

Abstract
The effects of chloroethylclonidine on α1-adrenoceptor-mediated contraction in endothelium-denuded caudal arteries and aorta from normotensive Wistar and Wistar Kyoto (WKY), and from spontaneously hypertensive (SHR) rats were evaluated.
Chloroethylclonidine elicited concentration-dependent contractions. Maximal contraction was similar in caudal arteries among strains (≈40% of noradrenaline effect). However, chloroethylclonidine elicited a higher contraction in aorta from SHR than from normotensive rats. In Wistar aorta chloroethylclonidine produced the smallest contractile response.
In SHR aorta, BMY 7378 and 5-methylurapidil blocked chloroethylclonidine-elicited contraction, while (+)-cyclazocine did not inhibit it; while in caudal arteries, 5-methylurapidil blocked chloroethylclonidine action; the other antagonists had no effect.
In chloroethylclonidine-treated aorta noradrenaline elicited biphasic contraction-response curves, indicating a high affinity (pD2, 8.5–7.5) chloroethylclonidine-sensitive component and a low affinity (pD2, 6.3–5.2) chloroethylclonidine-insensitive component. The high affinity component was blocked by chloroethylclonidine; while in caudal arteries noradrenaline elicited monophasic contraction-response curves with pD2 values (6.5–5.7) similar to the low affinity component in aorta.
Chloroethylclonidine inhibition of noradrenaline response was greater in aorta than in caudal arteries. Chloroethylclonidine increased the EC50 values of noradrenaline ≈1000 fold in aorta and ≈10 fold in caudal arteries.
In SHR aorta BMY 7378 protected α1D-adrenoceptors and in caudal arteries 5-methylurapidil protected α1A-adrenoceptors from chloroethylclonidine alkylation, allowing noradrenaline to elicit contraction.
These results show marked strain-dependent differences in the ability of chloroethylclonidine to contract aorta; moreover, chloroethylclonidine stimulates α1D-adrenoceptors in aorta and α1A-adrenoceptors in caudal arteries. The higher contraction observed in aorta from SHR and WKY suggests an augmented number of α1D-adrenoceptors in these strains.
Keywords: α1D-adrenoceptors, α1A-adrenoceptors, aorta, caudal artery, chloroethylclonidine, spontaneously hypertensive rats, Wistar rats
Introduction
Chloroethylclonidine has been considered a selective α1B-adrenoceptor irreversible antagonist, since it distinguishes between α1A- and α1B-adrenoceptors (Han et al., 1987; Minneman et al., 1988). However, it is now clear that chloroethylclonidine binds to all subtypes of α1- and α2-adrenoceptors, although α1A- and α2B-adrenoceptors are relatively insensitive to irreversible inactivation (Michel et al., 1993). The preferential alkylation of some α1- and α2-adrenoceptors is not due to an affinity difference of chloroethylclonidine for any of these sites, since the compound competes with radioligands for α1-adrenoceptors with affinities of 10–20 μM, and 1–3 μM for α2 sites (Michel et al., 1993), but rather to receptor accessibility in the cell membrane (Hirasawa et al., 1997).
Chloroethylclonidine was originally introduced as an irreversible α-adrenoceptor agonist, that produced phentolamine-sensitive contraction in rat aorta (Leclerc et al., 1980). However, most of the studies did not report this chloroethylclonidine-elicited contraction (Han et al., 1990; Oriowo & Ruffolo, 1992; O'Rourke et al., 1995; 1997; Piascik et al., 1997), even though direct contractions have been reported (Muramatsu et al., 1990; 1991). In addition, chloroethylclonidine produces a sustained contraction in the dog saphenous vein (Nunes & Guimaraes, 1993), rat vas deferens (Bultmann & Starke, 1993) and caudal artery (Piascik et al., 1995), by activation of α2-adrenoceptors. Therefore, the effects of chloroethylclonidine on α-adrenoceptor function have been mainly related to its ability to alkylate and inactivate the α1-adrenoceptor subtypes (i.e., it is an irreversible antagonist), with the following order of sensitivity: α1B⩾ α1D >> α1A (Perez et al., 1994), or being an irreversible agonist at α2-adrenoceptors (Bultmann & Starke, 1993; Docherty & O'Rourke, 1997; Nunes & Guimaraes, 1993; Piascik et al., 1995). In contrast, recent reports indicate that chloroethylclonidine also behaves as a partial α1-adrenoceptor agonist in α1A-adrenoceptor transfected cells (Villalobos-Molina et al., 1997), or in isolated left atrial and papillary muscles of rats (Williamson et al., 1994). Furthermore, we have reported that chloroethylclonidine elicited contraction by α1-adrenoceptor stimulation in aorta and caudal arteries from young Wistar Kyoto (WKY) rats (Villalobos-Molina et al., 1998). These controversial results suggest that a difference in rat strain sensitivity to chloroethylclonidine may exist. The current study was designed to determine the actions of chloroethylclonidine in Wistar, WKY and spontaneously hypertensive (SHR) rats caudal arteries and aorta, which predominantly express the α1A- and α1D-adrenoceptors, respectively (Goetz et al., 1995; Kenny et al., 1995; Lachnit et al., 1997; Piascik et al., 1997; Villalobos-Molina & Ibarra, 1996; Xu et al., 1998).
Methods
Systolic blood pressure measurement
Male Wistar (355±10 g, body weight), WKY (361±17 g) and SHR (324±5 g) rats of 6 months of age (±1 week) were reared in our animal facility under controlled light/dark conditions and fed ad libitum on standard diet. Systolic blood pressure was measured in the conscious state by a tail-cuff method (average values, Wistar, 115±20; WKY, 105±10 and SHR, 185±10 mmHg). All experimental procedures were approved by the Institutional Animal Care and Use Committee.
Contraction of aortic and caudal arterial rings
Rats were anaesthetized with ether, aorta and caudal arteries dissected and cleaned of surrounding tissue. The arteries were cut into rings (3–5 mm in length), denuded of endothelium and placed in 10 ml chambers filled with Krebs solution of the following composition (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, EDTA 0.026 and glucose 11.1, at 37°C and pH 7.4 with constant oxygenation (95% O2, 5% CO2 ). Arterial rings were attached to the bottom of the chamber and to a TSD105 physiological force transducer recorded by MP100WS for Windows (Biopac Systems Inc., CA, U.S.A.). Arterial rings were subjected to an initial optimal tension of 3 g for aorta and 2 g for caudal, obtained from preliminary experiments using increments in initial tension until the contractile response to noradrenaline was optimal. Aorta was challenged with 100 nM and caudal arteries with 3 μM noradrenaline, every 30 min and washed every 15 min for 2 h. In the last stimulation by noradrenaline, the arterial rings were exposed to acetylcholine (1 μM) to verify the functionality of endothelium. The absence of endothelium was confirmed by the lack of relaxing response to acetylcholine (Furchgott & Zawadski, 1980).
Experimental protocols
In order to determine α1-adrenoceptor independent muscular contraction arteries were incubated with a high KCl (80 mM) concentration, in the presence of (±)-propranolol (100 nM), rauwolscine (100 nM) and phentolamine (1 μM) to block β, α2 and α1-adrenoceptors, respectively, that could be activated by release of endogenous noradrenaline. A second challenge with potassium was done 30 min after washing the KCl off the first incubation.
Contraction of aortic and caudal arterial rings by chloroethylclonidine
In order to test the agonist action of chloroethylclonidine cumulative concentration-response curves to the alkylating agent were done as follows: in caudal arteries increasing concentrations of chloroethylclonidine were added every 5 min, while in aorta the agent was added every 30 min due to the different kinetics of chloroethylclonidine effect in each artery (Villalobos-Molina et al., 1998). Antagonists were used to block chloroethylclonidine (1–100 μM) contractile effect in both arteries; in this set of experiments 5-methylurapidil (100 nM), (+)-cyclazocine (10 nM) or BMY 7378 (8-(2-(4-(2-methoxyphenyl)-1-piperazinyl) ethyl)-8-azaspiro(4,5)decane-7,9-dione dihydrochloride) (100 nM), antagonists of α1A/D-, α1B- and α1D-adrenoceptors, respectively, were in contact with arteries 30 min before and during chloroethylclonidine incubation.
Antagonist effects of chloroethylclonidine on noradrenaline induced contraction and α1-adrenoceptor protection experiments
Cumulative concentration-response curves to noradrenaline (0.3 nM–100 μM) were obtained for each artery. Once the maximal response to noradrenaline was obtained, the arteries were washed every 15 min for 60 min. After the first noradrenaline curve, arteries were incubated with several concentrations of chloroethylclonidine (1–100 μM, one concentration per ring) for 45 min, washed thoroughly every 10 min for 60 min, and a second concentration-response curve to noradrenaline was obtained. In some experiments tissues were in contact with BMY 7378 (100 nM) or with 5-methylurapidil (100 nM) for 30 min before and during chloroethylclonidine incubation to protect α1-adrenoceptors from alkylation. Arterial rings were incubated with (±)-propranolol (100 nM) and rauwolscine (100 nM), to block β- and α2-adrenoceptors, before noradrenaline or chloroethylclonidine addition. Control experiments with noradrenaline were run in parallel to check the reproducibility over time between the two curves performed under the above conditions.
Data analysis
Data represent the means±s.e.mean of arterial rings from 3–6 animals per group. In all cases the responses to noradrenaline or the contractile response elicited by chloroethylclonidine were expressed as a percentage of the maximal response to noradrenaline obtained in the first concentration-response curve. Each concentration-response curve of noradrenaline was fitted to a logistic equation for one component,
or to an equation for two components,
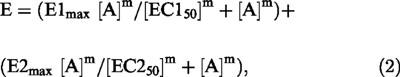 |
where E and [A] are the pharmacological effect and the concentration of agonist, respectively. Emax, EC50 and m are the asymptote, location and slope (fixing m value=1 and assuming no cooperative behaviour in the system) parameters, respectively. Location parameters were actually estimated as pD2 (i.e., the negative logarithm of EC50). Analysis of variance and Student Newman-Keuls tests were used to determine statistical differences between strains of rats and BMY 7378 and 5-methylurapidil treatments. Means were considered statistically different when the P value was smaller than 0.05.
Drugs and solutions
Acetylcholine, (−)-noradrenaline, rauwolscine, (±)-propranolol, chloroethylclonidine, 5-methylurapidil, (+)-cyclazocine and BMY 7378 were obtained from Research Biochemicals Int. (Natick, MA, U.S.A.). All stock solutions were prepared in distilled water except noradrenaline and 5-methylurapidil, which were dissolved in ascorbic acid solution (5 mg ml−1). Further dilutions were prepared in Krebs solution. All other chemicals were analytical grade from local sources.
Results
Contractile effect of noradrenaline and high KCl in arteries from normotensive and spontaneously hypertensive rats
Noradrenaline (0.3 nM–3 μM) elicited concentration-dependent contraction in both caudal arteries and aorta from Wistar, WKY and SHR rats. In caudal arteries the sensitivity to noradrenaline was significantly higher in Wistar (pD2, 6.97±0.03) than in WKY and SHR (pD2, 6.53±0.10 and 6.33±0.04, respectively, P<0.05). The maximal contraction induced by noradrenaline was higher in caudal arteries from Wistar>WKY>SHR (2.36±0.09, 1.15±0.05 vs 0.73±0.04 g, respectively, P<0.05). In aorta no differences in reactivity were observed between WKY and SHR. However, noradrenaline was more potent in aorta from WKY and SHR than in those from Wistar rats. Thus, pD2 values for noradrenaline were 8.47±0.05, 8.46±0.04 and 8.05±0.04 in WKY, SHR and Wistar rats, respectively (P<0.05). On the contrary, the maximal effects were lower in aorta from WKY (1.01±0.04 g) and SHR (0.96±0.03 g) than in those from Wistar rats (1.74±0.04 g, P<0.05). Aorta was more sensitive to noradrenaline than caudal arteries, irrespective of the strain used, suggesting that different α1-adrenoceptors are involved in the contractile response in these arteries.
Regarding KCl, the ion (80 mM) elicited a higher contraction in both aorta (1.94±0.10) and caudal (1.62±0.09 g) arteries from Wistar rats compared to WKY (1.46±0.15 and 1.24±0.03 g, respectively) and SHR (1.13±0.03 and 1.08±0.08 g, respectively).
Contractile effect of chloroethylclonidine in arteries from normotensive and spontaneously hypertensive rats
To investigate whether chloroethylclonidine induces α1-adrenoceptor-mediated contraction in blood vessels from normotensive and hypertensive rats (caudal arteries and aorta), the rings were incubated with the alkylating agent at 1–100 μM. Chloroethylclonidine consistently elicited a contractile response in caudal arteries from Wistar, WKY and SHR. The maximal contraction elicited by chloroethylclonidine in caudal arteries from Wistar, WKY and SHR showed no difference: maximal effects were 38.7±2.9% of maximal noradrenaline response in Wistar, 37.0±1.7% in WKY and 44.4±5.0% in SHR (Figure 1).
Figure 1.
Concentration-response of chloroethylclonidine-induced contraction in caudal arteries from Wistar, WKY and SHR rats. Arterial rings were incubated with (±)-propranolol and rauwolscine (100 nM, each) prior to and during chloroethylclonidine (1–100 μM) exposure. Contraction is expressed as percentage of maximal noradrenaline effect (2.36±0.09, 1.15±0.05 and 0.73±0.04 g for Wistar, WKY and SHR, respectively). Results represent the means±s.e.mean of 4–6 different animals.
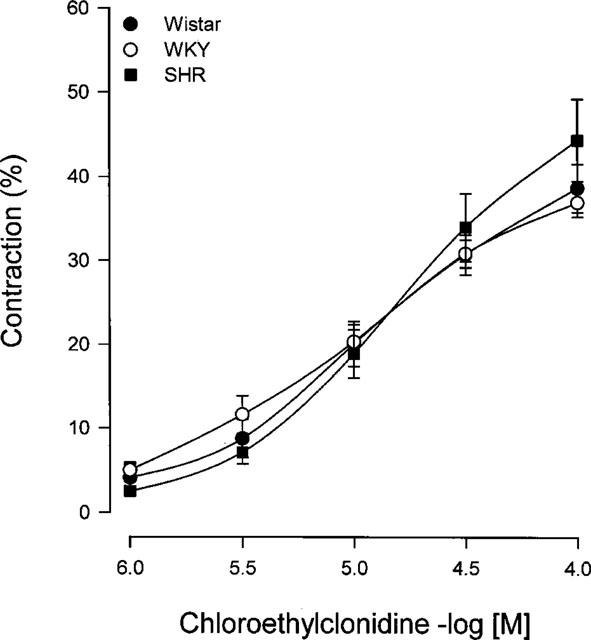
In order to characterize the interaction between chloroethylclonidine and the α1-adrenoceptors present in the vessel, caudal arteries from SHR were incubated with BMY 7378 (100 nM), (+)-cyclazocine (10 nM) or with 5-methylurapidil (100 nM), α1D-, α1B- and α1A/D-adrenoceptor antagonists, respectively (Giardina et al., 1996; Goetz et al., 1995; Hancock, 1996), before the alkylating agent. As observed, chloroethylclonidine-induced contraction in the caudal artery was significantly decreased by 5-methylurapidil but neither by (+)-cyclazocine nor by BMY 7378 (Figure 2), indicating the activation of α1A-adrenoceptors present in caudal arteries by chloroethylclonidine.
Figure 2.
Effect of 5-methylurapidil, (+)-cyclazocine and BMY 7378 on chloroethylclonidine-induced contraction in caudal arteries of spontaneously hypertensive rats. Caudal rings were incubated with BMY 7378, (+)-cyclazocine or 5-methylurapidil prior to and during chloroethylclonidine 1–100 μM. Results represent the means± s.e.mean of three different animals. *P<0.05 vs chloroethylclonidine.
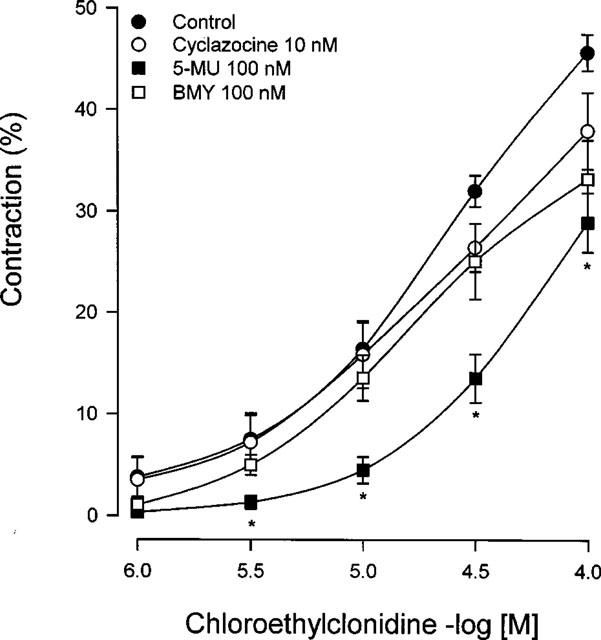
In aorta chloroethylclonidine induced concentration-dependent contractions, indicating an irreversible agonist effect of the alkylating agent. Interestingly, we found a marked difference in the ability of chloroethylclonidine to cause contraction in this vessel among the rat strains. The contractile response at low concentrations of this agonist was higher in hypertensive rats (Figure 3), while in aorta from Wistar rats chloroethylclonidine elicited a smaller contractile response compared to that observed in aortas from WKY and SHR (Figure 3).
Figure 3.
Concentration-response of chloroethylclonidine-induced contraction in aorta from Wistar, WKY and SHR rats. Aortic rings were incubated with (±)-propranolol and rauwolscine (100 nM, each) prior to and during chloroethylclonidine (1–100 μM) exposure. Data shows the percentage of maximal noradrenaline effect (1.74±0.04, 1.01±0.04, 0.96±0.03 and g for Wistar, WKY and SHR, respectively), and represent the means±s.e.mean of 4–6 different animals. +P<0.05 vs WKY; *P<0.05 vs Wistar.
In order to characterize chloroethylclonidine interaction with α1-adrenoceptors, aortas from SHR were incubated with BMY 7378, (+)-cyclazocine or 5-methylurapidil for 30 min before and during chloroethylclonidine pretreatment. BMY 7378 and 5-methylurapidil blocked chloroethylclonidine-induced contraction (Figure 4), while (+)-cyclazocine did not block it, suggesting an important role of α1D-adrenoceptors, present in aorta, in the contractile response elicited of chloroethylclonidine.
Figure 4.
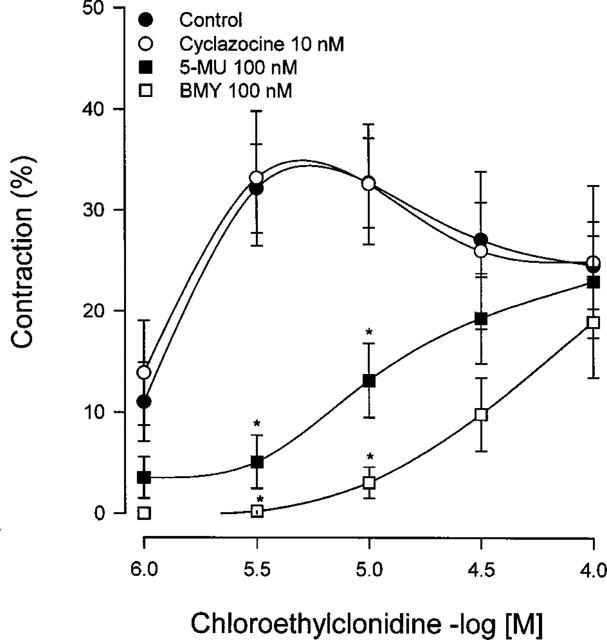
Effect of BMY 7378, 5-methylurapidil and (+)-cyclazocine on chloroethylclonidine-induced contraction in aorta from spontaneously hypertensive rats. Aortic rings were incubated with BMY 7378, 5-methylurapidil and (+)-cyclazocine prior to and during chloroethylclonidine (1–100 μM) exposure. Results represent the means±s.e.mean of 3–4 different animals. *P<0.05 vs chloroethylclonidine.
α1-Adrenoceptor antagonist effect of chloroethylclonidine
The effect of chloroethylclonidine on noradrenaline response was measured in caudal arteries and aorta from Wistar, WKY and SHR after 45 min incubation with the alkylating agent, followed by four washouts over a 60 min period. At this time the rapid transient contraction induced by chloroethylclonidine had vanished in caudal arteries. Chloroethylclonidine (3–100 μM) shifted the noradrenaline response to the right in a non-parallel manner in caudal arteries. However, as illustrated in Figure 5, caudal arteries from Wistar and WKY were more affected by chloroethylclonidine than those from SHR. Chloroethylclonidine (100 μM) increased the EC50 value of noradrenaline ≈10 fold in caudal arteries from Wistar and WKY (Figure 5), whereas in caudal vessels from SHR the increase was ≈3 fold (Figure 5). It should be noticed that low chloroethylclonidine concentrations did not block the contractile response to noradrenaline in caudal arteries from Wistar or SHR (Figure 5), but there was a clear concentration-dependent response in vessels from WKY (Figure 5).
Figure 5.
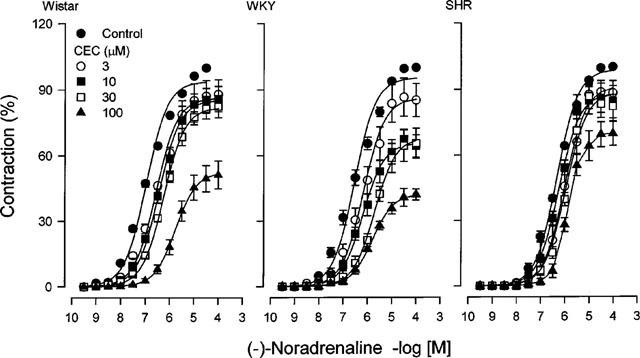
Effect of chloroethylclonidine on the concentration-response curves to noradrenaline in caudal arteries from Wistar, WKY and SHR rats. Arterial rings were incubated with increasing concentrations of chloroethylclonidine, after 45 min arteries were extensively washed and then subjected to a concentration-response curve to noradrenaline. Results are expressed as percentage of maximal noradrenaline-induced contraction and represent the means±s.e.mean of 4–6 different animals. The effect of 1 μM chloroethylclonidine is omitted for clarity.
Chloroethylclonidine caused a progressive loss in the maximal response of aorta to noradrenaline (in the range 0.3–100 nM), which was coincident with the emergence of a noradrenaline response in the range of 300 nM–100 μM, irrespective of the strain used. It is important to mention that chloroethylclonidine-induced contraction was not accounted for in these experiments, since we were studying chloroethylclonidine antagonism (Figure 6).
Figure 6.
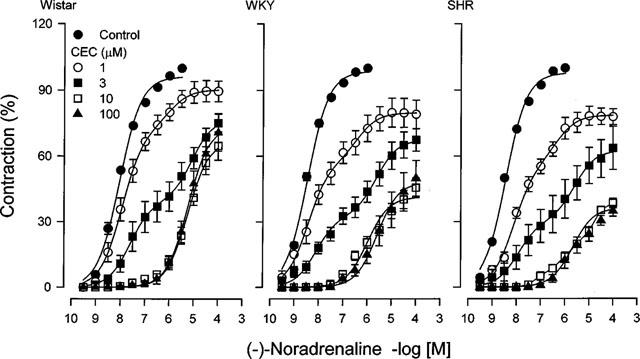
Effect of chloroethylclonidine on the concentration-response curve to noradrenaline in aorta from Wistar, WKY and SHR rats. Aortic rings from Wistar, WKY or SHR rats were incubated with increasing concentrations of chloroethylclonidine, after 45 min vessels were thoroughly washed and then subjected to a concentration-response curve to noradrenaline. Results represent the means±s.e.mean of 4–6 different animals. The effect of 30 μM chloroethylclonidine is omitted for clarity.
At low concentrations of chloroethylclonidine (1 and 3 μM), a biphasic response to noradrenaline was evident, indicating a high affinity (pD2, 8.5–7.5) chloroethylclonidine-sensitive component and a low affinity (pD2, 6.3–5.2) chloroethylclonidine-insensitive component. Thus, the actual rightward shift in the noradrenaline concentration-response curves is probably due to a progressive displacement in the relative contribution of the two distinctive affinity states. In aorta from Wistar (Figure 6) high chloroethylclonidine concentrations produced a smaller inhibition than in aorta from WKY and SHR rats (Figure 6). Furthermore, chloroethylclonidine (100 μM) increased the EC50 value to noradrenaline ≈1000 fold in WKY and SHR, and ≈670 fold in Wistar rats.
α1-Adrenoceptor protection experiments
We assessed the effect of chloroethylclonidine on vascular smooth muscle contraction induced by noradrenaline, in the presence or in the absence of the α1D-adrenoceptor antagonist BMY 7378 (0.01, 0.1 and 1 μM), or the α1A/D-adrenoceptor antagonist, 5-methylurapidil (1 μM). Since the affinity of α1D-adrenoceptors for BMY 7378 is the highest, this ligand is expected to protect the receptor against chloroethylclonidine modification in the aorta, whereas the other subtypes should be alkylated. With the same reasoning, 5-methylurapidil should protect α1A-adrenoceptors in the caudal artery. As anticipated chloroethylclonidine significantly inhibited the contractile response to noradrenaline in aorta and caudal arteries from SHR (Figures 7, 8). In the presence of BMY 7378 (10 or 100 nM), the inhibitory actions of chloroethylclonidine (1 μM) were totally abolished (Figure 7) in aorta, while BMY 7378 (1 μM) did not significantly protect the receptor in the caudal artery, but 5-methylurapidil (1 μM) did (Figure 8). The selective protection exerted by BMY 7378 and by 5-methylurapidil suggests that noradrenaline response in rat aorta is due to activation of α1D-adrenoceptors and in caudal artery is due to activation of α1A-adrenoceptors.
Figure 7.
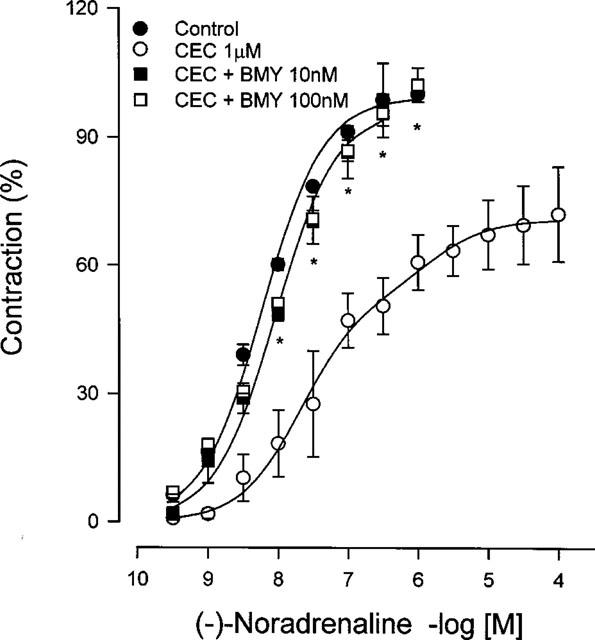
Protection by BMY 7378 of noradrenaline-induced contraction after chloroethylclonidine treatment in aorta from spontaneously hypertensive rats. Aortic rings were incubated with BMY 7378 prior to and during chloroethylclonidine exposure, then subjected to a concentration-response curve to noradrenaline. Data represent the means±s.e.mean of 4–6 different animals. *P<0.05 vs chloroethylclonidine.
Figure 8.
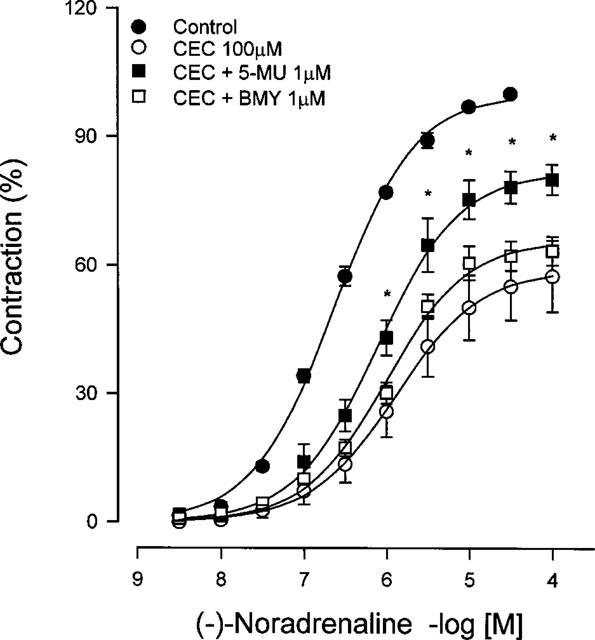
Protection by 5-methylurapidil and BMY 7378 of noradrenaline-induced contraction after chloroethylclonidine treatment in caudal arteries from spontaneously hypertensive rats. Caudal rings were incubated with 5-methylurapidil or with BMY 7378 before chloroethylclonidine exposure, then subjected to a concentration-response curve to noradrenaline. Data represent the means±s.e.mean of three different animals. *P<0.05 vs chloroethylclonidine.
Discussion
Chloroethylclonidine as a partial α1-adrenoceptor agonist
The effects of chloroethylclonidine on α-adrenoceptor function have been mainly related to its ability to alkylate and inactivate the α1-adrenoceptor subtypes (Hirasawa et al., 1997; Michel et al., 1993; Perez et al., 1994) or being an irreversible agonist of the α2-adrenoceptors (Bultmann & Starke, 1993; Docherty & O'Rourke, 1997; Nunes & Guimaraes, 1993; Piascik et al., 1995). The present study shows that chloroethylclonidine elicits contraction in both aorta and caudal arteries from normotensive and hypertensive rats. This is consistent with previous reports in rat aorta (Leclerc et al., 1980; Muramatsu et al., 1990; 1991), and with the fact that chloroethylclonidine is an imidazoline; therefore, it was expected to exhibit mixed antagonist/partial agonist properties, a distinctive characteristic of this chemical class (Ruffolo et al., 1980; Tian et al., 1990). In fact, we showed those mixed properties in chloroethylclonidine-induced Ca2+ mobilization in α1A-adrenoceptor transfected fibroblasts, these effects being related to receptor occupation rather than to alkylation of the receptor (Villalobos-Molina et al., 1997). Furthermore, we recently reported that chloroethylclonidine-induced contraction in aorta and in caudal arteries from WKY was blocked by phentolamine but not by rauwolscine, indicating that the contractility induced by chloroethylclonidine is mediated by α1-adrenoceptors (Villalobos-Molina et al., 1998). In contrast, other authors observed either a small contractile effect of chloroethylclonidine (Muramatsu et al., 1990; 1991; this study) or no effect at all (Docherty & O'Rourke, 1997; Kenny et al., 1995; O'Rourke et al., 1995; 1997) in the aorta from Wistar rats. These controversial reports suggest strain-dependent differences in the ability of chloroethylclonidine to induce aortic contraction. In addition, there were marked differences in the contractile response elicited by chloroethylclonidine between aorta and caudal arteries. In aorta, chloroethylclonidine-elicited contraction showed a much slower time-course than in caudal arteries, but aorta was much more sensitive to the alkylating agent than caudal arteries (data not shown; Villalobos-Molina et al., 1998). These differences, both in the time-course and in the sensitivity to chloroethylclonidine, could be due to different subtypes of α1-adrenoceptors involved in contraction. In this regard, it has been reported that α1D-adrenoceptors mediate contraction in rat aorta (Goetz et al., 1995; Kenny et al., 1995; Xu et al., 1998), while in the caudal artery it is the α1A subtype (Lachnit et al., 1997; Piascik et al., 1997; Villalobos-Molina & Ibarra, 1996). Furthermore, our data show that the contractile response to chloroethylclonidine in aorta from SHR was blocked by BMY 7378 (α1D-adrenoceptor antagonist) and by 5-methylurapidil (α1A-adrenoceptor antagonist with some affinity for α1D-adrenoceptors), suggesting that chloroethylclonidine behaves as an α1D-adrenoceptor partial agonist in rat aorta, and as an α1A-adrenoceptor partial agonist in caudal arteries, where the antagonist 5-methylurapidil but not BMY 7378, significantly blocked chloroethylclonidine-induced contraction. In support of these results, we described chloroethylclonidine as a partial α1A-adrenoceptor agonist in Ca2+ mobilization in transfected cells (Villalobos-Molina et al., 1997). In contrast, the α1B-adrenoceptor antagonist (+)-cyclazocine (at 10 nM a concentraction 15–75 fold the pKi for this receptor; Giardina et al., 1996) was unable to block chloroethylclonidine-induced contraction in none of the two arteries, suggesting that this receptor is not involved in chloroethylclonidine agonist action.
Chloroethylclonidine-induced contraction in arteries from normotensive and hypertensive rats showed very interesting aspects, (a) the contractile effect of chloroethylclonidine was not significantly different in caudal arteries from Wistar, WKY and SHR; (b) chloroethylclonidine elicited a significantly higher contraction in aorta from WKY than from Wistar rats and (c) chloroethylclonidine (3 and 10 μM) elicited a significantly higher contraction in aorta from SHR than in WKY and Wistar rats suggesting that α1D-adrenoceptors could be more abundant in SHR and WKY than in Wistar rats. In support of this contention, a recent report described a higher amount of α1D-adrenoceptor mRNA in aorta from SHR compared to WKY (Xu et al., 1998), indicating that hypertension and/or genetic factors seem to be involved in this phenomenon, as has been demonstrated in aorta from WKY where more α1D-adrenoceptors were found than in aorta from Wistar rats (Stassen et al., 1997). It is also possible that changes at post-receptor events may be involved in the higher reactivity to chloroethylclonidine, such as altered levels of G proteins in hypertension (Kanagy & Webb, 1996; Li et al., 1994). Studies using cells expressing the cloned receptors have shown that noradrenaline is a selective α1D-adrenoceptor agonist (Hancock, 1996; Horie et al., 1995; Minneman et al., 1994); however, we did not find differences in the contractile response to noradrenaline in aorta between SHR (pD2, 8.46±0.04) and WKY (pD2, 8.47±0.05), whereas the aorta from Wistar was hyporeactive to the catecholamine (pD2, 8.05±0.04) similar to the agonist effect of chloroethylclonidine per se in this strain. In contrast, the maximal effects with KCl or noradrenaline stimulation were smaller in both arteries from SHR and WKY than from Wistar, suggesting that these changes are not related to changes in α1-adrenoceptor density. We do not have a clear explanation in the lack of difference to noradrenaline-induced contraction between SHR and WKY, but it is possible that chloroethylclonidine may activate different signalling effector pathways upon the occupation of α1-adrenoceptor subtypes. Very recently, it has been suggested that agonists may interact with different sites in the same receptor and activate different G protein/effector pathways (Perez et al., 1996). In this regard, Tian et al. (1990) reported that chloroethylclonidine elicited contraction only in the presence of protein kinase activation in rabbit aorta. In addition, there is evidence indicating that activity of protein kinase is increased during hypertension (Takata & Kato, 1996).
Chloroethylclonidine as an irreversible α1-adrenoceptor antagonist
Analysis of the noradrenaline concentration-response curves after preincubation with different chloroethylclonidine concentrations show that in aorta the best fit was obtained with the two-site model for α1-adrenoceptors (i.e., α1D- and α1A- subtypes) with a high affinity (pD2, 8.5–7.5) chloroethylclonidine-sensitive and a low affinity (pD2, 6.3–5.2) chloroethylclonidine-insensitive components; while in caudal arteries (Wistar, WKY and SHR) the one-site model (i.e., α1A-adrenoceptors) produced the best fit with a chloroethylclonidine-insensitive, low affinity (pD2, 6.5–5.7) component. These results could be explained in the following way: chloroethylclonidine, in a concentration-dependent manner, alkylates and activates the α1D-adrenoceptor population responsible for the contraction of aorta (Goetz et al., 1995; Kenny et al., 1995; Villalobos-Molina & Ibarra, 1996; Xu et al., 1998); this causes a loss in the ability of noradrenaline, a selective α1D-adrenoceptor agonist (Hancock, 1996; Horie et al., 1995; Minneman et al., 1994), to elicit contraction through its interaction with α1D-adrenoceptors, which results in a non parallel rightward shift and a decrease in the maximal effect with increasing chloroethylclonidine concentrations (Figure 6). However, at higher concentrations, noradrenaline stimulates the contraction of the aorta suggesting its interaction with the α1A-adrenoceptors present in the vessel (Piascik et al., 1994; 1995). In contrast, noradrenaline triggers the contraction of the caudal artery despite chloroethylclonidine treatment, except for a non parallel rightward shift and a decrease in the maximal effect, at the highest concentrations of the alkylating agent (Figure 5), suggesting that α1A-adrenoceptors are the main population of this artery (Lachnit et al., 1997; Piascik et al., 1995; Villalobos-Molina & Ibarra, 1996). The response to noradrenaline in aorta from Wistar rats treated with different chloroethylclonidine concentrations, was greater than in the SHR or the WKY indicating a smaller antagonistic effect of the alkylating agent, consistent with a suggested reduced number of α1D-adrenoceptors in this strain (Stassen et al., 1997; Figure 6).
α1-Adrenoceptor protection experiments
The lack of chloroethylclonidine effect in eliciting contraction by previous exposure of aorta to BMY 7378 or 5-methylurapidil, strongly suggest that α1D-adrenoceptors are occupied by the alkylating agent in the SHR (Figure 3). Further support to this suggestion comes from experiments in which protection of α1D-adrenoceptors by BMY 7378 allowed noradrenaline-induced contraction in SHR aorta, despite chloroethylclonidine (1 μM) treatment (Figure 7); however, protecting the receptors with BMY 7378 was partial when the alkylating agent was at 100 μM (not shown), indicating that besides α1D-adrenoceptors, other α1 subtypes (i.e., α1A-adrenoceptors) could be involved in the response. A similar reasoning could explain what we observed in the caudal artery, where protecting the receptor with the α1A/D-adrenoceptor antagonist 5-methyl-urapidil precluded, significantly, chloroethylclonidine antagonism in noradrenaline-induced contraction, while BMY 7378 did not protect the receptor in this vessel (Figure 8).
To our knowledge, this is the first study that describes a strain-dependent difference in sensitivity to chloroethylclonidine-elicited contraction in rat aorta. Hypertension increased such process, since contractile response to chloroethylclonidine was higher in aorta from SHR rats. Chloroethylclonidine exerts its actions through a partial agonism at α1A- (caudal arteries) of Wistar, WKY and SHR and at α1D-adrenoceptors (aorta) of SHR⩾WKY>>Wistar. The fact that chloroethylclonidine effect was higher in aorta from SHR and WKY, suggests that α1D-adrenoceptors could be involved in the increased sensitivity of aorta from these strains to noradrenaline.
Acknowledgments
This work was supported in part by Grant No. 28553-M from CONACYT.
Abbreviations
- BMY 7378
(8-(2-(4-(2-methoxyphenyl)-1-piperazinyl) ethyl)-8-azaspiro(4,5)decane-7,9-dione dihydrochloride)
References
- BULTMANN R., STARKE K. Chloroethylclonidine: an irreversible agonist at prejunctional α2-adrenoceptors in rat vas deferens. Br. J. Pharmacol. 1993;108:336–341. doi: 10.1111/j.1476-5381.1993.tb12806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOCHERTY J.R., O'ROURKE M. The α-adrenoceptor-mediated actions of chloroethylclonidine. Gen. Pharmac. 1997;28:197–201. doi: 10.1016/s0306-3623(96)00187-5. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GIARDINA D., CRUCIANELLI M., ROMANELLI R., LEONARDI A., POGESSI E., MELCHIORRE C. Synthesis and biological profile of the enantiomers of [4-(4-amino-6, 7-dimethoxyquinazolin-2-yl) -cis- octahydroquinoxalin-1-yl] furan-2-ylmethanone (Cyclazocin), a potent competitive α1B-adrenoceptor antagonist. J. Med. Chem. 1996;39:4602–4607. doi: 10.1021/jm960510x. [DOI] [PubMed] [Google Scholar]
- GOETZ A.S., KING H.K., WARD S.D.C., TRUE T.A., RIMELE T.J., SAUSSY D.L. BMY 7378 is a selective antagonist of the D subtype of α1-adrenoceptors. Eur. J. Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- HAN C., ABEL P.W., MINNEMAN K.P. Heterogeneity of α1-adrenergic receptors revealed by chlorethylclonidine. Mol. Pharmacol. 1987;32:505–510. [PubMed] [Google Scholar]
- HAN C., LI J., MINNEMAN K.P. Subtypes of α1-adrenoceptors in rat blood vessels. Eur. J. Pharmacol. 1990;190:97–104. doi: 10.1016/0014-2999(90)94116-f. [DOI] [PubMed] [Google Scholar]
- HANCOCK A.A. α1-Adrenoceptor subtypes: a synopsis of their pharmacology and molecular biology. Drug Dev. Res. 1996;39:54–107. [Google Scholar]
- HIRASAWA A., SUGAWARA T., AWAJI T., TSUMAYA K., ITO H., TSUJIMOTO G. Subtype-specific differences in subcellular localization of α1-adrenoceptors: Chloroethylclonidine preferentially alkylates the accessible cell surface α1-adrenoceptors irrespective of the subtype. Mol. Pharmacol. 1997;52:764–770. doi: 10.1124/mol.52.5.764. [DOI] [PubMed] [Google Scholar]
- HORIE K., OBIKA K., FOGLAR R., TSUJIMOTO G. Selectivity of the imidazoline α-adrenoceptor agonists (oxymetazoline and cirazoline) for human cloned α1-adrenoceptor subtypes. Br. J. Pharmacol. 1995;116:1611–1618. doi: 10.1111/j.1476-5381.1995.tb16381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANAGY N.L., WEBB R.C. Increased responsiveness and decreased expression of G proteins in deoxycorticosterone hypertension. Hypertension. 1996;27:740–745. doi: 10.1161/01.hyp.27.3.740. [DOI] [PubMed] [Google Scholar]
- KENNY B.A., CHALMERS D.H., PHILPOTT P.C., NAYLOR A.M. Characterization of an α1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br. J. Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACHNIT W.G., TRAN A.M., CLARKE D.E., FORD A.P.D.W. Pharmacological characterization of an α1A-adrenoceptor mediating contractile responses to noradrenaline in isolated caudal artery of rat. Br. J. Pharmacol. 1997;120:819–826. doi: 10.1038/sj.bjp.0700983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LECLERC G., ROUOT B., SCHWARTZ J., VELLY J., WERMUTH C.G. Studies on some parasubstituted clonidine derivatives that exhibit an α-adrenoceptor stimulant activity. Br. J. Pharmacol. 1980;71:5–9. doi: 10.1111/j.1476-5381.1980.tb10902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI P., ZOU A.-P., AL-KAYED N.J., RUSCH N.J., HARDER D.R. Guanine nucleotide-binding proteins in aortic smooth muscle from hypertensive rats. Hypertension. 1994;23:914–918. doi: 10.1161/01.hyp.23.6.914. [DOI] [PubMed] [Google Scholar]
- MINNEMAN K.P., HAN C., ABEL P.W. Comparison of α1-adrenergic receptor subtypes distinguished by chlorethylclonidine and WB 4101. Mol. Pharmacol. 1988;33:509–514. [PubMed] [Google Scholar]
- MINNEMAN K.P., THEROUX T.L., HOLLINGER S., HAN C., ESBENSHADE T.A. Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol. Pharmacol. 1994;46:929–936. [PubMed] [Google Scholar]
- MICHEL M.C., KERKER J., BRANCHEK T.A., FORRAY C. Selective irreversible binding of chloroethylclonidine at α1- and α2-adrenoceptor subtypes. Mol. Pharmacol. 1993;44:1165–1170. [PubMed] [Google Scholar]
- MURAMATSU I., OHMURA T., KIGOSHI S., HASHIMOTO S., OSHITA M. Pharmacological subclassification of α1-adrenoceptors in vascular smooth muscle. Br. J. Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAMATSU I., KIGOSHI S., OHMURA T. Subtypes of α1-adrenoceptors involved in noradrenaline induced contractions of thoracic aorta and dog carotid artery. Jpn. J. Pharmacol. 1991;57:535–544. doi: 10.1254/jjp.57.535. [DOI] [PubMed] [Google Scholar]
- NUNES J.P., GUIMARAES S. Chloroethylclonidine irreversibly activates postjunctional alpha2-adrenoceptors in the dog saphenous vein. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;348:264–268. doi: 10.1007/BF00169154. [DOI] [PubMed] [Google Scholar]
- ORIOWO M.A., RUFFOLO R.R. Heterogeneity of postjunctional α1-adrenoceptors in mammalian aortae: subclassificcation based on chloroethylclonidine, WB 4101 and nifedipine. J. Vasc. Res. 1992;29:33–40. doi: 10.1159/000158929. [DOI] [PubMed] [Google Scholar]
- O'ROURKE M., GAVIN K., DOCHERTY J.R. Further investigation of the α-adrenoceptor-mediated actions of chloroethylclonidine in rat aorta. Eur. J. Pharmacol. 1997;336:37–42. doi: 10.1016/s0014-2999(97)01257-0. [DOI] [PubMed] [Google Scholar]
- O'ROURKE M., KEARNS S., DOCHERTY J.R. Investigation of the actions of chloroethylclonidine in rat aorta. Br. J. Pharmacol. 1995;115:1399–1406. doi: 10.1111/j.1476-5381.1995.tb16630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREZ D.M., HWA J., GAIVIN R., MATHUR M., BROWN F., GRAHAM R.M. Constitutive activation of a single effector pathway: evidence for multiple activation states of a G protein-coupled receptor. Mol. Pharmacol. 1996;49:112–122. [PubMed] [Google Scholar]
- PEREZ D.M., PIASCIK M.T., MALIK N., GAIVIN R., GRAHAM R.M. Cloning, expression, and tissue distribution of the rat homolog of the bovine α1C-adrenergic receptor provide evidence for its classification as the α1A subtype. Mol. Pharmacol. 1994;46:823–831. [PubMed] [Google Scholar]
- PIASCIK M.T., GUARINO R.D., SMITH M.S., SOLTIS E.E., SAUSSY D.L., Jr, PEREZ D.M. The specific contribution of the novel alpha-1D adrenoceptor to the contraction of vascular smooth muscle. J. Pharmacol. Exp. Ther. 1995;275:1583–1589. [PubMed] [Google Scholar]
- PIASCIK M.T., HROMETZ S.L., EDELMANN S.E., GUARINO R.D., HADLEY R.W., BROWN R.D. Immunocytochemical localization of the alpha-1B adrenergic receptor and the contribution of this and the other subtypes to vascular smooth muscle contraction: analysis with selective ligands and antisense oligonucleotides. J. Pharmacol. Exp. Ther. 1997;283:854–868. [PubMed] [Google Scholar]
- PIASCIK M.T., SMITH M.S., SOLTIS E.E., PEREZ D.M. Identification of the mRNA for the novel α1D-adrenoceptor and two other α1-adrenoceptors in vascular smooth muscle. Mol. Pharmacol. 1994;46:30–40. [PubMed] [Google Scholar]
- RUFFOLO R.R., Jr, WADDELL J.E., YADEN E.L. Receptor interactions of imidazolines. IV. Structural requirements for alpha adrenergic receptor occupation and receptor activation by clonidine and a series of structural analogs in rat aorta. J. Pharmacol. Exp.Ther. 1980;213:267–272. [PubMed] [Google Scholar]
- STASSEN F.R.M., WILLEMSEN M.J.J.M.F., JANSSEN G.M.J., DEMEY J.G.R. α1-Adrenoceptor subtypes in rat aorta and mesenteric small arteries are preserved during left ventricular dysfunction post-myocardial infarction. Cardiovasc. Res. 1997;33:706–713. doi: 10.1016/s0008-6363(96)00261-1. [DOI] [PubMed] [Google Scholar]
- TAKATA Y., KATO H. Adrenoceptors in SHR: alterations in binding characteristics and intracellular signal transduction pathways. Life. Sci. 1996;58:91–106. doi: 10.1016/0024-3205(95)02213-9. [DOI] [PubMed] [Google Scholar]
- TIAN W.N., GUPTA S., DETH R.C. Species differences in chloroethylclonidine antagonism at vascular alpha-1 adrenergic receptors. J. Pharmacol. Exp. Ther. 1990;253:877–883. [PubMed] [Google Scholar]
- VILLALOBOS-MOLINA R., IBARRA M. α1-Adrenoceptors mediating contraction in arteries of normotensive and spontaneously hypertensive rats are of the α1D or α1A subtypes. Eur. J. Pharmacol. 1996;298:257–263. doi: 10.1016/0014-2999(95)00781-4. [DOI] [PubMed] [Google Scholar]
- VILLALOBOS-MOLINA R., LÓPEZ-GUERRERO J.J., IBARRA M. Chloroethylclonidine is a partial α1-adrenoceptor agonist in aorta and caudal arteries of the Wistar Kyoto rat. Eur. J. Pharmacol. 1998;351:49–52. doi: 10.1016/s0014-2999(98)00275-1. [DOI] [PubMed] [Google Scholar]
- VILLALOBOS-MOLINA R., VÁZQUEZ-PRADO J., GARCÍA-SÁINZ J.A. Chloroethylclonidine is a partial α1A-adrenoceptor agonist in cells expressing recombinant α1-adrenoceptor subtypes. Life. Sci. 1997;61:PL391–PL395. doi: 10.1016/s0024-3205(97)00987-9. [DOI] [PubMed] [Google Scholar]
- WILLIAMSON A.P., SEIFEN E., LINDEMANN J.P., KENNEDY R.H. WB 4101-and CEC-sensitive positive inotropic actions of phenylephrine in rat cardiac muscle. Am. J. Physiol. 1994;266:H2462–H2467. doi: 10.1152/ajpheart.1994.266.6.H2462. [DOI] [PubMed] [Google Scholar]
- XU K., LU Z., WEI H., ZHANG Y., HAN C. Alteration of α1-adrenoceptor subtypes in aortas of 12-month-old spontaneously hypertensive rats. Eur. J. Pharmacol. 1998;344:31–36. doi: 10.1016/s0014-2999(97)01559-8. [DOI] [PubMed] [Google Scholar]