muscle cell via gap junctions between the endothelium and smooth muscle (Chaytor et al., 1998; Taylor et al., 1998; Hutcheson et al., 1999; Dora et al., 1999). Interestingly, endothelial cell hyperpolarization in response to the additions of potassium was shown by Edwards et al. (1998).
Taken together, evidence from our study suggests that the external addition of potassium does not mimic EDHF activity in small mesenteric arteries from Wistar rats. Additionally the characteristics of relaxation responses to potassium differ from those reported for stimulation of KIR activity. This suggests that potassium does not act directly as an endothelium-derived hyperpolarizing factor in these arteries.
Acknowledgments
These studies were supported by a grant awarded from the British Diabetic Association and the Medical Research Council.
Abbreviations
- EDHF
endothelium-derived hyperpolarizing factor
- HPSS
HEPES-buffered physiological saline solution
- L-NAME
Nω-nitro-L-arginine methyl ester
- L-NOARG
Nω-nitro-L-arginine
- ODQ
oxadiazolo-quinoxalin-1-one
- PSS
physiological saline solution
- SNP
sodium nitroprusside
References
- ADEAGBO A.S. Endothelium-derived hyperpolarizing factor: characterization as a cytochrome P450 1A-linked metabolite of arachidonic acid in perfused rat mesenteric preartiolar bed. Am. J. Hypertens. 1997;10:763–771. doi: 10.1016/s0895-7061(97)00057-5. [DOI] [PubMed] [Google Scholar]
- CHATIAGNEAU T., FELETOU M., DUHAULT J., VANHOUTTE P.M. Epoxyeicosanoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxation of rabbit arteries. J. Physiol (Lond) 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOU T.-C., TALALAY P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., MARTIN P.E.M., CHAYTOR A.T., EVANS W.H., GARLAND C.J., GRIFFITH T.M. Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: inhibition by a connexin-mimetic peptide. Biochem. Biophys. Res. Commun. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterisation of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTCHESON I.R., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Nitric oxide-independent relaxations to acetylcholine and A23187 involve different routes of heterocellular communication. Role of gap junctions and phospholipase A2. Circ. Res. 1999;84:53–63. doi: 10.1161/01.res.84.1.53. [DOI] [PubMed] [Google Scholar]
- JOHNSON T.D., MARRELLI S.P., STEENBERG M.L., CHILDRES W.F., BRYAN R.M. Inward rectifier potassium channels in the rat middle cerebral artery. Am. J. Physiol. 1998;274:R541–R547. doi: 10.1152/ajpregu.1998.274.2.R541. [DOI] [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. 1996;492.2:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARRON J.G., HALPERN W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am. J. Physiol. 1990;259:H902–H908. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- MCCARRON J.G., QUAYLE J.M., HALPER W., NELSON M.T. Cromakalim and pinacidil dilate small mesenteric arteries but not small cerebral arteries. Am. J. Physiol. 1991;261:H287–H291. doi: 10.1152/ajpheart.1991.261.2.H287. [DOI] [PubMed] [Google Scholar]
- PARKINGTON H.C., TONTA M.A., COLEMAN H.A., TARE M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAYLE J.M., DART C., STANDEN N.B. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J. Physiol. 1996;494:715–726. doi: 10.1113/jphysiol.1996.sp021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Revs. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.-F., FELETOU M., THOLLON C., VILAINE J.-P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D., ALEXANDER S.P.H., BENNETT T., BOYD E.A., FRY J.R., GARDINER S.M., KEMP P.A., MCCULLOCH A., KENDALL D.A. An exogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem. Biophys. Res. Commun. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- RUBANYI G.M., VANHOUTTE P.M. Potassium-induced release of endothelium-derived relaxing factor from canine femoral arteries. Circ. Res. 1988;62:1098–1103. doi: 10.1161/01.res.62.6.1098. [DOI] [PubMed] [Google Scholar]
- TAYLOR H.J., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Inhibition of the gap junctional component of endothelium-dependent relaxation in rabbit iliac artery by 18-alpha glycerrhetinic acid. Br. J. Pharmacol. 1998;125:1–4. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE R., HILEY R. A comparison of EDHF-mediated and anandamide-induced relaxations in the rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1573–1584. doi: 10.1038/sj.bjp.0701546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HOGESTATT E.D. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]