

Abstract
Since cytochrome P450-derived metabolites of arachidonic acid and K+ have been implicated in endothelium-derived hyperpolarizing factor (EDHF)-dependent responses, the aim of this study was to determine whether such factors contribute to non-nitric oxide (NO), endothelium-dependent relaxation to bradykinin (BK) in bovine isolated coronary artery.
In rings of artery contracted with U46619 and treated with indomethacin (3 μM) and NG-nitro-L-arginine (L-NOARG; 100 μM), relaxation to BK (0.01 nM-0.3 μM) was blocked by ∼60% after inhibition of K+ channels with either high extracellular K+ (high [K+]o; 15–67 mM) or apamin (0.3 μM).
Ouabain (1 μM), an inhibitor of Na+/K+-ATPase, decreased the sensitivity to BK without affecting the maximum response. In L-NOARG-treated rings, ouabain had no further effect on the relaxation to BK. An inhibitor of inward-rectifying K+ channels, Ba2+ (30 μM), had no effect on relaxations to BK in the absence or presence of either L-NOARG or ouabain.
KCl (2.5–10 mM) elicited small relaxations (∼20%) that were abolished by nifedipine (0.3 μM) and ouabain.
Both the high [K+]o/apamin-sensitive relaxation to BK, and the relaxation to the KATP channel-opener, levcromakalim (0.6 μM), were unaffected by the cytochrome P450 inhibitor, 7-ethoxyresorufin (10 μM), or by co-treatment with a phospholipase A2 inhibitor, arachidonyl trifluoromethyl ketone (AACOCF3; 3 μM) and a diacylglycerol (DAG)-lipase inhibitor, 1,6-bis-(cyclohexyloximinocarbonylamino)-hexane (RHC 80267; 30 μM). The non-NO/high [K+]o-insensitive, ∼40% relaxation to BK was, however, abolished by these treatments.
Therefore, neither cytochrome P450-derived metabolites of arachidonic acid nor K+ appear to mediate the EDHF-like relaxation to BK (i.e the non-NO, high [K+]o/apamin-sensitive component) in bovine coronary arteries. Cytochrome P450-derived metabolites may be released at higher BK concentrations to act in parallel with NO and the high [K+]o/apamin-sensitive mechanism.
Keywords: Endothelium-dependent relaxation, EDHF, nitric oxide, bradykinin, bovine coronary artery, ouabain, cytochrome P450, K+, apamin, Ba2+
Introduction
Non-prostanoid, endothelium-dependent relaxation in many blood vessels is mediated not only by nitric oxide (NO) but also by a non-NO, high K+-sensitive mechanism which may be attributable to the release of an endothelium-derived hyperpolarizing factor (EDHF; Taylor & Weston, 1988; Kilpatrick & Cocks, 1994; Garland et al., 1995; Drummond & Cocks, 1996; Feletou & Vanhoutte, 1999). The mechanism of EDHF-mediated relaxation of vascular smooth muscle remains controversial. Two recent proposals are that EDHF is either K+ (Edwards et al., 1998) or a cytochrome P450-derived metabolite of arachidonic acid (Komori & Vanhoutte, 1990; Mombouli & Vanhoutte, 1997; Feletou & Vanhoutte, 1999; Fisslthaler et al., 1999).
In support of their claim that EDHF is K+, Edwards et al. (1998) showed that non-prostanoid, non-NO, endothelium-dependent hyperpolarization to acetylcholine (ACh) in rat resistance arteries was blocked by a combination of the small (SKCa) and large (BKCa) conductance Ca2+-activated K+ channel inhibitors, apamin and charybdotoxin, respectively. The response was also blocked by a combination of ouabain and Ba2+, inhibitors of Na+/K+-ATPase and inward-rectifying K+ channels (KIR), respectively. Edwards et al. (1998) found that low concentrations (<20 mM) of K+ applied exogenously caused ouabain- and Ba2+-sensitive smooth muscle hyperpolarization and relaxation, mimicking the non-NO, endothelium-dependent responses to ACh. Furthermore, using a K+ measuring electrode strategically positioned between the endothelium and smooth muscle, they were able to show that in response to ACh, the K+ concentration increased by approximately 5 mM in the myoendothelial space in an apamin- and charybdotoxin-sensitive manner. Thus, the authors concluded that EDHF is likely to be universally K+, which after being released from endothelial cells via SKCa and BKCa channels activates smooth muscle Na+/K+-ATPase and KIR to initiate hyperpolarization and relaxation (Edwards et al., 1998).
The conclusion by Edwards et al. (1998) that EDHF is K+ may not hold true for all arteries. For example, it is well established that in large epicardial arteries of the pig, an EDHF-like mechanism compensates for up to 80% of the relaxation to BK after inhibition of NO synthesis (Kilpatrick & Cocks, 1994). This is despite the fact that these large arteries express few functional KIR (Quayle et al., 1996). Also, Fisslthaler et al. (1999) have shown that while combined application of Ba2+ and ouabain had no effect on BK-induced, EDHF-mediated vascular responses in porcine large epicardial arteries, transfection with antisense oligonucleotides against cytochrome P450 2C inhibited the response. Thus, these findings not only argue against a role for KIR and Na+/K+-ATPase but also suggest that in porcine coronary arteries at least, EDHF represents a product of cytochrome P450 2C metabolism.
Earlier observations also support a role for cytochrome P450 in EDHF-dependent vasorelaxation. For instance, a variety of inhibitors of arachidonic acid formation and cytochrome P450 have been shown to partially inhibit endothelium-dependent relaxations in many isolated arteries (Singer et al., 1984; Pinto et al., 1987; Hecker et al., 1994; Bauersachs et al., 1997; Satake et al., 1997) and perfused vascular beds (Fulton et al., 1992; 1996; Bauersachs et al., 1994; Adeagbo & Henzel, 1998). Also, products of cytochrome P450-dependent metabolism of arachidonic acid including epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetraenoic acids (HETEs), have been shown to be released by cultured bovine coronary artery endothelial cells (Rosolowsky et al., 1996; Rosolowsky & Campbell, 1996; Gebremedhin et al., 1998) and to relax vascular smooth muscle (Proctor et al., 1987; Rosolowsky et al., 1990, 1996; Gebremedhin et al., 1992; Hecker et al., 1994; Campbell et al., 1996; Zou et al., 1996; Pratt et al., 1998). Also, EETs can activate smooth muscle calcium-dependent K+ channels to initiate hyperpolarization (Gebremedhin et al., 1992; Hu & Kim, 1993; Campbell et al., 1996; Zou et al., 1996).
In the bovine coronary artery, the non-prostanoid relaxation to BK, which is sensitive to high extracellular K+ and therefore likely attributable to an EDHF-like mechanism, compensates for up to 95% of the overall response after inhibition of NO (Drummond & Cocks, 1996). Also, a third mechanism independent of both NO and the EDHF-like mechanism is present in this tissue and acts as a second level backup behind NO and EDHF, accounting for ∼40% of the overall response to BK. Therefore, the aim of this study was to determine whether endothelium-derived K+ or cytochrome P450-derived metabolites of arachidonic acid mediate non-NO, endothelium-dependent relaxation to BK in the bovine coronary artery. Our data show that while cytochrome P450-derived metabolites of arachidonic acid may be involved, they do not mediate the EDHF-like response. Also, our results indicate that endothelium-derived K+ does not appear to have any role in non-prostanoid, non-NO endothelium-dependent relaxation in the bovine coronary artery.
Methods
Tissue preparation
Sections of bovine myocardium containing the left anterior descending coronary artery were obtained from a local abattoir and transported to the laboratory in ice cold Krebs solution (composition in mM; Na+ 143.1, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, Cl− 127.8, HCO3− 25.0, SO42− 1.2, H2PO4− 1.2 and glucose 11.0). Ring segments (3 mm) of coronary artery were prepared as previously described (Drummond & Cocks, 1996) and then suspended between two stainless steel wire hooks, one of which was connected to a force-displacement transducer (model FT03C, Grass, Quincy, MA, U.S.A.) and the other to a micrometer-adjustable support leg. Preparations were immersed in water-jacketed 30 ml organ baths containing warm (37°C), carbogen-bubbled (95% O2, 5% CO2) Krebs solution (pH 7.4). Changes in isometric force were amplified and displayed on dual-channel, flat-bed recorders (W & W Scientific Instruments, Basel, Switzerland).
Tissue equilibration
Rings of artery were allowed to equilibrate under zero tension for a period of 25 min, after which time they were passively stretched to a resting tension of 5 g. After a further 25 min, rings were stretched again to 5 g passive tension, allowed to equilibrate and then maximally contracted with an isotonic, high potassium physiological salt solution (KPSS; composition in mM; K+ 124.9, Na+ 25.0, Ca2+ 2.5, Mg2+ 1.2, Cl− 128.7, HCO3− 25.0, SO42− 1.2, H2PO4− 1.2 and glucose 6.1). Once a stable plateau of active force to KPSS was obtained (KPSSmax), tissues were washed with normal Krebs solution and allowed to return to resting levels of passive force. All tissues were then treated with the cyclo-oxygenase inhibitor, indomethacin (3 μM), for the remainder of the experiment. Some tissues were also treated with the L-type voltage-operated Ca2+ channel (VOCC) inhibitor, nifedipine (0.3 μM), to prevent excessive levels of pre-contraction in experiments where an isotonic 67 mM KCl bathing solution (high [K+]o) was used to inhibit K+ channels. Previously, we have shown that while nifedipine abolished contractions to a maximum depolarizing concentration of extracellular K+, it had no effect on either the ability of U46619 to cause contraction or any component of the relaxation to BK (Drummond & Cocks, 1996).
Experimental protocol
Twenty-five minutes after the addition of indomethacin and nifedipine, rings were either left untreated (control) or treated with either the NO synthase inhibitor, NG-nitro-L-arginine (L-NOARG; 100 μM), high [K+]o, the soluble guanylatecyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-α]quinoxaline-1-one (ODQ; 10 μM), the SKCa inhibitor, apamin (0.3 μM), the Na+/K+-ATPase inhibitor, ouabain (1 μM or 1 mM) or the KIR inhibitor, Ba2+ (30 μM). Some L-NOARG-treated tissues were further treated with either high [K+]o, ODQ, apamin, ouabain or Ba2+ or ouabain plus Ba2+. Other L-NOARG-treated rings were treated with the combination of apamin (0.3 μM), charybdotoxin (0.1 μM) and the KATP channel inhibitor, glibenclamide (10 μM). Also, separate rings were treated with either inhibitors of arachidonic acid formation including the phospholipase A2 inhibitors, quinacrine (30 μM; Bauersachs et al., 1994) and arachidonyl trifluoromethyl ketone (AACOCF3; 3 μM; Street et al., 1993; Riendeau et al., 1994) and the diacylglycerol (DAG) lipase inhibitor, 1,6-bis-(cyclohexyloximinocarbonylamino)-hexane (RHC 80267; 30 μM; Sutherland & Amin, 1982), or blockers of subsequent metabolism via the cytochrome P450 pathway such as N,N-diethylaminoethyl-2,2-diphenylvalerate (SKF-525a; 100 μM; Hecker et al., 1994), clotrimazole (100 μM; Hecker et al., 1994) or 7-ethoxyresorufin (10 μM; Tassaneeyakul et al., 1993). All tissues were then contracted with titrated concentrations of the thromboxane A2-mimetic, U46619, to ∼40% of their KPSSmax. Upon reaching a stable plateau, rings were exposed to cumulatively increasing half-log molar concentrations of either BK or NO-donors, S-nitroso-N-acetylpenicillamine (SNAP) and sodium nitroprusside (SNP), or with either single concentrations of levcromakalim (0.6 μM) or KCl (2.5–10 mM).
Statistics
All cumulative concentration-relaxation curves were normalized as percentages of relaxation from the initial U46619-induced contraction level. Each normalized curve was then computer-fitted (Graphpad Prism, version 1.00) with a sigmoidal regression curve of the following equation,
where X is the logarithm of the agonist concentration and Y is the response. BOTTOM is the lower response plateau, TOP is the upper response plateau and pD2 is the X value when the response is halfway between BOTTOM and TOP. The variable HILLSLOPE controls the slope of the curve. Mean sensitivity (pEC50 values), maximum relaxations (Rmax) and their standard errors (s.e.) were then calculated for each response curve. U46619-induced levels of contraction were expressed as percentages of the respective KPSSmax values for each tissue. Values of n represent number of rings of artery, each from different animals. Differences in mean pEC50 and Rmax values were tested for significance by means of one way analysis of variance (ANOVA) with multiple comparisons via Dunnett's test (when treatment groups were being compared with controls only) or Tukey-Kramer's test (when treatment groups were being compared amongst themselves as well as with controls). All differences were accepted as significant at the P<0.05 level.
Drugs and their sources
Ba2+ chloride, bradykinin triacetate, clotrimazole, indomethacin, NG-nitro-L-arginine (L-NOARG), quinacrine (Sigma, MO, U.S.A.); apamin, arachidonyl trifluoromethyl ketone (AACOCF3), 7-ethoxyresorufin, nifedipine, 1H-[1,2,4]oxadizolo[4,3-α]quinoxaline-1-one (ODQ), N,N-diethylaminoethyl-2,2-diphenylvalerate hydrochloride (SKF-525a), 1,5,5-hydroxy-11,9-(epoxymethano) prosta-5Z,13E-dienoic acid (U46619), S-nitroso-N-acetylpenicillamine (SNAP), 1,6-bis-(cyclohexyloximinocarbonylamino)-hexane (RHC 80267; Sapphire Bioscience, N.S.W., Australia); ouabain (Calbiochem); sodium nitroprusside (SNP; David Bull Laboratories, Australia) and levcromakalim (kind gift from Dr Grant McPherson).
Stock solutions of nifedipine (10 mM), ouabain (100 mM) and U46619 (1 mM) were made up in absolute ethanol, while those of indomethacin (100 mM) and L-NOARG (100 mM) were made up in Na2CO3 (1 M) and NaHCO3 (1 M), respectively. Stock solutions of AACOCF3 (1 mM), clotrimazole (100 mM), 7-ethoxyresorufin (10 mM), ODQ (10 mM), RHC 80267 (10 mM) and levcromakalim (10 mM) were made up in dimethyl sulphoxide. All subsequent dilutions of these drugs were in distilled water. All other drug stocks were made up in distilled water. Note that none of the vehicles used in this study had an effect on any component of the response to BK or on the responses to SNAP, SNP, levcromakalim and KCl.
Results
Confirmation that an EDHF-like mechanism acts as a backup relaxation mechanism for NO
BK caused concentration-dependent (pEC50, 9.74±0.16) and maximum (Rmax, 103.6±0.9%) relaxation in endothelium-intact rings of bovine isolated coronary artery contracted to ∼40% KPSSmax with U46619 (Figure 1). L-NOARG (100 μM) caused a small, significant reduction in Rmax to BK (95.8±1.6%) and although there was a trend for L-NOARG to cause a reduction in sensitivity (pEC50, 9.37±0.13), it did not reach significance (Figure 1). Also, a near-identical pattern of inhibition of responsiveness to BK was observed with ODQ (10 μM; pEC50, 9.25±0.23; Rmax, 96.2±2.1%; Figure 1). The combination of L-NOARG and ODQ was no more effective at inhibiting the response to BK than was either compound alone (pEC50, 9.29±0.12; Rmax, 97.8±2.3%; Figure 1). ODQ caused an ∼100-fold increase in the concentration of the NO donor, SNAP, required to elicit a threshold response (Figure 1).
Figure 1.
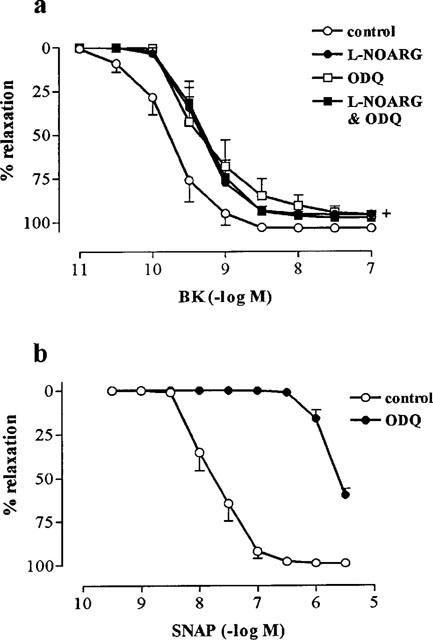
Effect of (a) the NO synthase inhibitor, L-NOARG (100 μM) and guanylate cyclase inhibitor, ODQ (10 μM), on endothelium-dependent relaxations to BK in rings of bovine coronary artery contracted with U46619. (b) Effect of ODQ (10 μM) on endothelium-independent relaxations to the NO-donor, SNAP in similarly contracted rings of bovine coronary artery. Values (mean±s.e.mean from n=6 experiments) are expressed as a percentage reversal of the initial U46619-induced contraction. (+) indicates that Rmax values for all treatments significantly different from those obtained in control tissues (P<0.05 for Tukey-Kramer's t-statistic after one way ANOVA).
In the presence of L-NOARG, the response to BK was markedly inhibited by high [K+]o (67 mM; pEC50, 8.07±0.23; Rmax, 38.6±8.6%; Figure 2). By contrast, high [K+]o had no effect on the response to BK in the absence of L-NOARG (Figure 2). A similar degree of inhibition of L-NOARG-resistant relaxation to BK was observed with a lower concentration of [K+]o (15 mM) (n=5; data not shown). Like high [K+]o, apamin (0.3 μM) inhibited the L-NOARG-resistant response to BK (pEC50, 8.35±0.13; Rmax, 51.5±10.0%) and had no effect on the control response (Figure 2). A combination of apamin (0.3 μM), glibenclamide (10 μM) and charybdotoxin (0.1 μM) was no more effective at inhibiting the L-NOARG-resistant response to BK than was apamin on its own (n=4; data not shown).
Figure 2.
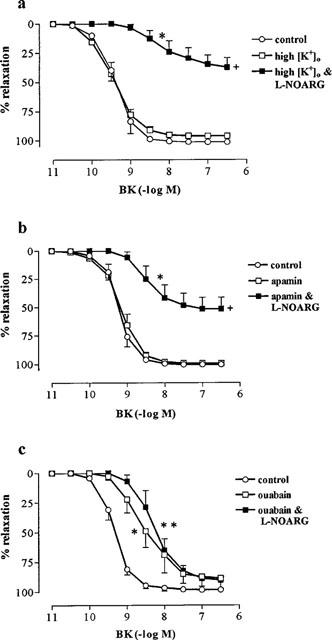
Effect of (a) 67 mM extracellular K+ (high [K+]o), (b) apamin (0.3 μM) and (c) ouabain (1 μM) on endothelium-dependent relaxations to BK in the absence and presence of L-NOARG (100 μM) in rings of bovine coronary artery contracted with U46619. Values (mean±s.e.mean from n=5–6 experiments) are expressed as a percentage reversal of the initial U46619-induced contraction. (*P<0.05 and **P<0.001) and (+P<0.001) indicate that pEC50 and Rmax values, respectively were significantly different from those obtained in control tissues (for Tukey-Kramer's t-statistic after one way ANOVA).
Effect of ouabain and Ba2+
The pEC50 (8.4±0.15; n=6) and Rmax (92.3±1.7%) to BK in tissues treated with a combination of ouabain (1 mM) and Ba2+ (30 μM) were significantly (P<0.05) reduced compared to controls. The relaxation to BK was further reduced by ouabain and Ba2+ (30 μM) in L-NOARG-treated tissues (pEC50, 7.9±0.15; Rmax, 62.1±5.5%; n=6). Ouabain alone caused the same degree of inhibition of BK-induced relaxations in the absence and presence of L-NOARG as that observed with the combination of ouabain and Ba2+ (n=6; data not shown). Ba2+ (30 μM) had no effect on control or L-NOARG-resistant responses to BK (n=6; data not shown). Ouabain (1 μM) caused a significant inhibition of the pEC50 (8.58±0.22) but unlike the higher concentration, had no effect on Rmax (88.9±8.3%) to BK (Figure 2). The subsequent addition of L-NOARG to tissues treated with 1 μM ouabain, caused no further inhibition of relaxations to BK (pEC50, 8.30±0.21; Rmax, 88.46±2.5%; Figure 2).
Combined treatment with ouabain (1 mM) and Ba2+ (30 μM) had no effect on relaxation to the NO donor, SNP (control: pEC50, 7.65±0.2; Rmax, 91.5±4.6; ouabain and Ba2+: pEC50, 7.28±0.12; Rmax, 76.3±9.3%; n=6).
Response to KCl
Increasing the extracellular concentration of K+ from 5.3 to 10.3 mM with KCl caused relaxation of 18.0±3.7% (n=5) followed by contraction (Figure 3). No further relaxation to K+ was observed when the concentration was raised to 15.3 mM (data not shown). Both relaxation (Rmax, 4.8±1.2%) and contraction to K+ were inhibited by nifedipine (0.3 μM; Figure 3). Also, the relaxation to K+ was abolished by ouabain (1 μM; Figure 3).
Figure 3.
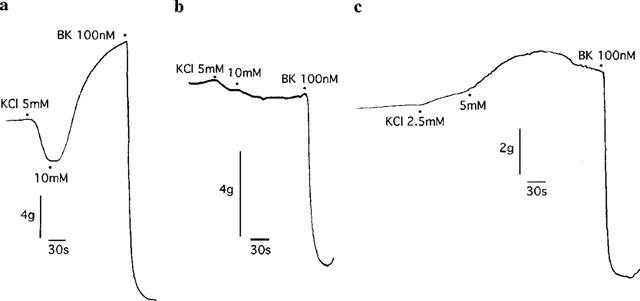
Digitized recordings showing responses to exogenous administration of 2.5–10 mM KCl in rings of bovine coronary artery contracted with U46619 in the (a) absence or presence of either (b) nifedipine (0.3 μM) or (c) ouabain (1 μM). Also shown are maximum relaxations to bradykinin (BK; 100 nM).
Effect of inhibitors of arachidonic acid formation and cytochrome P450 activity on NO-independent relaxations to BK and responses to levcromakalim
Quinacrine (30 μM), SKF 525a (100 μM) and clotrimazole (100 μM), each significantly reduced both the Rmax (63.0±8.6%, 45.8±7.0%, 42.1±15.2%, respectively) and sensitivity (pEC50, 8.21±0.18, 8.09±0.14, 8.64±0.17, respectively) of the L-NOARG-resistant response to BK (pEC50, 8.98±0.08; Rmax, 94.7±2.1%; Figure 4). By contrast, AACOCF3 (3 μM) and RHC 80267 (30 μM), either alone (n=6; data not shown) or in combination (pEC50, 8.77±0.23; Rmax, 88.9±2.2%) and 7-ethoxyresorufin (10 μM; pEC50, 9.15 ±0.17; Rmax, 81.8±4.2%) had no significant effect on the L-NOARG-resistant response to BK (Figure 4).
Figure 4.
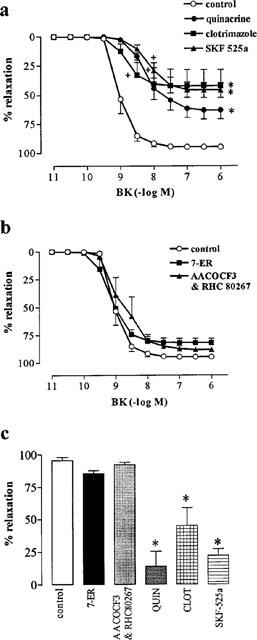
Effect of inhibitors of free arachidonic acid formation [quinacrine (QUIN; 30 μM); AACOCF3 (3 μM); RHC 80267 (30 μM)] and cytochrome P450 activity [7-ethoxyresorufin (7-ER; 10 μM); clotrimazole (CLOT; 100 μM); SKF 525a (100 μM)] on L-NOARG-resistant responses to BK (a and b) and on relaxations to levcromakalim (0.6 μM; c) in rings of bovine coronary artery contracted with U46619. All values (mean±s.e.mean from n=4–6 experiments) are expressed as a percentage reversal of the initial U46619-induced contraction. * (P<0.01) and (+) (P<0.01) indicate Rmax and pEC50 values significantly different from control (Dunnett's modified t-statistic after a single one-way ANOVA).
The ATP-sensitive K+ channel opener, levcromakalim (0.6 μM), caused a near-maximum relaxation (Rmax, 95.3±2.5%) which was unaffected by either 7-ethoxyresorufin (10 μM) or the combination of RHC 80267 (30 μM) and AACOCF3 (3 μM; Figure 4). By contrast, quinacrine (30 μM), clotrimazole (100 μM) and SKF-525a (100 μM) significantly reduced the Rmax to levcromakalim to 13.8±11.5% 45.0±13.7% and 21.8±5.1%, respectively (Figure 4).
Effect of inhibitors of arachidonic acid formation and cytochrome P450 activity on non-NO, high K+-resistant relaxations to BK
While both AACOCF3 (n=5) and RHC 80267 (n=6) alone had no effect on non-NO, K+-resistant relaxation to BK, in combination they virtually abolished this response (Rmax, 6.1±1.7%; Figure 5). Similarly, 7-ethoxyresorufin (Rmax, 5.4±4.0%) abolished the L-NOARG/high [K+]o-resistant relaxations to BK (Figure 5), despite the fact that the latter of these compounds had no effect on the L-NOARG-resistant responses to BK.
Figure 5.
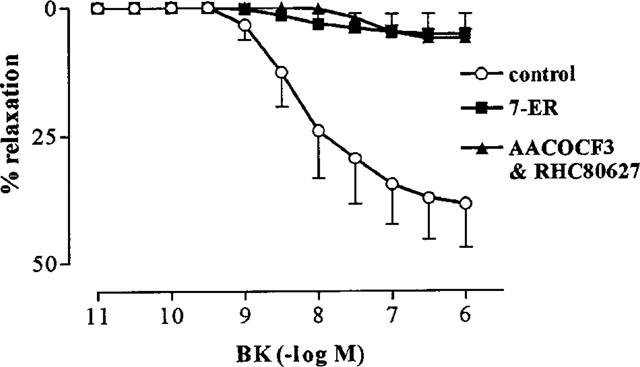
Effect of 7-ethoxyresorufin (7-ER; 10 μM) and a combination of AACOCF3 (3 μM) and RHC 80267 (30 μM) on L-NOARG/high [K+]o-resistant responses to BK in rings of bovine coronary artery contracted with U46619. All responses were obtained in the presence of L-NOARG (100 μM) and 67 mM [K+]o. Values (mean±s.e.mean from n=6 experiments) are expressed as a percentage reversal of the initial U46619-induced contraction.
Effect of the combination of AACOCF3 and RHC 80267 and 7-ethoxyresorufin on control and high [K+]o-resistant relaxation to BK
In the absence of any treatments, the combination of AACOCF3 (3 μM) and RHC 80267 (30 μM) had no effect on the control response to BK (pEC50, 9.3±0.2; Rmax, 99.9±2.0%; n=6). Under the same conditions, however, 7-ethoxyresorufin (10 μM) caused a significant (P<0.01) reduction in Rmax to BK (82.8±3.9%; n=5) without affecting the pEC50.
Similar to its effects on the control response, the combination of AACOCF3 (3 μM) and RHC 80267 (30 μM) also had no effect on the high [K+]o-resistant response to BK (pEC50, 9.2±0.09; Rmax, 95.7±2.3%; n=6). By contrast, 7-ethoxyresorufin (10 μM) abolished this response (Rmax, 4.8±3.7%; n=5; P<0.01). Concentration-dependent relaxations to SNAP (pEC50, 7.80±0.11; Rmax, 98.4±0.7%; n=12) were unaffected by the combination of AACOCF3 and RHC 80267 (n=5) but were markedly inhibited by 7-ethoxyresorufin (pEC50, 5.65±0.64; Rmax, 20.3±6.6; n=6, P<0.01 for both values).
Discussion
The results of this study suggest that a cytochrome P450-derived metabolite of arachidonic acid is involved in endothelium-dependent relaxation in the bovine coronary artery, but that it does not mediate the component of the response attributable to an EDHF-like mechanism. The cytochrome P450-dependent mechanism appeared to be activated only by high concentrations of BK, to function as a backup with the EDHF-like mechanism for NO, the predominant non-prostanoid, endothelium-derived relaxing factor (EDRF) in this tissue. Also, our findings do not support a role for K+ as an EDRF in the bovine coronary artery.
Recently, Fisslthaler et al. (1999) reported that cytochrome P450 2C is an EDHF synthase in the porcine coronary artery. Our findings that both the PLA2 inhibitor, quinacrine, and the cytochrome P450 inhibitors, SKF-525a and clotrimazole, all attenuated the L-NOARG-resistant response to BK in the bovine coronary artery appear to support the notion that a cytochrome P450-derived metabolite of arachidonic acid is involved in a smooth muscle relaxation mechanism attributable to EDHF (Komori & Vanhoutte, 1990; Fulton et al., 1992; Hecker et al., 1994; Bauersachs et al., 1994; Mombouli & Vanhoutte, 1997; Feletou & Vanhoutte, 1999). However, the inhibitory effects of these compounds most likely resulted from non-specific actions on smooth muscle K+ conductance since each compound also blocked relaxation to the K+ channel opener, levcromakalim. Similar inhibition of relaxations to the KATP channel openers, levcromakalim and pinacidil, has been reported in rat hepatic (Zygmunt et al., 1996) and mesenteric (Fukao et al., 1997) arteries by quinacrine and SKF-525a. By contrast, neither AACOCF3, an inhibitor of PLA2 which is structurally and mechanistically distinct from quinacrine (Gelb et al., 1994), nor 7-ethoxyresorufin, a xenobiotic cytochrome P450 inhibitor (Tassaneeyakul et al., 1993) which inhibits endothelium-dependent relaxations to arachidonic acid in the perfused rat kidney (Oyeken et al., 1991), had any effect on relaxations to either levcromakalim or BK after NO inhibition. Also, combined PLA2 and DAG lipase inhibition with AACOCF3 and RHC 80267, respectively, failed to have any effect on the response to BK after NO inhibition. Therefore, relaxations to the EDHF-like mediator do not appear to involve cytochrome P450-derived metabolites of arachidonic acid, at least in bovine coronary arteries.
A cytochrome P450-derived metabolite of arachidonic acid may, however, have mediated the component of the response to BK not attributable to either NO or EDHF, since inhibition of the two main sources of arachidonic acid with AACOCF3 and RHC 80267, as well as inhibition of cytochrome P450 with 7-ethoxyresorufin abolished this L-NOARG/high [K+]o-resistant response. 7-ethoxyresorufin is a relatively specific competitive substrate inhibitor of 1A isoforms of cytochrome P450 (Tassaneeyakul et al., 1993). Also, cytochrome P4501A isozymes have been immunohistochemically localized in both mammalian and non-mammalian endothelial cells. Thus, the block by 7-ethoxyresorufin of L-NOARG/high [K+]o-resistant relaxations to BK may implicate a role for members of the cytochrome P4501A family in this low efficacy component of the response.
While 7-ethoxyresorufin inhibited relaxations to BK in the presence of L-NOARG and high K+, it also blocked the NO-dependent component of the response. This effect most likely occurred downstream of NO synthase since relaxations to the NO-donor, SNAP, were also markedly attenuated by 7-ethoxyresorufin. These findings are consistent with previous studies which have shown 7-ethoxyresorufin inhibits endothelium-dependent relaxation known to be due solely to NO (Rees et al., 1990) as well as relaxation to exogenous NO in the rat isolated aorta (Bennett et al., 1992; Oyeken et al., 1994; Li & Rand, 1996) and guinea-pig taenia coli (Selemidis et al., 1997). One possible mechanism underlying these inhibitory actions of 7-ethoxyresorufin involves the generation of superoxide anions which are known to inactivate NO (Ignarro et al., 1988). Irrespective of the mechanism involved, however, the findings here that 7-ethoxyresorufin abolished all the response to BK in the presence of high K+ indicates that it may prove to be a useful compound to study responses due to EDHF-like mechanisms in isolation from other non-prostanoid relaxing factors.
Although the nature of the cytochrome P450-derived metabolite of arachidonic acid which appeared to be involved in the response to BK is unknown, two possible classes of compounds are EETs and HETEs. Such compounds are not only released from bovine endothelial cells in culture, but also have been shown to relax isolated segments of bovine coronary arteries (Rosolowsky et al., 1996; Rosolowsky & Campbell, 1996; Gebremedhin et al., 1998; Pratt et al., 1998). If these metabolites did play a role in mediating the non-NO/[K+]-resistant component, they need not have acted in a paracrine manner. For example, inhibitors of cytochrome P450 are known to prevent the influx of Ca2+ that follows agonist-induced depletion of intracellular Ca2+ stores in many cell types including bovine coronary artery endothelial cells (Alvarez et al., 1992; Graier et al., 1995). Furthermore, the exogenous application of 5,6-EET to bovine coronary and human umbilical artery endothelial cells was shown to cause a sustained increase in intracellular Ca2+ which was similar in time course and magnitude to the second phase increase in Ca2+ observed in response to BK (Graier et al., 1995). Taken together, these observations suggest that EETs can act as intracellular second messengers in endothelial cells, signalling transmembrane Ca2+ influx in response to agonist-induced depletion of internal Ca2+ stores (Baron et al., 1997). In the bovine coronary artery, non-NO/high [K+]o-resistant relaxations occurred over a higher concentration range of BK than that which stimulated NO and the EDHF-like mechanism. This may indicate that activation of the remaining non-prostanoid mechanism requires higher levels of intracellular endothelial cell Ca2+. Thus, cytochrome P450-derived metabolites of arachidonic acid like EETs and HETEs may act intracellularly to raise endothelial cell Ca2+ levels high enough to activate an as yet unidentified smooth muscle relaxing mechanism.
The present study extends our earlier findings in the same tissue (Drummond & Cocks, 1996) that NO is the predominant mediator of endothelium-dependent relaxation with the EDHF-like and the low-efficacy, cytochrome-P450-dependent mechanisms, both acting as backup vasodilator systems for NO. Thus, the NO synthase inhibitor, L-NOARG, and the guanylate cyclase inhibitor, ODQ (Garthwaite et al., 1995), when used alone or in combination with each other, inhibited relaxations to BK to the same small but significant degree. Also, the NO-independent relaxation to BK was blocked by ∼60% by either high [K+]o (Drummond & Cocks, 1996) or the SKCa channel inhibitor, apamin, neither of which had any effect in the absence of L-NOARG. Furthermore, the observation that combined treatment with AACOCF3 and RHC 80267, as well as 7-ethoxyresorufin, all had no effect in L-NOARG-treated tissues when the EDHF-like mechanism was active (i.e. in the absence of high [K+]o) indicates that the cytochrome P450-dependent mechanism also acts as backup for the EDHF-like mechanism.
This study also shows that EDHF-like relaxation to BK in the bovine coronary artery is unlikely to be mediated by endothelial cell K+ (Edwards et al., 1998). Thus, while the EDHF-like mechanism mediated near maximum relaxation (∼95%), 5–10 mM extracellular K+ caused only small relaxations (∼20%). Also, relaxations to K+ appeared to be mediated by activation of Na+/K+-ATPase since they were abolished by low concentrations of ouabain. By contrast, ouabain had no effect on the EDHF-like component of the response to BK. This finding supports those of Quignard et al. (1999) who showed that endothelium-dependent hyperpolarization to BK in the pig coronary artery was unaffected by ouabain. Finally, we have previously demonstrated that EDHF-like relaxations to BK in bovine and pig coronary arteries are unaffected by nifedipine in concentrations (>0.3 μM) that abolished contractions to KCl (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996). Thus, EDHF-like relaxations to BK in these tissues can occur independently of the need to close L-type VOCCs. This was not the case for the small relaxations to K+ since they were abolished by nifedipine.
Since our studies appear to exclude a role for cytochrome P450 and K+, it remains a possibility that direct communication between endothelial cells and smooth muscles cells, via heterocellular gap junctions, was involved in the EDHF-like relaxations to BK (Kúhberger et al., 1994; Chaytor et al., 1998; Dora et al., 1999; Yamamoto et al., 1999). As described for similar EDHF-like responses in the guinea-pig mesenteric artery (Yamamoto et al., 1999), we have preliminary data which shows that the proposed gap junctional uncoupling agent, 18-β-glycyrrhetinic acid (Goldberg et al., 1996), appears to inhibit EDHF-like relaxation in the bovine isolated coronary artery (Selemidis & Cocks, unpublished observations). The pattern of inhibition we observed with 18-β-glycyrrhetinic acid was similar to that with the high (1 mM) concentration of ouabain used here. That is, 18-β-glycyrrhetinic acid not only inhibited the response to BK, but it also significantly improved the degree of block by L-NOARG. High concentrations of ouabain (0.1–1 mM) are known to prevent the formation of gap junctions and conversely 18-β-glycyrrhetinic acid is known to inhibit Na+/K-ATPase (Rabito et al., 1987; Watsky et al., 1990). Since our findings with lower concentrations of ouabain suggest that Na+/K+-ATPase is not involved in the EDHF-like response in the bovine coronary artery, the shared inhibitory effects of 18-β-glycyrrhetinic and high concentrations of ouabain may implicate a role for gap junctions. It remains to be determined whether such gap junctions are involved in transfer of the EDHF-like response from endothelium to smooth muscle (i.e. myoendothelial gap junctions) or if they co-ordinate the spread of hyperpolarization within a single layer of the vessel wall (i.e. endothelial-endothelial or muscle-muscle).
In conclusion, our studies show that three non-prostanoid factors operate in parallel to mediate endothelium-dependent relaxations to BK in the bovine coronary artery. NO is the dominant factor and is responsible for most of the response. After block of NO, an EDHF-like mechanism which does not involve either endothelial K+ or a cytochrome P450-derived metabolite of arachidonic acid, compensates for nearly all of the maximum relaxation, whilst inhibition of both NO and the EDHF-like mechanism reveals a second level of backup perhaps mediated by a cytochrome P450-dependent mechanism.
Acknowledgments
This work was supported by grants from the National Health & Medical Research Council and the National Heart Foundation of Australia.
Abbreviations
- AACOCF3
arachidonyl trifluoromethyl ketone
- ACh
acetylcholine
- Ba2+
barium
- BK
bradykinin
- BKCa
large conductance Ca2+-activated K+ channel
- DAG
diacylglycerol
- EDHF
endothelium-derived hyperpolarizing factor
- EDRF
endothelium-derived relaxing factor
- EET
epoxyeicosatrienoic acid
- HETE
hydroxyeicosatetranoic acid
- high [K+]o
high extracellular K+
- KIR
inward-rectifying K+ channel
- L-NOARG
NG-nitro-L-arginine
- NO
nitric oxide
- ODQ
1H-[1,2,4]-oxadiazolo[4,3-α]quinoxaline 1-one
- PLA2
phospholipase A2
- RHC 80267
1,6-bis-(cyclohexyloximinocarbonylamino)-hexane
- SKCa
small conductance Ca2+-activated K+ channel
- SKF-525a
N,N-diethylaminoethyl-2,2-diphenylvalerate
- SNAP
S-nitroso-N-acetylpenicillamine
- SNP
sodium nitroprusside
- U46619
1,5,5-hydroxy-11,9-(epoxymethano)prosta-5Z,13E-dienoic acid
References
- ADEAGBO A.S.O., HENZEL M.K. Calcium-dependent phopholipase A2 mediates the production of endothelium-derived hyperpolarizing factor in perfused rat mesenteric prearteriolar bed. J. Vasc. Res. 1998;35:27–35. doi: 10.1159/000025562. [DOI] [PubMed] [Google Scholar]
- ALVAREZ J., MONTERO M., GARCIA-SANCHO J. Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB J. 1992;6:786–792. doi: 10.1096/fasebj.6.2.1537469. [DOI] [PubMed] [Google Scholar]
- BARON A., FRIEDEN M., BÉNY J.L. Epoxyeicosatrienoic acids activate a high-conductance, Ca2+-dependent K+ channel on pig coronary artery endothelial cells. J. Physiol. 1997;504:537–543. doi: 10.1111/j.1469-7793.1997.537bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUERSACHS J., FLEMING I., SCHOLZ D., POPP R., BUSSE R. Endothelium-derived hyperpolarizing factor, but not nitric oxide, is reversibly inhibited by brefeldin A. Hyperten. 1997;30:1598–1605. doi: 10.1161/01.hyp.30.6.1598. [DOI] [PubMed] [Google Scholar]
- BAUERSACHS J., HECKER M., BUSSE R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid metabolite in the coronary microcirculation. Br. J. Pharmacol. 1994;113:1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENNETT B.M., MCDONALD B.J., NIGAM R., LONG P.G., SIMON W.C. Inhibition of nitrovasodilator- and acetylcholine-induced relaxation and cyclic GMP accumulation by the cytochrome P-450 substrate, 7-ethoxyresorufin. Can. J. Physiol. Pharmacol. 1992;70:1297–1303. doi: 10.1139/y92-181. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., MARTIN P.E.M., CHAYTOR A.T., EVANS W.H., GARLAND C.J., GRIFFITH T.M. Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarisation: inhibition by a connexin-mimetic peptide. Biochem. Biophys. Res. Commun. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Evidence for mediation by endothelium-derived hyperpolarizing factor of relaxation to bradykinin in the bovine isolated coronary artery independently of voltage-operated Ca2+ channels. Br. J. Pharmacol. 1996;117:1035–1040. doi: 10.1111/j.1476-5381.1996.tb16693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–271. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelial dysfunction: a novel therapeutic target. The alternative: EDHF. J. Mol. Cell Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULTON D., MCGIFF J.C., QUILLEY J. Contribution of NO and cytochrome P450 to the vasodilator effect of bradykinin in the rat kidney. Br. J. Pharmacol. 1992;107:722–725. doi: 10.1111/j.1476-5381.1992.tb14513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULTON D., MCGIFF J.C., QUILLEY J. Role of phospholipase C and phospholipase A2 in the nitric oxide-independent vasodilator effect of bradykinin in the rat perfused heart. J. Pharmacol. Exp. Ther. 1996;278:518–526. [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends. Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one. J. Pharmacol. Exp. Ther. 1995;48:184–188. [PubMed] [Google Scholar]
- GEBREMEDHIN D., HARDER D.R., PRATT P.F., CAMPBELL W.B. Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries:role of a cytochrome P450 metabolite. J Vascular Res. 1998;35:274–284. doi: 10.1159/000025594. [DOI] [PubMed] [Google Scholar]
- GEBREMEDHIN D., MA Y-H., FALCK J.R., ROMAN R.J., VANROLLINS M., HARDER D.R. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am. J. Physiol. 1992;263:H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- GELB M.H., JAIN M.K., BERG O.G. Inhibition of phospholipase A2. FASEB J. 1994;8:916–924. doi: 10.1096/fasebj.8.12.8088457. [DOI] [PubMed] [Google Scholar]
- GOLDBERG G.S., MORENO A.P., BECHBERGER J.F., HEARN S.S., SHIVERS R.R., MACPHEE D.J., ZHANG Y.C., NAUS C.C.G. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp. Cell Res. 1996;222:48–53. doi: 10.1006/excr.1996.0006. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., STUREK M. Cytochrome P450-mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU S., KIM H.S. Activation of K+ channel in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. Eur. J. Pharmacol. 1993;230:215–221. doi: 10.1016/0014-2999(93)90805-r. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., BYRNS R.E., BUGA G.M., WOOD K.S., CHAUDHURI G. Pharmacological evidence that endothelium-derived relaxing factor is nitric oxide: Use of pyrogallol and superoxide dismutase to study endothelium-dependent and nitric oxide-elicited vascular smooth muscle relaxation. J. Pharmacol. Exp. Ther. 1988;244:181–189. [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMORI K., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor. Blood Vessels. 1990;27:238–245. doi: 10.1159/000158815. [DOI] [PubMed] [Google Scholar]
- KÚHBERGER E., GROSCHNER K., KUKOVETZ W.R., BRUNNER F. The role of myoendothelial cell contact in non-nitric oxide-, non-prostanoid-mediated endothelium-dependent relaxation of porcine coronary artery. Br. J. Pharmacol. 1994;113:1289–1294. doi: 10.1111/j.1476-5381.1994.tb17138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI C.G., RAND M.J. Inhibition of NO-mediated responses by 7-ethoxyresorufin, a substrate and competitive inhibitor of cytochrome P450. Br. J. Pharmacol. 1996;118:57–62. doi: 10.1111/j.1476-5381.1996.tb15366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOMBOULI J-V, VANHOUTTE P.M. Endothelium-derived hyperpolarising factor(s): updating the unknown. Trends Pharmacol. Reviews. 1997;18:252–256. [PubMed] [Google Scholar]
- OYEKEN A.O., MCGIFF J.C., QUILLEY J. Cytochrome P-450-dependent vasodilator responses to arachidonic acid in the isolated, perfused kidney of the rat. Circ. Res. 1991;68:958–965. doi: 10.1161/01.res.68.4.958. [DOI] [PubMed] [Google Scholar]
- OYEKEN A.O., MCGIFF J.C., ROSENCRANTZ-WEISS P., QUILLEY J. Relaxant responses of rabbit aorta: Influence of cytochrome P450 inhibitors. J. Pharmacol. Exp. Ther. 1994;268:262–269. [PubMed] [Google Scholar]
- PINTO A., ABRAHAM N.G., MULLANE K.M. Arachidonic acid-induced endothelial-dependent relaxations of canine coronary arteries: Contribution of a cytochrome P-450-dependent pathway. J. Pharmacol. Exp. Ther. 1987;240:856–863. [PubMed] [Google Scholar]
- PRATT P.F., FALCK J.R., REDDY K.M., KURIAN J.B., CAMPBELL W.B. 20-heTE relaxes bovine coronary arteries through the release of prostacyclin. Hypertension. 1998;31:237–241. doi: 10.1161/01.hyp.31.1.237. [DOI] [PubMed] [Google Scholar]
- PROCTOR K.G., FALCK J.R., CAPDEVILA J. Intestinal vasodilation by epoxyeicosatrienoic acids: Arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ. Res. 1987;60:50–59. doi: 10.1161/01.res.60.1.50. [DOI] [PubMed] [Google Scholar]
- QUAYLE J.M., DART C., STANDEN N.B. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J. Physiol. (London) 1996;494:715–726. doi: 10.1113/jphysiol.1996.sp021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperolarising factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RABITO C.A., JARRELL J.A., SCOTT J.A. Gap junctions and synchronization of polarization process during epithelial reorganization. Am. J. Physiol. 1987;253:C329–C336. doi: 10.1152/ajpcell.1987.253.2.C329. [DOI] [PubMed] [Google Scholar]
- REES D.D., PALMER R.M.J., SCHULZ R., HODSON H.F., MONCADA S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIENDEAU D., GUAY J., WEECH P.K., LALIBERTE F., YERGEY J., LI C., DESMARAIS S., PERRIER H., LIU S., NICOLL-GRIFFITH D. Arachidonyl trifluromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetranoic acid by calcium ionophore-challenged platelets. J. Biol. Chem. 1994;269:15619–15624. [PubMed] [Google Scholar]
- ROSOLOWSKY M., CAMPBELL W.B. Synthesis of hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Biochim. Biophys. Acta. 1996;1299:267–277. doi: 10.1016/0005-2760(95)00216-2. [DOI] [PubMed] [Google Scholar]
- ROSOLOWSKY M., FALCK J.R., CAMPBELL W.B. Synthesis and biological activity of epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Adv. Prostagl. Thrombox. Leukot. Res. 1996;21:213–216. [PubMed] [Google Scholar]
- ROSOLOWSKY M., FALCK J.R., WILLERSON J.T., CAMPBELL W.B. Synthesis of lipoxygenase and epoxygenase products of arachidonic acid by normal and stenosed canine coronary arteries. Circ. Res. 1990;66:608–621. doi: 10.1161/01.res.66.3.608. [DOI] [PubMed] [Google Scholar]
- SATAKE N., SHIBATA M., SHIBATA S. Endothelium- and cytochrome P450-dependent relaxation induced by isoproterenol in rat aortic rings. Eur. J. Pharmacol. 1997;319:37–41. doi: 10.1016/s0014-2999(96)00822-9. [DOI] [PubMed] [Google Scholar]
- SELEMIDIS S., SATCHELL D.G., COCKS T.M. Evidence that NO acts a redundant NANC inhibitory neurotransmitter in the guinea-pig taenia coli. Br. J. Pharmacol. 1997;121:604–611. doi: 10.1038/sj.bjp.0701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGER H.A., SAYE J.A., PEACH M.J. Effects of cytochrome P-450 inhibitors on endothelium-dependent relaxation in rabbit aorta. Blood Vessels. 1984;21:223–230. doi: 10.1159/000158515. [DOI] [PubMed] [Google Scholar]
- STREET I.P., LIN H.K., LALIBERTE F., GHOMASHCHI F., WANG Z., PERRIER H., TREMBLAY N.M., HUANG Z., WEECH P.K., GELB M.H. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- SUTHERLAND C.A., AMIN D. Relative activities of rat and dog platelet phospholipase A2 and diglyceride lipase. J. Biol. Chem. 1982;257:14006–14010. [PubMed] [Google Scholar]
- TASSANEEYAKUL W., BIRKETT D.J., VERONESE M.E., MCMANUS M.E., TUKEY R.H., QUATTROCHI L.C., GELBOIN H.V., MINERS J.O. Specificity of substrate and inhibitor probes for human cytochromes P450 1A1 and 1A2. J. Pharmacol. Exp. Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- TAYLOR S.G., WESTON A.H. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends. Pharmacol. Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- WATSKY M.A., MCCARTNEY M.D., MCLAUGHLIN B.J., EDELHAUSER H.F. Corneal endothelial junctions and the effect of ouabain. Invest. Opthalmol. Vis. Sci. 1990;31:933–941. [PubMed] [Google Scholar]
- YAMAMOTO Y, , IMAEDA K, SUZUKI H. Endothelium-dependent hyperpolarisation and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZOU A-P., FLEMING J.T., FALCK J.R., JACOBS E.R., GEBREMEDHIN D., HARDER D.R., ROMAN R.J. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K+-channel activity. Am. J. Physiol. 1996;270:F822–F832. doi: 10.1152/ajprenal.1996.270.5.F822. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS S.C., HOGESTATT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]