

In a recent article Colquhoun (1998) showed how measurements of agonist affinity at receptors were difficult to interpret owing the conformational change that occurs in the receptor upon agonist binding. It is often assumed, however, for the G-protein coupled receptors that measurements of agonist affinity in the presence of guanine nucleotides (which uncouple receptor and G-protein) represent ground state affinities. As I have argued elsewhere (Strange 1998, 1999) this is unlikely to be true and agonist binding, even to the uncoupled receptor, may involve a conformational change. This can be represented in the scheme shown in Figure 1 where the agonist binds with different affinities to different states of the receptor, R and R*, the latter being the species that couples to the G-protein (Samama et al., 1993). Agonist affinities observed (Kd) will not be ground state affinities and will depend on KA, KA* and KR as in equation 1. It is important to determine values for the different equilibrium constants and in principle this is possible if mutant receptors are available locked in to the R* or R states.
Mutants favouring the R* state have been reported for several receptors although only in the case of the β2-adrenergic receptor have these been fully characterized (Lefkowitz et al., 1993; Samama et al., 1993). These show increased agonist affinities independent of G-protein coupling, increased functional potencies for agonists to activate signalling systems and increased agonist-independent activation of signalling systems. There is also some increase in the ability of the receptor to couple to G-proteins so this mutation is not entirely clean. In these mutant receptors KR (Figure 1) will be favourable and Kd will approximate to 1/KA*.
Figure 1.
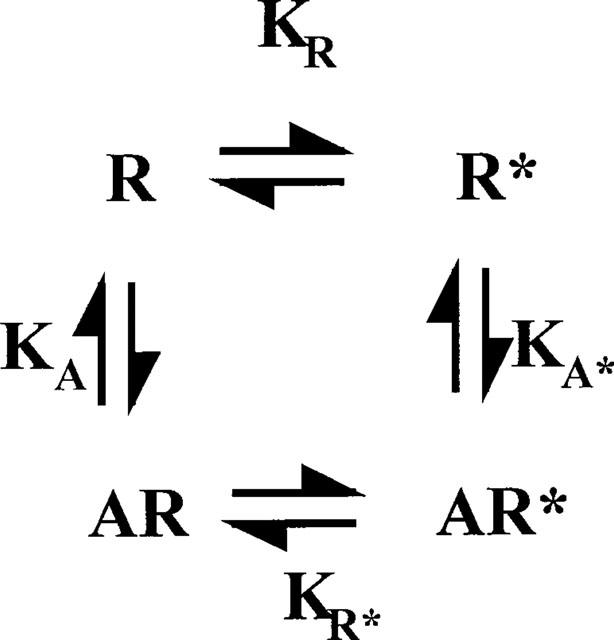
Agonist binding to G-protein coupled receptors under conditions where coupling to G-proteins is suppressed. Receptor exists in a ground state (R) and a partially activated state (R*), that can normally couple to the G-protein to form R*G as in Samama et al., 1993. KA and KA* are association constants for binding of the agonist to R and R* and the observed equilibrium dissociation constant for agonist binding (Kd) is given by equation 1.
No explicit description of a mutant receptor locked in to the R state has been made but mutants in the conserved aspartate about two-thirds down TMII may provide such a model. This amino acid residue is one of the most conserved in all the GPCR's (Van Rhee & Jacobsen, 1996) suggesting an important structural role or a role in the activation of the receptor. The residue has been mutated to Ala or Asn in several receptors (references in Van Rhee & Jacobsen, 1996 and Chung et al., 1988; Fraser et al., 1990; Horstman et al., 1990; Neve et al., 1991; Wang et al., 1991; 1993). In the mutant receptors agonists are unable (except in two cases) to activate signalling systems and, where this has been measured, coupling of receptor to G-protein is impaired and the regulation of agonist binding by sodium ions prevented. These characteristics are those of a receptor that is unable to form the activated state and indeed it has been suggested that the TMII Asp is important in receptor activation for forming a salt bridge with the Arg at the base of TMIII released from the AspArgTyr sequence following the protonation of the TMIII Asp (Ballasteros et al., 1998). This mutant receptor is then a candidate for a receptor that cannot undergo the R/R* transition and so should exhibit a decrease in agonist affinity independent of G-protein coupling i.e. KR is unfavourable and Kd reduces to KA (Equation 1). The effects seen may be quite small if the extent to which R* is favoured in the presence of the agonist is not great and this may account for some of the variability in results reported (see below). Nevertheless, if this speculation about the role of the TMII Asp is correct then the effects on agonist affinity of the mutation of the TMII Asp should be a rough index of the extent to which conformational effects can contribute to the effects of other mutations of the receptor.
There are difficulties in interpreting some of the data on the TMII Asp mutants in the literature as the characterization of the mutants is not extensive, usually involving only one or two agonists. In some cases the mutation causes a modest decrease in agonist affinity under conditions where G-protein coupling has been suppressed, in other cases there is no effect and sometimes the affinity is slightly increased. This has been characterized in two studies for the β2-adrenergic receptor (Strader et al., 1987; 1988; Chung et al., 1988) and the affinity for the full agonist, isoprenaline, is lower (∼9 fold) in the mutant in the absence of G-protein coupling as predicted by the theory proposed above. For this receptor, it is possible to estimate values of KA, KA*, KR, KR* as follows. As stated above, in the TMII Asp mutant the R* state will be disfavoured and agonist affinity will approximate KA. In the constitutively active mutant receptors R* will be strongly favoured and agonist affinity will approximate KA*. These values may then be used in Equation 1 with the affinity for agonist at the native receptor to determine values for the different equilibrium constants. For the full agonist isoprenaline (average native Kd 165 nM (Chung et al., 1988; Samama et al., 1993); average effect of TMII Asp mutation 9.06 fold reduction in affinity (Strader et al., 1987; 1988; Chung et al., 1988); effect of constitutively active mutation 25 fold increase in affinity (Samama et al., 1993)) these values are: KA (6.7 105 M−1), KA* (1.52108 M−1), KR (0.037), KR* (8.39). The value for Kd for the native receptor is 165 nM (equivalent to 6.05 106 M−1) so that there is a discrepancy of ∼9 fold between the affinity of the native receptor and the affinity of the ground state.
This calculation shows that for the β2-adrenergic receptor the observed Kd for a full agonist may not be a true measure of the affinity of the agonist for binding to the ground state of the receptor and that there is a contribution from the R/R* transition. The extent of this problem for other GPCR's is unclear at present and further work needs to be done on this. If discrepancies are found between determinations of Kd and ground state affinities in other GPCR's then this has implications for the interpretation of the results of mutations of residues that are thought to interact directly with ligands. In the case of the mutation of a residue that may form a hydrogen bond to the ligand the energetic effect of the deletion of the hydrogen bond is expected to be a reduction of up to ∼20 fold in affinity (Strange, 1996). If the mutation were to impair the R/R* transition then this effect could contribute up to ∼10 fold to affinity changes based on the above calculation. Given that the ligand/receptor interaction and R/R* transition are likely to be linked events it will be difficult to disentangle the individual contributions to the overall affinity change. If, however, the effects of the mutation were evaluated in a receptor locked in to the R* state (using the mutations considered earlier) then the size of the effect would reflect deletion of the hydrogen bond alone. Interpretation of mutation of a hydrophobic interaction may be easier as such a mutation should reduce binding affinity by up to 104 fold (Strange, 1996). In this case effects on the conformation of the receptors will be small by comparison.
References
- BALLASTEROS J., KITANOVIC S., GUARNIERI F., DAVIES P., FROMME B.J., KONVICKA K., CHI L., MILLAR R.P., DAVIDSON J.S., WEINSTEIN H., SEALFON S.C. Functional microdomains in G-protein coupled receptors. J. Biol. Chem. 1998;273:10445–10453. doi: 10.1074/jbc.273.17.10445. [DOI] [PubMed] [Google Scholar]
- CHUNG F., WANG C., POTTER P.C., VENTER J.C., FRASER C.M. Site directed mutagenesis and continuous expression of the human β-adrenergic receptor. J. Biol. Chem. 1988;263:4052–4055. [PubMed] [Google Scholar]
- COLQUHOUN D. Binding, gating and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRASER C.M., WANG C., ROBINSON D.A., GOCAYNE J.D., VENTER J.C. Site directed mutagenesis of m1 muscarinic acetylcholine receptors: conserved aspartic acids play important roles in receptor function. Mol. Pharmacol. 1990;36:840–847. [PubMed] [Google Scholar]
- HORSTMAN D.A., BRANDON S., WILSON A.L., GUYER C.A., CRAGO E.J., LIMBIRD L.E. An aspartate conserved among G-protein coupled receptors confers allosteric regulation of α2 adrenergic receptors by sodium ions. J. Biol. Chem. 1990;265:21590–21595. [PubMed] [Google Scholar]
- LEFKOWITZ R.J., COTECCHIA S., SAMAMA P., COSTA T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol. Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- NEVE K.A., COX B.A., HENNINGSEN R.A., SPANOYANNIS A., NEVE R.L. Pivotal role for aspartate 80 in the regulation of dopamine D2 receptor affinity for drugs and inhibition of adenylyl cyclase. Mol. Pharmacol. 1991;39:733–739. [PubMed] [Google Scholar]
- SAMAMA O., COTECCHIA S., COSTA T., LEFKOWITZ R.J. A mutation induced activated state of the β2-adrenergic receptor. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- STRADER C.D., SIGAL I.S., CANDELORE M.R., RANDS E., HILL W.S., DIXON R.A.F. Conserved aspartic acid residues 79 and 113 of the β-adrenergic receptor have different roles in receptor function. J. Biol. Chem. 1988;263:10267–10271. [PubMed] [Google Scholar]
- STRADER C.D., SIGAL I.S., REGISTER R.B., CANDELORE M.R., RANDS E., DIXON R.A.F. Identification of residues required for ligand binding to the β-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 1987;84:4384–4388. doi: 10.1073/pnas.84.13.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRANGE P.G. The energetics of ligand binding at catecholamine receptors. Trends. Pharmacol. Sci. 1996;17:238–244. doi: 10.1016/0165-6147(96)10025-0. [DOI] [PubMed] [Google Scholar]
- STRANGE P.G. Three state and two state models. Trends. Pharmacol. Sci. 1998;19:85–86. doi: 10.1016/s0165-6147(98)01175-4. [DOI] [PubMed] [Google Scholar]
- STRANGE P.G. G-protein coupled receptors: conformations and states. Biochem. Pharmacol. 1999;58:1081–1088. doi: 10.1016/s0006-2952(99)00144-6. [DOI] [PubMed] [Google Scholar]
- VAN RHEE A.M., JACOBSEN K.A. Molecular architecture of G-protein coupled receptors. Drug Dev. Res. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG C., BUCK M.A., FRASER C.M. Site directed mutagenesis of α2A adrenergic receptors: identification of amino acids involved in ligand binding and receptor activation by agonist. Mol. Pharmacol. 1991;40:168–179. [PubMed] [Google Scholar]
- WANG C., GALLAGHER T.K., SHIH J.C. Site directed mutagenesis of the serotonin 5HT2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol. Pharmacol. 1993;43:931–940. [PubMed] [Google Scholar]