

Abstract
Clotrimazole (CLT) is an antimycotic agent with a potential role in the treatment of cancer. Whole-cell patch clamp recordings and Fura-2 AM fluorescence measurements were used to investigate the inhibition by CLT of recombinant human cardiac L-type Ca2+ channel α1C subunits, stably expressed in human embryonic kidney (HEK 293) cells.
CLT (100 nmol l−1 to 25 μmol l−1) reduced Ca2+ channel currents in a concentration-dependent manner. Inhibition was neither use- or voltage-dependent. The effects of CLT were rapid and maximal effects were attained within 3 min. Application of CLT also caused an acceleration of apparent Ca2+ channel current inactivation.
Basal current density and the degree of inhibition due to CLT were not significantly altered by pretreating cells with 3 mmol l−1 1-aminobenzotriazole for 1 h, or by dialysing cells for 10 min with 2 mmol l−1 α-napthoflavone via the patch pipette, suggesting that the inhibitory action of CLT was not due to inhibition of cytochrome P-450.
CLT (10 μmol l−1) did not influence [Ca2+]i, as determined by Fura-2 AM fluorescence measurements.
Dialysing cells for 10 min with the non-specific serine/threonine kinase inhibitor H-7 (10 μmol l−1) was without effect on basal current density or on the inhibitory response to 10 μmol l−1 CLT, indicating that CLT is not acting via an indirect effect on these kinases.
These data suggest that CLT exerts a direct blocking effect on the α1C subunit at therapeutic concentrations. This effect may explain the abbreviation of the action potential duration by CLT observed in cardiac myocytes.
Keywords: L-type Ca2+ channels, α1C subunit, clotrimazole, cytochrome P-450
Introduction
Clotrimazole (CLT) is one of a family of imidazole-derived antimycotic agents which inhibit cytochrome P-450 mediated reactions, an effect which accounts for its potent antifungal effects (Sheets et al., 1986; Ayub & Levell, 1988). More recently, CLT has been shown to be of benefit in the treatment of sickle cell anaemia (Brugnara et al., 1995; 1996) and of β-thalassemia (De Franchesci et al., 1996), an effect attributed to the suppression of erythrocyte Ca2+-activated K+ (KCa) channels (Alvarez et al., 1992; Brugnara et al., 1993; Ishii et al., 1997). Inhibition of the KCa channel by CLT may be independent of effects on cytochrome P-450 (Brugnara et al., 1995; Coupry et al., 1996), and direct inhibitory effects of CLT on KCa channels have also been demonstrated in pituitary GH3 cells (Wu et al., 1999) and in carotid body type I cells (Hatton & Peers, 1996).
CLT also has a potential clinical role in the treatment of cancer. It can inhibit the in vitro and in vivo proliferation of both normal and cancerous cells (Benzaquen et al., 1995). It is well documented that CLT affects intracellular Ca2+ homeostasis in a variety of cell types, including thymocytes (Alvarez et al., 1992), platelets (Alonso et al., 1991; Sargeant et al., 1992), neutrophils (Montero et al., 1991) and in GH3 and chromaffin cells (Villalobos et al., 1992). This effect may contribute to its antiproliferative properties.
In both pituitary GH3 and pancreatic B-cells, CLT has been shown to have a direct inhibitory effect on voltage-gated Ca2+ channels in the plasma membrane (Welker & Drews, 1997; Wu et al., 1999). In the myocardium, L-type voltage-gated Ca2+ channels are the major pathway through which Ca2+ entry occurs during excitation. Ca2+ influx through these channels triggers the release of Ca2+ from intracellular stores. This initiates and regulates contractile activity, and is therefore of major importance in controlling excitation-contraction coupling. In a recent study (Thomas et al., 1999), CLT was shown to have a potent inhibitory effect on native L-type Ca2+ channels in isolated guinea-pig ventricular myocytes. This effect appeared to cause significant suppression of the plateau and abbreviation of the cardiac action potential. The mechanism of this effect of CLT was unclear, although the authors proposed either a direct effect on the channel protein, or an effect secondary to regulation of intracellular Ca2+ levels. With respect to the former suggestion, the direct inhibition of voltage-gated Ca2+ entry by CLT has been demonstrated in pituitary GH3 and in chromaffin cells, although again this effect may only occur secondary to depletion of intracellular Ca2+ stores (Alonso et al., 1991; Villalobos et al., 1992; Benzaquen et al., 1995). Such inhibition of cardiac ICa by CLT could also occur by the regulation of cytochrome P-450, due to changes in cyclic adenosine 3′5′-monophosphate (cyclic AMP) levels by P-450 mediated metabolites of arachidonic acid (Xiao et al., 1998).
Even if the inhibitory actions of CLT on ICa are considered to result from direct modulation of the Ca2+ channel protein itself, it still remains unclear whether this regulation occurs on the pore-forming α1C subunit itself, or is due to regulation of the α1C in combination with other auxiliary subunits. In addition, there are notable species differences in the responses of ICa recorded in cardiac myocytes to pharmacological modulators (compare, for example, Campbell et al., 1996; Lacampagne et al., 1995). Results obtained in other mammalian species cannot be assumed to be similar in human tissue (and therefore have potential clinical implications). In these studies we have examined the effects of CLT on the function of recombinant human cardiac L-type Ca2+ channel α1C subunit (Schultz et al., 1993) expressed alone without auxiliary subunits in human embryonic kidney (HEK 293) cells, and explored the mechanism underlying the effects of CLT on this channel. We demonstrate that CLT inhibits the α1C subunit in the absence of auxiliary subunits, and this inhibition is independent of any effect on [Ca2+]i or on cytochrome P-450 inhibition.
Methods
All experiments were carried out in HEK 293 cells stably expressing the human cardiac L-type calcium channel α1C subunit (Schultz et al., 1993). The construction of this cell line has been previously described (Fearon et al., 1997). Cells were grown in minimum essential medium with Earle's salts (Gibco, Paisley, U.K.), containing 9% (v v−1) foetal calf serum (Globepharm, Esher, Surrey, U.K.), 1% (v v−1) non-essential amino acids, gentamicin (50 mg l−1), 10,000 μl−1 penicillin G, 10 mg l−1 streptomycin, 0.25 mg l−1 amphotericin and 400 mg l−1 G418 (all Gibco) at 37°C in a humidified atmosphere of air/CO2 (19 : 1). Cells were harvested from their culture flasks by trypsinization and plated out onto coverslips 24–48 h before use in electrophysiological studies.
Pieces of coverslip with attached cells were transferred to a continually perfused (approximately 2 ml min−1) recording chamber (volume 80 μl) and whole-cell patch-clamp recordings (Hamill et al., 1981) were made using patch pipettes of resistance 4–7 MΩ. Cells were perfused with a solution composed of (in mmol l−1): NaCl 95, CsCl 5, MgCl2 0.6, BaCl2 20, HEPES 5, D-glucose 10, TEA-Cl 20 (21–24°C, pH 7.4) and patch electrodes were filled with a solution of composition (in mmol l−1): CsCl 120, TEA-Cl 20, MgCl2 2, EGTA 10, HEPES 10, ATP 2 (pH 7.2). Cells were voltage clamped at −80 mV, and whole-cell currents (termed IBa since Ba2+ was used as charge carrier) were evoked by step depolarizing the membrane to various test potentials for 100 ms at a frequency of 0.1 Hz. Series resistance compensation of 70–85% was applied. Current traces were filtered at 1 kHz, digitized at 2 kHz and stored on computer for later analysis. Capacitative transients were minimized by analogue means (residual transients have been truncated for illustrative purposes) and corrections for leak current were made by the appropriate scaling and subtraction of the average leak current evoked by small hyperpolarizing and depolarizing steps (⩽20 mV). Current amplitudes were measured over the last 10–15 ms of each step depolarization. All analysis and voltage protocols were performed using an Axopatch 200A amplifier in combination with a Digidata 1200 and pCLAMP 6.0.3 software (Axon Instruments).
To monitor [Ca2+]i in HEK 293 cells, they were pre-incubated for 1 h at 21–24°C in control solution (composition given below) containing 4 μmol l−1 Fura 2-AM. Samples were then placed in the perfusion chamber and changes in [Ca2+]i were indicated from the fluorescence emitted at 510 nm due to alternative excitation at 340 and 380 nm using Joyce Loebl PhoCal apparatus (Applied Imaging). For these studies, the control perfusate was of composition (in mmol l−1): NaCl 135, KCl 5, MgSO4 1.2, CaCl2 10, HEPES 5, and glucose 10 (pH 7.4, osmolarity adjusted to ca. 300 mOsm with sucrose, 21–24°C). Since calibration of fluorescence into absolute [Ca2+]i values can be subject to artefactual inaccuracies (Duchen, 1992), data are presented as ratio signals.
Clotrimazole (CLT; Sigma) and 1-aminobenzotriazole (1-ABT; Sigma) were prepared by dissolution in the extracellular perfusate. A stock solution of CLT (10 mmol l−1) was prepared in acetone before dilution to the final bath concentration. In experiments with 1-ABT cells were incubated for 1 h at room temperature in the drug solution immediately after dissolving the drug. Cells were then transferred to the recording chamber and recordings were made as described. Experiments with 1-ABT were carried out under low light intensity. In experiments using α-napthoflavone (Sigma) or H-7 (Tocris Cookson), cells were dialysed for 10 min with a pipette solution containing the compound at the required concentration. Following such dialysis, recordings were made as described above.
Results are expressed as means±s.e.mean, and statistical comparisons were made using paired or unpaired Student's t-tests, as appropriate.
Results
Inhibition of L-type Ca2+ channel α1C subunits by clotrimazole
All data presented here were obtained from a clonal cell line in HEK 293 cells stably expressing the human cardiac L-type calcium channel α1C subunit (Fearon et al., 1997). Figure 1A (representative of six such recordings), illustrates the inhibitory action of bath applied clotrimazole (CLT; 10 μmol l−1). Whilst cells were repeatedly step depolarized to +10 mV (holding potential −80 mV) to evoke inward barium currents (IBa), the extracellular perfusate was switched to one containing CLT. This always caused a reversible reduction in IBa; mean current reduction at this concentration of CLT was 70.2±3.0% (n=6). Typical current traces obtained before and during the application of CLT are illustrated in Figure 1B (left). Note that in the presence of CLT, currents displayed pronounced inactivation. In Figure 1B (right), the trace obtained following the application of 10 μmol l−1 CLT has been rescaled by the factor indicated, such that the peak current amplitude is the same as that obtained under control conditions. Clearly, the apparent rate of channel inactivation was increased by CLT. To quantify this effect, a single exponential curve was fitted to the inactivating section of current traces under control conditions and following the application of 10 μmol l−1 CLT. The time constant for this function (τinact) was significantly (P<0.002, Student's paired t-test) decreased following the application of CLT (τinact control, 273.2±41.2 ms, τinact CLT, 146.8±31.0 ms; n=13). The inhibitory effect of CLT was not due to inhibition of the channel by acetone (used as a solvent for CLT), since acetone (1 : 1000 v v−1) was without effect on IBa when applied alone in the extracellular perfusate (Figure 1C; representative of six cells examined).
Figure 1.

(A) Time-series plot of Ca2+ channel current amplitudes. Each plotted point represents the current amplitude evoked by repeated step depolarisations (100 ms, 0.1 Hz) to +10 mV from a holding potential of −80 mV. Amplitudes were measured over the last 10–15 ms of the depolarizing step. The period of exposure of cells to 10 μmol l−1 CLT is indicated by the horizontal bars. (B) Example current recordings taken from the time series in (A) obtained prior to (control) and following (CLT) a 3 min bath application of CLT. Right, current observed following CLT application has been scaled by multiplying the current trace by 2.17 such that the peak current amplitude is the same as that obtained under control conditions. (C) as in (A) except cells were perfused with a solution containing 1 : 000 (v v−1) acetone for the period indicated by the horizontal bar. Inset, individual traces taken from the time series prior to (control) and following (acetone) the bath application of acetone.
Current-voltage (I–V) relationships were constructed in nine cells following the application of 1 μmol l−1 CLT (Figure 2A). It was apparent that CLT inhibited currents to a similar degree at all activating test potentials studied, indicating a lack of voltage-dependence. The apparent shift in reversal potential is likely due to a background outward current, since when currents are fully blocked (e.g. with Cd2+), the leak conductance is outwardly rectifying (not shown). A concentration-response relationship was also determined for CLT (Figure 2B), measuring currents either at their peak (open symbols) or towards the end of each step depolarization (filled symbols). The plotted percentage of current inhibition was calculated following correction of current amplitudes for run-down (which was a commonly observed phenomenon; see Figure 1A) as previously described (Hatton & Peers, 1997). Inhibition could be detected at concentrations greater than 100 mmol l−1, and increased in a graded manner with increasing concentration, with currents almost completely inhibited at a concentration of 25 μmol l−1 (as measured at the end of the step depolarizations).
Figure 2.
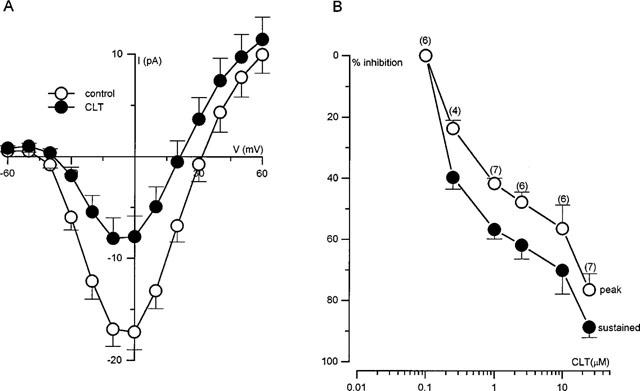
(A) Mean (with vertical s.e.m. bars) Ca2+ current density versus voltage relationships obtained before (n=9 cells) and after (n=9 cells) the bath application of 1 μmol l−1 CLT. (B) Concentration-dependent inhibition of IBa by CLT. Plot of percentage inhibition of Ca2+ current caused by bath application of a range of concentrations. Each point is the measured percentage inhibition with vertical s.e.m. bars taken from the number of cells indicated above each point. Inhibition was calculated from measurements of currents evoked by step depolarizations from −80 mV to +10 mV. Since currents evoked in the presence of CLT were transient in nature, current amplitudes were measured at their peaks and at the end of the depolarizing steps.
The above-described experiments were all conducted by bath applying CLT whilst cells were repeatedly step depolarized. Therefore, the inhibitory effect could have arisen by an interaction of CLT with a site on the channel protein accessible only whilst the channel was in its open configuration. To investigate this possibility, CLT (10 μmol l−1) was bath applied for 3 min whilst cells were held constantly at −80 mV. Following this period, current amplitudes evoked by step depolarizations were examined and compared to those obtained prior to the application of CLT. An immediate inhibition of channel activity was still seen when this depolarizing protocol was applied to cells (see Figure 3) and the inhibited currents also showed the accelerated inactivation described above. Mean percentage inhibition due to 10 μmol l−1 CLT, when applied without step depolarizations, was 72.8±5.4% (n=4), a value extremely similar to that seen in control cells when continually pulsed during the application of CLT (70.2±3.0%, n=6). This demonstrates that CLT does not require the channel to be in its open configuration in order to inhibit channel activity.
Figure 3.
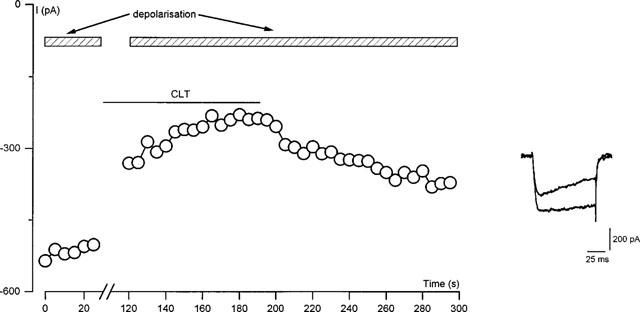
Inhibition by CLT is not use-dependent. Time-series plot of Ca2+ current amplitudes evoked in a representative HEK 293 cell by repeated step depolarizations (100 ms, 0.1 Hz) to +10 mV from a holding potential of −80 mV. Period of bath application of 10 μmol l−1 CLT is indicated by the lower horizontal bar. Cells were step depolarized for the period indicated by the upper horizontal bars. Inset, individual current traces from the time-series evoked before (control) and following (CLT) the bath application of CLT.
Lack of involvement of cytochrome P-450 in the inhibitory actions of CLT
To investigate possible mechanisms underlying the inhibitory effect of CLT, we first examined whether this effect was mediated by cytochrome P-450 inhibition, by comparing the actions of CLT with those of the suicide substrate P-450 inhibitor, 1-aminobenzotriazole (1-ABT). This compound is structurally unrelated to the imidazole antimycotics and inhibits P-450 by a completely different mechanism of action (Murray & Reidy, 1990; Halpert, 1995). Following incubation of cells for 1 h in 3 mmol l−1 1-ABT (Mathews et al., 1985), basal current density (determined by step depolarizing cells from −80 to +10 mV) was 9.3±2.4 pA pF−1 (n=6), a value not significantly different to that seen in unincubated controls (14.6±3.7 pA pF−1, n=6; P>0.08, Student's unpaired t-test). Furthermore following such incubation, bath application of 10 μmol l−1 CLT still caused a reduction in channel activity (Figure 4), and this reduction was also associated with an acceleration of channel inactivation (e.g. Figure 4, lower inset). The mean degree of inhibition by CLT was 71.7±2.8% (n=6), a value not significantly different to the degree of inhibition seen at this concentration in untreated controls (70.2±3.0%, n=6; P>0.2, unpaired Student's t-test). To further substantiate the finding that the effects of CLT are independent of an effect on cytochrome P-450, cells were dialysed for 10 min with a pipette solution containing 2 mmol l−1 α-napthoflavone, a structurally distinct inhibitor of the cytochrome (Alvarez et al., 1991). Basal current density, evoked by a step depolarization from −80 to +10 mV, was 11.7±4.8 pA pF−1 (n=6) following dialysis with α-napthoflavone, a value not significantly different to that seen in cells dialysed for 10 min with a drug-free pipette solution (14.6±3.7 pA pF−1, n=6; P>0.06, unpaired Student's t-test). Furthermore, cell dialysis with this P-450 inhibitor was without effect on the ability of 10 μmol l−1 CLT to reduce calcium channel activity. The mean degree of inhibition in untreated control cells was 70.2±3.0% (n=6); following dialysis with α-napthoflavone inhibition was 65.9±2.7% (n=6; P>0.06, unpaired Student's t-test).
Figure 4.
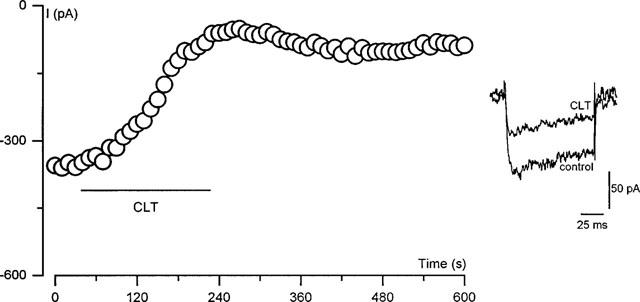
Inhibition of IBa due to CLT is independent of effects on cytochrome P-450. Each plotted point represents the current amplitude evoked by repeated step depolarizations (100 ms, 0.1 Hz) to +10 mV from a holding potential of −80 mV. Cells were pre-incubated in 3 mmol l−1 1-ABT for 1 h prior to recording. The period of exposure of cells to 10 μmol l−1 CLT is indicated by the horizontal bar. Inset shows individual traces obtained before (control) and after the application (CLT) of CLT.
CLT does not alter [Ca2+]i homeostasis
It was noteworthy that the inhibitory actions of CLT were associated with an accelerated inactivation (e.g. Figure 1). This raised the possibility that inhibition arose due to CLT causing release of Ca2+ from intracellular stores, since Ca2+-dependent inactivation of Ca2+ currents is similarly associated with accelerated inactivation (see e.g. Eckert & Tillotson, 1981). Further experiments were therefore carried out to investigate whether changes in [Ca2+]i were involved in the inhibitory effect of CLT on IBa. As exemplified in Figure 5 (representative of six such recordings), the bath application 10 μmol l−1 CLT was without effect on [Ca2+]i levels, as determined by Fura-2 fluorescence. This demonstrates that the reduction of IBa due to CLT is not secondary to a rise of [Ca2+]i due to release from intracellular stores. To demonstrate that [Ca2+]i signals were obtainable from these cells, exposure to solutions containing 100 mmol l−1 K+ (Na+ substitution) evoked large, reversible rises of [Ca2+]i.
Figure 5.
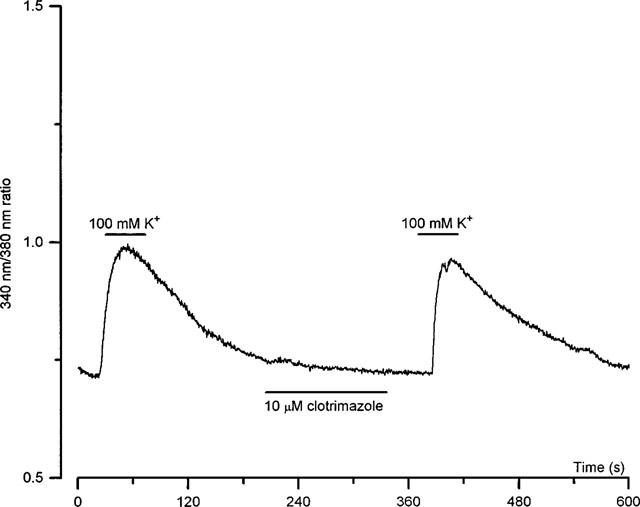
Lack of effect of CLT on [Ca2+]i in HEK 293 cells. Representative continuous recording of [Ca2+]i (expressed as the uncalibrated 340 nm/380 nm ratio) in a field of HEK 293 cells stably expressing the L-type Ca2+ channel α1C subunit. For the periods indicated by the horizontal bars, the cells were exposed to a perfusate containing either 100 mmol l−1 K+, or 10 μmol l−1 clotrimazole, as indicated. This example is representative of six such recordings.
CLT inhibition of Ca2+ channel activity is not due to modulation of protein kinases
It is well documented that Ca2+ channels can be modulated by a wide range of serine and threonine kinases, in both native tissue and in recombinant expression systems (Hosey et al., 1996; Sperelakis et al., 1996). It therefore remains possible that the inhibitory effect of CLT on the Ca2+ channel is due to indirect modulation of such kinases. To investigate this, cells were dialysed for 10 min with the non-specific serine/threonine kinase inhibitor H-7 (10 μmol l−1; Hidaka et al., 1984) via the patch pipette. Following this incubation, basal current density was −12.35±2.1 pA pF−1 (n=6), a value not significantly different to that seen in unincubated controls (14.6±3.7 pA pF−1, n=6; P>0.6). Moreover, dialysis with H-7 was without effect on the ability of 10 μmol l−1 CLT to inhibit Ca2+ channel activity. The mean degree of inhibition following such dialysis was 62.4±8.0% (n=6), a value not significantly different to that seen in controls (70.2±3.0%, n=6; P>0.6, unpaired Student's t-test).
Discussion
The present study demonstrates that exposure of stably transfected HEK 293 cells to CLT results in a reduction in IBa through the recombinant human cardiac L-type Ca2+ channel α1C subunit. This inhibitory effect was associated with an acceleration of channel inactivation kinetics, an effect reported previously for CLT on native Ca2+ channels in pancreatic β-cells (Welker & Drews, 1997). In order to inhibit the channel, CLT did not require the channel to be in its open state, since inhibition could be attained whilst cells were held closed at a constant membrane potential in the presence of CLT. Furthermore, the effects of CLT were not voltage-dependent, since a similar degree of current inhibition was seen at all activating test potentials (Figure 2). These inhibitory effects are in good agreement with the effects of CLT recently described on native L-type Ca2+ channels in guinea-pig ventricular myocytes (Thomas et al., 1999), an effect which may be responsible for the shortening of the cardiac action potential duration in these cells. It is, however, noteworthy that the inhibitory action of CLT described here is more than 10 fold more potent than its effect in guinea-pig myocytes. Whether this is due to species variation (human vs guinea-pig), or to the absence of auxiliary subunits in the present study, remains to be determined.
The effects of CLT on the cytochrome P-450 enzyme system are well documented (Sheets et al., 1986; McGiff, 1991). A recent study has demonstrated the involvement of this system in mediating the effects of CLT on native cardiac L-type Ca2+ channels (Xiao et al., 1998). The effect was mediated by modulation of intracellular levels of cyclic AMP by P-450 mediated metabolites of arachidonic acid. The role of the cyclic AMP-dependent protein kinase in regulating cardiac L-type Ca2+ channel activity is also well documented (see McDonald et al., 1994), and a reduction in intracellular cyclic AMP levels by CLT would cause inhibition of such channel activity. In the studies presented here, a role for cytochrome P-450 in regulating the Ca2+ channel can be discounted since incubation of cells for 1 h in 1-ABT, a structurally dissimilar P-450 inhibitor (Murray & Reidy, 1990; Halpert, 1995), was without effect on basal Ca2+ channel activity. More importantly, following this incubation, CLT was still able to exert its inhibitory effect on channel function (Figure 3). This conclusion discounting the involvement of cytochrome P-450 in mediating the effects of CLT is further supported by the finding that following cell dialysis with the P-450 inhibitor α-napthoflavone, both the basal current density and the inhibitory response to CLT were unaffected. This compound has been previously shown to modulate plasma membrane Ca2+ channels in rat thymocytes via a P-450 dependent mechanism at the concentration used in our studies (Alvarez et al., 1991).
In the present study, the effects of CLT were examined in cells expressing the pore-forming α1C subunit alone, in the absence of any auxiliary subunits. Though this subunit is an in-vitro substrate for the cyclic AMP-dependent protein kinase (Yoshida et al., 1992; Gerhardstein et al., 1996), there is conflicting functional evidence that this subunit can be modulated by cyclic AMP-dependent phosphorylation when expressed alone (Hosey et al., 1996); the presence of the auxiliary β subunit is required for such phosphorylation to occur (Klockner et al., 1992). This evidence argues further against a role for cytochrome P-450 in mediating the effects of CLT on this channel. The present study further rules out a non-specific effect of CLT on cellular protein kinases which may regulate the α1C subunit since cell dialysis with the non-specific serine/threonine protein kinase inhibitor H-7 (Hidaka et al., 1984) was without effect on either basal Ca2+ channel function or on the inhibitory response to CLT.
The inhibition of plasmalemmal voltage-gated Ca2+ channels by CLT has been demonstrated in both chromaffin and pituitary GH3 cells (Villalobos et al., 1992; Wu et al., 1999) and also in pancreatic β-cells (Welker & Drews, 1997), an effect which may be attributable to the regulation of [Ca2+]i by the antimycotic agent (Alonso et al., 1991; Montero et al., 1991; Alvarez et al., 1992; Sargeant et al., 1992; Benzaquen et al., 1995). Such a rise in [Ca2+]i could have explained the inhibitory effects of CLT reported here, since elevation of [Ca2+]i is well known to have an inhibitory effect on Ca2+ channel function (Brehm & Eckert, 1978; Tillotson, 1979; Kramer et al., 1991). However, our microfluorimetric studies (Figure 5) indicated that CLT, at a concentration seen to cause around 70% inhibition of Ca2+ channel activity in the voltage clamp experiments, had no effect on [Ca2+]i in HEK 293 cells. This lack of effect was observed despite the fact that cells were perfused with a solution containing 10 mmol l−1 Ca2+. Furthermore, in the electrophysiological studies described here, all recordings were made whilst dialysing cells with a high concentration of the Ca2+ chelator EGTA, and the inhibitory actions of CLT were sustained as long as the drug was present. Indeed, CLT could be applied repeatedly to the same cell with no loss in the degree of inhibition observed (Figure 1A). This would not be the case if CLT discharged intracellular Ca2+ stores since we used Ba2+ as charge carrier during patch-clamp recordings, and stores would therefore have no means of refilling in the absence of extracellular Ca2+. Taken together, these data demonstrate that the inhibitory effects of CLT are unlikely to be mediated by modulation of cellular Ca2+ homeostasis.
The findings of this study point to a direct action of CLT on the expressed α1C subunit to reduce channel activity and accelerate channel inactivation during the depolarizing step. CLT could act as an open channel blocker since in its presence the initial rate of channel activation appeared unchanged (see e.g. Figure 1B), but as the channels open they become blocked and currents do not reach the amplitudes seen under control conditions. Block of the channel could then continue during the depolarizing step such that increasing numbers of channels become blocked, giving the reduced current amplitudes observed throughout the rest of the depolarization.
To summarize, CLT causes a reduction in inward Ca2+ channel current through the recombinant human cardiac L-type Ca2+ channel α1C subunit, independently of either cytochrome P-450 or modulation of intracellular Ca2+ levels, and does not require the presence of auxiliary subunits. This suggests that CLT causes blockade of the expressed Ca2+ channel by a direct effect on the pore-forming α1C subunit itself. Further studies are required in order to determine whether such a direct action on the Ca2+ channel protein is responsible for the shortening of the cardiac action potential observed previously in isolated myocytes.
Acknowledgments
This work was supported by the British Heart Foundation.
Abbreviations
- 1-ABT
1-aminobenzotriazole
- CLT
clotrimazole
References
- ALONSO M.T., ALVAREZ J., MONTERO M., SANCHEZ A., GARCIA-SANCHO J. Agonist-induced Ca2+ influx into human platelets is secondary to the emptying of intracellular Ca2+ stores. Biochem. J. 1991;280:783–789. doi: 10.1042/bj2800783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVAREZ J., MONTERO M., GARCIA-SANCHO J. Cytochrome P-450 may link intracellular Ca2+ stores with plasma membrane Ca2+ influx. Biochem. J. 1991;274:193–197. doi: 10.1042/bj2740193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVAREZ J., MONTERO M., GARCIA-SANCHO J. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P-450 inhibitors. J. Biol. Chem. 1992;267:11789–11793. [PubMed] [Google Scholar]
- AYUB M., LEVELL M.J. Structure-activity relationships of the inhibition of human placental aromatase by imidazole drugs including ketoconazole. J. Steroid. Biochem. 1988;31:65–72. doi: 10.1016/0022-4731(88)90207-5. [DOI] [PubMed] [Google Scholar]
- BENZAQUEN L.R., BRUGNARA C., BYERS H.R., GATTON-CELLI S., HALPERIN J.A. Clotrimazole inhibits cell proliferation in vitro and in vivo. Nature Med. 1995;1:534–540. doi: 10.1038/nm0695-534. [DOI] [PubMed] [Google Scholar]
- BREHM P., ECKERT R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- BRUGNARA C., ARMSBY C.C., SAKAMOTO M., RIFAI N., ALPER S.L., PLATT O. Oral administration of clotrimazole and blockade of human erythrocyte Ca2+-activated K+ channel: the imidazole ring is not required for inhibitory activity. J. Pharmacol. Exp. Ther. 1995;273:266–272. [PubMed] [Google Scholar]
- BRUGNARA C., DE FRANCESCHI L., ALPER S.L. Inhibition of Ca2+-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Invest. 1993;92:520–526. doi: 10.1172/JCI116597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUGNARA C., GEE B., ARMSBY C.C., KURTH S., SAKAMOTO M., RIFAI N., ALPER S.L., PLATT O.S. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J. Clin. Invest. 1996;97:1227–1234. doi: 10.1172/JCI118537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPBELL D.L., STAMLER J.S., STRAUSS H.C. Redox modulation of L-type calcium channels in ferret ventricular myocytes–dual mechanism regulation by nitric-oxide and s-nitrosothiols. J. Gen. Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUPRY I., ARMSBY C.C., ALPER S.L., BRUGNARA C., PARINI A. Clotrimazole and efaroxan inhibit red cell Gardos channel independently of imidazoline I1 and I2 binding sites. Eur. J. Pharmacol. 1996;295:109–112. doi: 10.1016/0014-2999(95)00642-7. [DOI] [PubMed] [Google Scholar]
- DE FRANCESCHI L., ROUYER-FESSARD P., ALPER S.L., JOUAULT H., BRUGNARA C., BEUZARD Y. Combination therapy of erythropoietin, hydroxyurea, and clotrimazole in a beta thalassemic mouse: a model for human therapy. Blood. 1996;87:1188–1195. [PubMed] [Google Scholar]
- DUCHEN M.R.Fluorescence–Monitoring cell chemistry in vivo Monitoring Neuronal Activity; A Practical Approach 1992Oxford University Press: Oxford; 231–260.Stamford, J.A. (ed.) [Google Scholar]
- ECKERT R., TILLOTSON D.L. Calcium-mediated inactivation of the calcium conductance in caesium-loaded giant neurones of Aplysia californica. J. Physiol. 1981;314:265–280. doi: 10.1113/jphysiol.1981.sp013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEARON I.M., PALMER A.C.V., BALMFORTH A.J., BALL S.G., MIKALA G., SCHWARTZ A., PEERS C. Hypoxia inhibits the recombinant α1C subunit of the human cardiac L-type Ca2+ channel. J. Physiol. 1997;500:551–556. doi: 10.1113/jphysiol.1997.sp022041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERHARDSTEIN B., PURI T., ZHAO X.L., HOSEY M. In-vitro phosphorylation of subunits of cardiac L-type Ca channels by protein kinase A and protein kinase C. Biophys. J. 1996;70:A186. [Google Scholar]
- HALPERT J.R. Structural basis of selective cytochrome P-450 inhibition. Ann. Rev. Pharmacol. Toxicol. 1995;35:29–53. doi: 10.1146/annurev.pa.35.040195.000333. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv–Eur. J. Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HATTON C.J., PEERS C. Effects of cytochrome P-450 inhibitors on ionic currents in isolated rat type I carotid body cells. Am. J. Physiol. 1996;271:C85–C92. doi: 10.1152/ajpcell.1996.271.1.C85. [DOI] [PubMed] [Google Scholar]
- HATTON C.J., PEERS C. Multiple effects of nordihydroguaiaretic acid on ionic currents in rat isolated type I carotid body cells. Br. J. Pharmacol. 1997;122:923–929. doi: 10.1038/sj.bjp.0701452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIDAKA H., INAGAKI M., KAWAMOTO S., SASAKI Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- HOSEY M.M., CHIEN A.J., PURI T.S. Structure and regulation of L-type calcium channels. A current assessment of the properties and roles of channel subunits. Trends Cardiovasc. Med. 1996;6:265–273. doi: 10.1016/S1050-1738(96)00109-0. [DOI] [PubMed] [Google Scholar]
- ISHII T.M., SILVIA C., HIRSCHBERG B., BOND C.T., ADELMAN J.P., MAYLIE J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLOCKNER U., ITAGAKI K., BODI I., SCHWARTZ A. β-subunit expression is required for cAMP-dependent increase of cloned cardiac and vascular calcium channel currents. Pflugers Arch–Eur. J. Physiol. 1992;420:413–415. doi: 10.1007/BF00374479. [DOI] [PubMed] [Google Scholar]
- KRAMER R.H., KACZMAREK L.K., LEVITAN E.S. Neuropeptide inhibition of voltage-gated calcium channels mediated by mobilization of intracellular calcium. Neuron. 1991;6:557–563. doi: 10.1016/0896-6273(91)90058-8. [DOI] [PubMed] [Google Scholar]
- LACAMPAGNE A., DUITTOZ A., BOLANOS P., PEINEAU N., ARGIBAY J.A. Effect of sulfhydryl oxidation on ionic and gating currents associated with L-type calcium channels in isolated guinea-pig ventricular myocytes. Cardiovasc. Res. 1995;30:799–806. [PubMed] [Google Scholar]
- MATHEWS J.M., DOSTAL L.A., BEND J.R. Inactivation of rabbit pulmonary cytochrome P-450 in microsomes and isolated perfused lungs by the suicide substrate 1-aminobenzotriazole. J. Pharmacol. Exp. Ther. 1985;235:186–190. [PubMed] [Google Scholar]
- MCDONALD T.F., PELZER S., TRAUTWEIN W., PELZER D.J. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MCGIFF J.C. Cytochrome P-450 metabolism of arachidonic acid. Ann. Rev. Pharmacol. Toxicol. 1991;31:339–369. doi: 10.1146/annurev.pa.31.040191.002011. [DOI] [PubMed] [Google Scholar]
- MONTERO M., ALVAREZ J., GARCIA-SANCHO J. Agonist-induced Ca2+ influx in human neutrophils is secondary to the emptying of intracellular calcium stores. Biochem. J. 1991;2:73–79. doi: 10.1042/bj2770073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY M., REIDY G.F. Selectivity in the inhibition of mammalian cytochromes P-450 by chemical agents. Pharmacol. Rev. 1990;42:85–101. [PubMed] [Google Scholar]
- SARGEANT P., CLARKSON W.D., SAGE S.O., HEEMSKERK J.W. Calcium influx evoked by Ca2+ store depletion in human platelets is more susceptible to cytochrome P-450 inhibitors than receptor-mediated calcium entry. Cell Calcium. 1992;13:553–564. doi: 10.1016/0143-4160(92)90035-q. [DOI] [PubMed] [Google Scholar]
- SCHULTZ D., MIKALA G., YATANI A., ENGLE D.B., ILES D.E., SEGER B., SINKE R.J., WEGHUIS D.O., KLOCKNER U., WAKAMORI M., WANG J.J., MELVIN D., VARADI G., SCHWARTZ A. Cloning, chromosomal localization, and functional expression of the α1 subunit of the L-type voltage-dependent calcium channel from normal human heart. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6228–6232. doi: 10.1073/pnas.90.13.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEETS J.J., MASON J.I., WISE C.A., ESTABROOK R.W. Inhibition of rat liver microsomal cytochrome P-450 steroid hydroxylase reactions by imidazole antimycotic agents. Biochem. Pharmacol. 1986;35:487–491. doi: 10.1016/0006-2952(86)90224-8. [DOI] [PubMed] [Google Scholar]
- SPERELAKIS N., KATSUBE Y., YOKOSHIKI H., SADA H., SUMII K. Regulation of the slow Ca++ channels of myocardial cells. Mol. Cell. Biochem. 1996;164:85–98. doi: 10.1007/BF00408644. [DOI] [PubMed] [Google Scholar]
- THOMAS G.P., KARMAZYN M., ZYGMUNT A.C., ANTZELEVITCH C., NARAYANAN N. The antifungal antibiotic clotrimazole potently inhibits L-type calcium current in guinea-pig ventricular myocytes. Br. J. Pharmacol. 1999;126:1531–1533. doi: 10.1038/sj.bjp.0702475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TILLOTSON D. Inactivation of Ca conductance dependent on entry of Ca ions in molluscan neurones. Proc. Natl. Acad. Sci. U.S.A. 1979;76:1497–1500. doi: 10.1073/pnas.76.3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLALOBOS C., FONTERIZ R., LOPEZ M.G., GARCIA A.G., GARCIA-SANCHO J. Inhibition of voltage-gated Ca2+ entry into GH3 and chromaffin cells by imidazole antimycotics and other cytochrome P450 blockers. FASEB J. 1992;6:2742–2747. doi: 10.1096/fasebj.6.9.1319362. [DOI] [PubMed] [Google Scholar]
- WELKER S., DREWS G. Imidazole antimycotics affect the activity of various ion channels and insulin secretion in mouse pancreatic B-cells. Naunyn-Schmiedebergs Arch. Pharmacol. 1997;356:543–550. doi: 10.1007/pl00005089. [DOI] [PubMed] [Google Scholar]
- WU S.N., LI H.F., JAN C.R., SHEN A.Y. Inhibition of Ca2+ activated K+ current by clotrimazole in rat anterior pituitary GH3 cells. Neuropharmacology. 1999;38:979–989. doi: 10.1016/s0028-3908(99)00027-1. [DOI] [PubMed] [Google Scholar]
- XIAO Y.F., HUANG L., MORGAN J.P. Cytochrome P450: a novel system modulating Ca2+ channels and contraction in mammalian heart cells. J. Physiol. 1998;508:777–792. doi: 10.1111/j.1469-7793.1998.777bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIDA A., TAKAHASHI M., NISHIMURA S., TAKESHIMA H., KOKUBUN S. Cyclic AMP-dependent phosphorylation and regulation of the cardiac dihydropyridine-sensitive Ca channel. FEBS Lett. 1992;309:343–349. doi: 10.1016/0014-5793(92)80804-p. [DOI] [PubMed] [Google Scholar]