

Abstract
An increased sensitivity to glucose was observed in islets pre-exposed for 1 h to glibenclamide (0.1 μmol 1−1), but not to tolbutamide (100 μmol l−1), as indicated by a shift to the left of the dose-response curve (EC50 at 5.8±0.3 mmol l−1 glucose vs 10.6±0.8 in control islets; n=11, P<0.005).
According to this secretory pattern also glucose utilization at 2.5 and 5.0 mmol l−1 glucose was higher in islets exposed to glibenclamide.
Since binding to mitochondria results in an increased enzyme activity, we measured hexokinase (HK) and glucokinase (GK) activity both in a cytosolic and in a mitochondrion-enriched fractions. Cytosolic hexokinase activity was similar in islets exposed to glibenclamide and in control islets but mitochondrial hexokinase activity was significantly increased after exposure to glibenclamide (124±7 vs 51±9 nmol μg prot−1 90 min−1, P<0.01), with no change in the enzyme protein content. In contrast, glucokinase activity in the two groups of islets was similar.
When in islets < exposed to glibenclamide hexokinase binding to mitochondria was inhibited by the addition of 20 nmol l−1 dicyclohexylcarbodiimide (DCC), no increase of glucose sensitivity was observed (EC50 10.9±1.3 mmol l−1 glucose, n=3, similar to that of control islets).
These data indicate that a 1 h exposure to glibenclamide causes the beta cell to become more sensitive to glucose. This increased sensitivity is associated with (and may be due to) an increased hexokinase activity, in particular the mitochondrial-bound, more active, form. This mechanism may contribute to the hypoglycemic action of this drug.
Keywords: Pancreatic islets, glibenclamide, sulphonylurea, insulin release, glucose sensitivity, glucose metabolism, hexokinase, glucokinase, dicyclohexylcarbodiimide (DCC), mitochondria
Introduction
Sulphonylureas are widely used for the treatment of type 2 Diabetes Mellitus. Their hypoglycemic action is due to the stimulation of insulin release, after binding to a receptor located on the beta cell plasma membrane. Following this binding, closure of ATP-dependent potassium channels leads to opening of voltage-gated Ca2+ channels, an increase in cytoplasmic calcium concentration, and the subsequent stimulation of insulin containing granules exocytosis (Boyd, 1988; Groop, 1992).
This sequence of events is more evident after an acute administration of the drug. A more prolonged exposure to sulphonylureas elicits, both in vivo and in vitro, a series of less studied and understood events that deserve further investigations. In vitro studies showed that, after binding to its receptor, glibenclamide (and probably other sulphonylureas such as gliclazide or glipizide) is internalized inside the beta cell (Carpentier et al., 1986), and direct or indirect interactions may occur with intracellular targets. As a consequence, the biologic activity of glibenclamide in vivo lasts several hours, although at that time the plasma insulin concentrations are not as greatly increased as after an acute stimulation. Therefore, different effects distinct from its action on the ATP-dependent K+ channels may occur.
The present study investigates the beta cell function in cells pre-exposed to glibenclamide and the mechanisms involved.
Methods
Materials
Crude collagenase was obtained from Boehringer Mannheim (Mannheim, Germany). Culture medium CMRL-1066, heat inactivated foetal calf serum (FCS), L-glutamine and gentamycin were obtained from Gibco (Glasgow, U.K.). All other chemicals were of analytical grade. Polyclonal antibody against hexokinase I was kindly provided by Dr John E. Wilson (Michigan State University, Michigan, U.S.A.).
Drugs
Glibenclamide and Tolbutamide were purchased from Sigma-Aldrich (St. Louis, Missouri, U.S.A.).
Islet preparation and culture conditions
Pancreatic islets were isolated by the collagenase method from 200 to 250 g fed male Wistar rats injected i.p. with 0.2 ml of a 0.2% pilocarpine solution 2 h before killing by decapitation. With this technique, 300–400 islets were isolated from each pancreas (Purrello et al., 1989). The whole procedure was completed within 120 min. Purified islets were cultured overnight at glucose 5.5 mmol l−1 in CMRL-1066 medium containing 10% FCS, 2 mmol l−1 L-glutamine and 10 μg ml−1 gentamycin at 37°C in a 95% air/5% CO2 atmosphere and, the following day, for 1 h in the presence or the absence of 0.1 μmol l−1 glibenclamide or 100 μmol l−1 tolbutamide.
Insulin secretion
Islets cultured in the presence or in the absence of glibenclamide or tolbutamide were washed twice in Krebs Ringer HEPES buffer (KRHB, containing (in mmol l−1): NaCl 134, KC 4.7, CaCl2 1, MgSO4 1.2, KH2PO4 1.2, HEPES 10, 0.5% BSA, pH 7.40. Groups of five purified islets were then incubated for 30 min at 37°C with glucose 0–16.7 mmol l−1 or, in some experiments, with KIC 0–40 mmol l−1; then insulin was measured by radioimmunoassay in the medium (Purrello et al., 1989). Results are expressed as insulin released in the medium (pg islet−1 30 min−1).
Glucose phosphorylating activity
Glucose phosphorylation rate was measured by measuring the rate of glucose-6-phosphate formation in a fluorimetric assay (Trus et al., 1981), in cytosolic and mitochondria-enriched fractions prepared as described by Giroix et al. (1990). Pancreatic islets (∼600) were washed twice in glucose-free KRHB and then homogenized (30 strokes) in 300 μl of ice cold HEPES (10 mmol l−1) buffer containing (mmol l−1): sucrose 250, EDTA 2, DTT 5 and MgCl2 5. Islet homogenates were centrifuged (5 min at 1000×g), the supernatants collected and centrifuged for 60 min at 100,000×g (Trus et al., 1981). The pellets from this second centrifugation were sonicated (3×10 s). All the procedure was carried out at 4°C. The assay volume contained 4 μl of supernatant in 100 μl of (mmol l−1): HEPES hydrochloride (pH 7.7) 50, potassium chloride (KCl) 100, magnesium chloride (MgCl) 7.4, β-mercaptoethanol 15, β-nicotinamide adenine dinucleotide (NAD+) 0.5, 0.05% BSA, 2.5 μg ml−1 glucose-6-phosphate dehydrogenase (G6PDH), 5 mmol l−1 adenosine triphosphate (ATP), and glucose concentrations ranging from 0.03 to 100 mmol l−1. In a typical experiment, glucose was added at 0.03, 0.06, 0.12, 0.25, and 0.5 mmol l−1 to measure hexokinase activity, and at 5, 7.5, 10, 15, 20, 25, 50, 65, 80, and 100 mmol l−1 to measure glucokinase activity. The reaction was stopped after 1 h at 30°C by adding 1 ml 500 mmol l−1 sodium bicarbonate buffer, pH 9.4. Fluorescence was then measured at 460 nm (excitation at 340 nm). A standard curve was obtained by incubating with assay reagents 0.3 to 1.0 nmol glucose-6-phosphate and 1 nmol NADH. In each assay, tissue blanks were also performed by incubating 0.5 or 100 mmol l−1 glucose in absence of adenosine triphosphate; in addition, at each glucose concentration, controls in the presence of 5 mmol l−1 ATP, but in absence of islet homogenate, were performed. The Vmax and Km of hexokinase and glucokinase activity were calculated in each experiment by the Eadie-Hofstee plot. To calculate the glucokinase activity, the Vmax for hexokinase was subtracted from the activities measured at concentrations higher than 5.0 mmol l−1 glucose. Protein was measured by the method of Bradford (1976).
Glucose utilization
The utilization of glucose was determined by measuring the formation of 3H2O from [5-3H]-glucose as previously described (Purrello et al., 1993). In brief, groups of 15 islets were incubated in 40 μl of Krebs Ringer Bicarbonate buffer containing 2 μCi D-[5-3H]-glucose, and glucose concentrations ranging from 2.5 to 20 mmol l−1. The incubation was carried out in 1 ml glass vials inside an airtight sealed 20 ml glass scintillation vial that contained 500 μl distilled water. After 2 h at 37°C the reaction was stopped by adding 0.5 mol l−1 HCl (100 μl injected through the rubber seal). Scintillation vials were then incubated overnight at 37°C, and the water radioactivity was measured.
Hexokinase protein levels
Hexokinase I proteins levels were measured by Western blot analysis in the cytosolic and mitochondrial fractions. Islets (∼1800) were homogenized in (mmol l−1): Tris-HCl (pH 6.8) 80, sucrose 250, MgCl2 5, EDTA 5, phenylmethylsulphonyl-fluoride 1, N-ethylmaleimide 0.2. To prepare the subcellular fractions the homogenates were centrifuged (5 min at 1000 g), the supernatants collected and centrifuged for 60 min at 100,000×g. The supernatant and the pellet of this second centrifugation were sonicated (3×10 s) in 100 μl of lysing solution containing 5% sodium dodecyl sulphate (SDS). All the procedure was carried out at 4°C. Protein content was measured by the BCA protein assay kit (Pierce, Rockford, IL, U.S.A.) using bovine serum albumin as standard. Before performing the Western blot protein content was measured, to be sure to use equivalent amount of proteins in the two experimental groups (islets exposed or not to glibenclamide). Proteins were separated by electrophoresis on a 12% polyacrilamide gels containing SDS, and transferred to nitrocellulose filters. Filters were washed at room temperature in PBS containing 0.2% Tween 20, 1% dried milk and then blocked with the same buffer containing 10% dried milk at room temperature for 1 h. Filters were then washed twice with PBS for 10 min, and incubated with polyclonal (rabbit) antibody against hexokinase I (1 : 4 000) for 1 h at room temperature. After the incubation, filters were washed and incubated with sheep anti-rabbit peroxidase-linked whole antibody (Amersham, Buckinghamshire, U.K.) at room temperature for 1 h. The filters were washed in PBS and incubated with ECL detection reagents (Amersham, U.K.) for 1 min at room temperature. They were wrapped with Saran Wrap and exposed to X-OMAT AR films (Kodak).
Statistical analysis
Statistical significance was assessed by Student's t-test for unpaired comparison.
Results
Insulin release
In pancreatic islets exposed to 0.1 μmol glibenclamide we observed an increased basal insulin secretion (196±16 vs 63±8 pg·islet−1·30 min−1 mean±s.e.mean, n=11, P<0.005) and an increased sensitivity for glucose-stimulated insulin release, as indicated by a shift to the left of the glucose dose-response curve in respect to control islets (EC50 at 5.8±0.3 and 10.6±0.8 mmol/l glucose, respectively; n=11, P<0.005). No significant difference in the maximal response was observed (Figure 1). Identical kinetics were obtained in control islets and in islets exposed for 1 h to glibenclamide. Insulin content was similar in control islets or in islets exposed for 1 h to glibenclamide (52±4 vs 49±6 ng islet−1, respectively, n=8). A more prolonged exposure to glibenclamide (12–24 h), in addition to these alterations, showed also a decrease in glucose-induced insulin release (glucose desensitization), as previously observed by several investigators (Zawalich, 1989; Henquin, 1980; Rabuazzo et al., 1992). The effect of glibenclamide on the glucose dose-response curve was not affected by the simultaneous presence of cycloheximide (5 μg l−1), an inhibitor of protein synthesis (data not shown).
Figure 1.
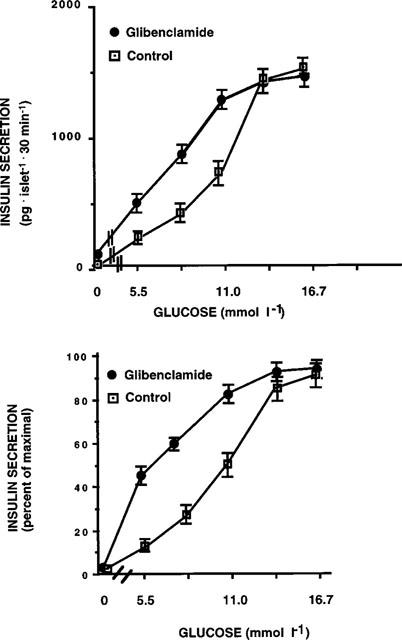
Glucose-induced insulin release in control pancreatic islets and in islets exposed for 1 h to 0.1 μmol glibenclamide (pg islet−1 30 min−1). Data are presented as absolute values in the upper panel, and as per cent of maximal release in the lower panel. Results represent the mean±s.e.mean of 11 separate experiments.
In islets exposed to 0.1 μmol l−1 glibenclamide, in contrast to what was observed with glucose, the sensitivity to alfa-ketoisocaproate (KIC) was unchanged (Figure 2). Moreover, in islets exposed for 1 h to a different sulphonylurea, tolbutamide (100 μmol l−1), the glucose dose-response curve was similar to control islets (Figure 3).
Figure 2.
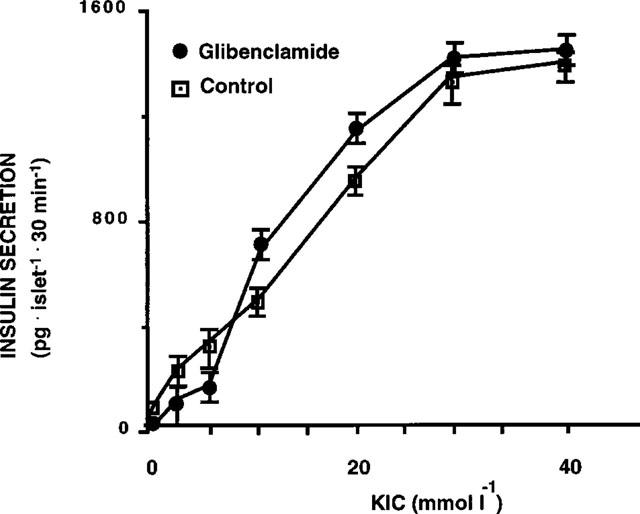
Insulin release in response to α-KIC in control pancreatic islets and in islets exposed for 1 h to 0.1 μmol l−1 glibenclamide (pg islet−1 30 min−1). Results represent the mean±s.e.mean of three separate experiments.
Figure 3.
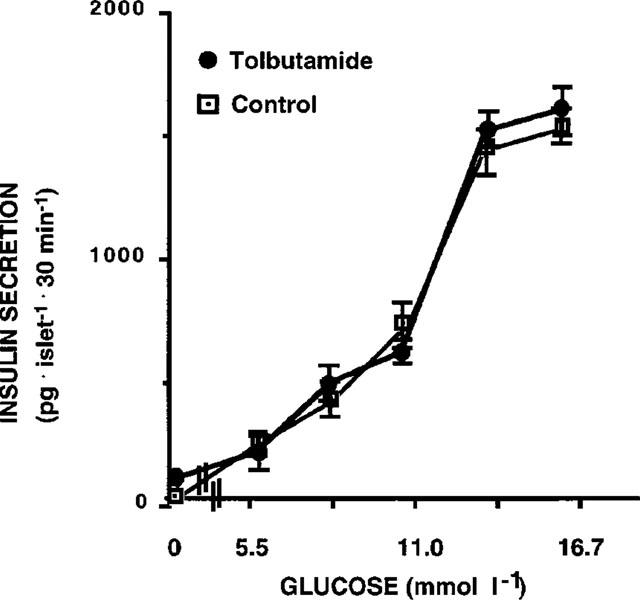
Glucose-induced insulin release in control pancreatic islets and in islets exposed for 1 h to 100 μmol l−1 tolbutamide (pg islet−1 30 min−1). Results represent the mean±s.e.mean of three separate experiments.
Glucose utilization
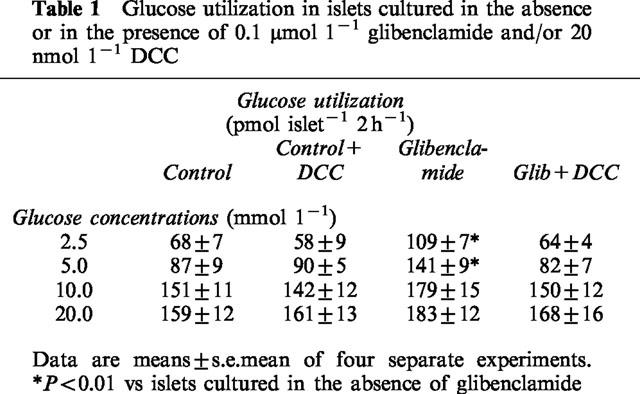
According to the insulin secretion data we found that glucose utilization at 2.5 and 5.0 mmol l−1 glucose was significantly higher in islets exposed to glibenclamide (Table 1).
Table 1.
Glucose utilization in islets cultured in the absence or in the presence of 0.1 μmol 1−1 glibenclamide and/or 20 nmol 1−1 DCC
Glucose phosphorylating activity
Hexokinase and glucokinase activity was measured in both the cytosolic fraction and in a mitochondrion enriched-fraction of pancreatic islets. In islets exposed to 0.1 μmol l−1 glibenclamide, the hexokinase activity in the cytosol was similar to control islets, but it was significantly increased in the mitochondrial fraction (Table 2). As a consequence, in islets cultured in the presence of glibenclamide the ratio between mitochondrial and cytosolic hexokinase was increased approximately 3 fold in comparison to control islets (Figure 4).
Table 2.
Glucose phosphorylating activity in islets cultured with or without 0.1 μmol 1−1 glibenclamide and/or 20 nmol 1−1 DCC
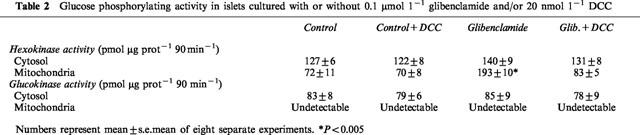
Figure 4.
Subcellular distribution of hexokinase activity in cytosolic and mitochondrial fractions of control rat islets and of islets exposed for 1 h to 0.1 μmol l−1 glibenclamide. Results represent the mean±s.e.mean of eight separate experiments.
Glucokinase activity was similar in the two groups of islets in the cytosolic fraction (83±8 and 85±9 pmol μg prot−1 90 min−1), and it was always undetectable in the mitochondrial fraction.
Effect of dicyclohexylcarbodiimide (DCC)
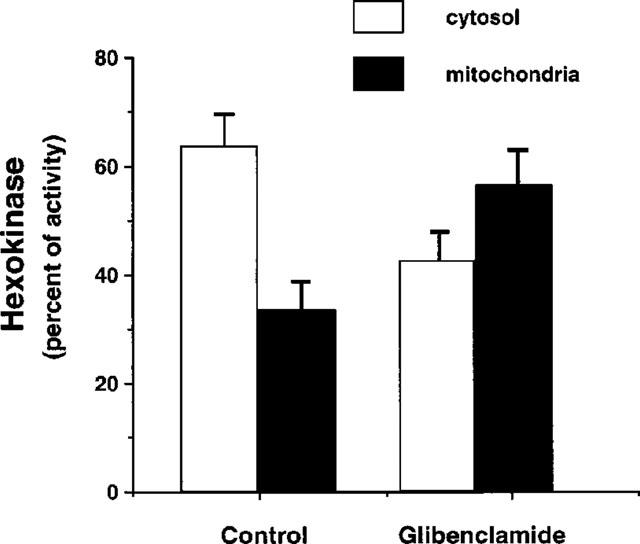
Dicyclohexylcarbodiimide (DCC) is a known inhibitor of hexokinase binding to the outer mitochondrial membrane (Nakashima et al., 1986). To further investigate in pancreatic islets the relationship between the hexokinase transfer to mitochondria and the lower secretory threshold to glucose, we measured insulin release in islets exposed for 1 h to glibenclamide in the presence of 20 nmol l−1 DCC. Incubation with DCC prevented the increased sensitivity of insulin release in response to glucose (Figure 5): in islets cultured with glibenclamide in the presence of DCC the EC50 of insulin release in response to glucose was 10.9±0.4 mmol l−1 (n=4), significantly higher than in islets incubated only with glibenclamide (5.8±0.3, P<0.05), and not different from control islets, incubated either in the presence or absence of DCC (EC50 10.9±1.3 and 10.6±0.8, respectively). Incubation with DCC also prevented the increased glucose utilization at 2.5 and 5.0 mmol l−1 glucose (Table 1), and the increase of hexokinase activity in the mitochondrial fraction (Table 2).
Figure 5.
Glucose-induced insulin release in control pancreatic islets and in islets exposed for 1 h to 0.1 μmol l−1 glibenclamide in the presence of 20 nmol l−1 dicyclohexylcarbodiimide (DCC), an inhibitor of hexokinase binding to the outer mitochondrial membrane (pg islet−1 30 min−1). Results represent the mean±s.e.mean of four separate experiments.
Hexokinase protein levels
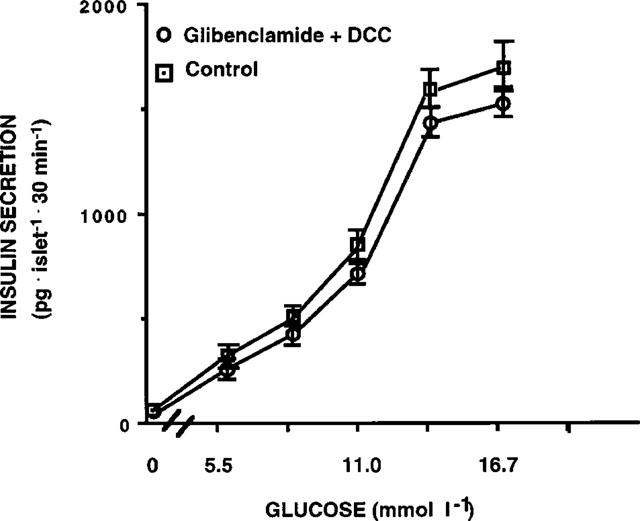
To investigate whether the difference observed in islet hexokinase activity was due to an increased enzyme content or to a different kinetic, we measured the enzyme protein content by Western blot analysis. Protein recovery was similar in control islets or in islets exposed for 1 h to glibenclamide. We found that both in the cytosolic and in the mitochondrial fraction enzyme content was similar in the two groups (Figure 6).
Figure 6.
Hexokinase protein levels measured by Western blot analysis in cytosolic and mitochondrial fractions of control rat islets and of islets exposed for 1 h to 0.1 μmol l−1 glibenclamide. A representative of three separate experiments is shown. In this experiment, protein aliquots of 20 μg of cytosol and of the mitochondria-enriched fraction were resolved by electrophoresis on a 12% polyacrylamide gel.
Discussion
These data show that a 1 h pre-exposure to glibenclamide makes pancreatic islets more sensitive to glucose. The dose-response to increasing glucose concentrations is shifted to the left, making islets responsive to glucose concentrations that normally are not stimulatory or less stimulatory. The increase in glucose sensitivity is accompanied by (and might be due to) an increase in hexokinase activity: in particular the activity of the fraction of the enzyme that is bound to mitochondria is increased, with no change in the enzyme protein content. Therefore, an increase in the enzyme specific activity is conceivable, that might be due to a direct interaction of glibenclamide with its mitochondrial receptors (Szewczyk et al., 1997). In contrast to the effects observed in islets pre-exposed to glibenclamide, pre-exposure to tolbutamide did not induce the same secretory alterations, indicating that the closure of ATP-sensitive potassium channels is not sufficient to elicit these changes. Glibenclamide effects were prevented by Dicyclohexylcarbodiimide (DCC), a known, although non-specific, inhibitor of hexokinase binding to the outer mitochondrial membrane (Nakashima et al., 1986). DCC at the same time prevented the increased sensitivity of insulin release in response to glucose and the changes in glucose utilization and hexokinase activity, making, therefore, likely the association between all these effects. This hypothesis is further supported by the observation that, in contrast to what was observed with glucose, the sensitivity to KIC (a secretagogue which enters glycolysis downstream of glucose phosphorylation) was unchanged in islets pre-exposed to glibenclamide.
Hexokinase binding to the outer mitochondrial membrane forms a receptor complex that includes the pore protein called VDAC (Nakashima et al., 1986). In this complex the enzyme forms a tetramer in contrast to the free cytosolic enzyme that maintains a monomeric form (Xie & Wilson, 1990). The enzyme may be found both free in the cytosol or bound to mitochondria (Flek et al., 1984; Linden et al., 1982). Reversible association to mitochondria represents an important mechanism for the hexokinase enzyme activity regulation. In the bound form the hexokinase sensitivity to glucose 6-phosphate inhibition is reduced compared to the soluble form (Rasschaert et al., 1990; Sener et al., 1986). In addition, a preferred access to ATP synthesized in the inner mitochondrial compartment also occurs (Brdiczka & Wallimann, 1994). The mitochondrial-bound enzyme, therefore, represents a more active form, capable of producing more G6P in a given time (Brdiczka & Wallimann, 1994). Since glucose phosphorylation is a key regulator of glucose-induced insulin release by pancreatic beta cells, the production of more G6P, by accelerating glucose metabolism, may represent a mechanism for increasing insulin secretion. Due to the low hexokinase Km for glucose, these events occur at low glucose concentrations. Our hypothesis, therefore, is that in islets exposed to glibenclamide the increased sensitivity to glucose is the consequence of the increased hexokinase activity caused by the enzyme shift to mitochondria. We have previously reported a similar finding in islets pre-exposed to 16.7 mmol glucose (Rabuazzo et al., 1997). Moreover, it has been previously reported that in RINm5F cells sulphonylureas may affect hexokinase binding to isolated porin, although a different drug was used (glimepiride) and a decreased binding was found under that experimental condition (Muller et al., 1994).
Other observations support this hypothesis. In many cultured insulinoma cells the glucose dose-response curve is markedly shifted to the left, with a maximal response reached at glucose concentrations lower than 1 mmol l−1. In these cells most hexokinase is bound to mitochondria (Shimizu et al., 1988). When hexokinase is overexpressed because of in vitro transfection (Ishihara et al., 1994) or in vivo transgene expression (Voss-McCowan et al., 1994) a shift to the left of glucose-stimulated insulin secretion is observed. Finally, overexpression of hexokinase in isolated islets through a recombinant adenovirus results in an increased secretory activity at low glucose concentrations. Also in this model, a large percentage of the overexpressed hexokinase is associated with mitochondria (Becker et al., 1994).
These findings, therefore, support the view that glibenclamide may have different effects on pancreatic beta cells distinct from its action on the ATP-dependent K+ channels. Previous data already showed that part of the enhancing effect of glibenclamide may be due to enhanced phosphoinositide hydrolysis (Zawalich, 1991). Finally, our results may help to elucidate the mechanism of action of glibenclamide in patients chronically treated with this drug. In these subjects the oral administration of one or two daily doses of glibenclamide is effective in lowering their plasma glucose concentration, but without an apparent increase of the fasting plasma insulin levels that instead return toward pretreatment levels after the initial elevation (O'Meara et al., 1990). This may be a consequence of the amelioration of blood glucose levels, and the increased sensitivity of beta cell to glucose that we have observed in this experimental model might explain the better secretory activity that these patients maintain in spite of the reduced glucose levels.
In conclusion our data show that beta cells become more sensitive to glucose after a short exposure to glibenclamide. This effect may contribute to the mechanism of action of this drug in stimulating insulin release.
Abbreviations
- ATP
adenosine triphosphate
- DCC
dicyclohexylcarbodiimide
- GK
glucokinase
- G6P
glucose-6-phosphate
- G6PDH
glucose-6-phosphate dehydrogenase
- HK
hexokinase
- KIC
alfa-ketoisocaproate
- NAD+
β-nicotinamide adenine dinucleotide
References
- BECKER T.C., BELTRANDELRIO H., NOEL R.J., JOHNSON J.H., NEWGARD C.B. Overexpression of hexokinase I in isolated islets of Langerhans via recombinant adenovirus. J. Biol. Chem. 1994;269:21234–21238. [PubMed] [Google Scholar]
- BOYD A.E. Sulfonylurea receptors, ion channels and fruit flies. Diabetes. 1988;37:847–850. doi: 10.2337/diab.37.7.847. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- BRDICZKA D., WALLIMANN T. The importance of the outer mitochondrial compartment in regulation of energy metabolism. Mol. Cell Biochem. 1994;133:69–83. doi: 10.1007/BF01267948. [DOI] [PubMed] [Google Scholar]
- CARPENTER J.L., SAWANO F., RAVAZZOLA M., MALAISSE W.J. Internalization of 3H-glibenclamide in pancreatic islet cells. Diabetologia. 1986;29:259–261. doi: 10.1007/BF00454887. [DOI] [PubMed] [Google Scholar]
- FLEK C.H., BENZ R., ROOS N., BRDICZKA D. Evidence for identity between the hexokinase binding protein of rat brain hexokinase to mitochondria. Arch. Biochem. Biophys. 1984;234:341–352. [Google Scholar]
- GIROIX M.H., SENER A., BAILBE D., PORTHA B., MALAISSE W.J. Impairment of the mitochondrial oxidative response to D-glucose in pancreatic islets from adult rats injected with streptozotocin during the neonatal period. Diabetologia. 1990;33:654–660. doi: 10.1007/BF00400566. [DOI] [PubMed] [Google Scholar]
- GROOP L.C. Sulfonylureas in NIDDM. Diabetes Care. 1992;15:737–754. doi: 10.2337/diacare.15.6.737. [DOI] [PubMed] [Google Scholar]
- HENQUIN J.C. Tolbutamide stimulation and inhibition of insulin release: studies of the underlaying ionic mechanisms in isolated rat islets. Diabetologia. 1980;18:151–160. doi: 10.1007/BF00290493. [DOI] [PubMed] [Google Scholar]
- ISHIHARA H., ASANO T., TSUKUDA K., KATAGIRI H., INUKAI K., ANAI M., KIKUCHI M., YAZAKI Y., MIYAZAKI J., OKA Y. Overexpression of hexokinase I but not Glut1 glucose transporter alters concentration dependence of glucose-stimulated insulin secretion in pancreatic β-cell line MIN6. J. Biol. Chem. 1994;269:3081–3087. [PubMed] [Google Scholar]
- LINDEN M., GELLERFORS P., NELSON B.D. Pore protein and the hexokinase binding protein from the outer membrane of rat liver mitochondria are identical. FEBS Lett. 1982;141:189–192. doi: 10.1016/0014-5793(82)80044-6. [DOI] [PubMed] [Google Scholar]
- MULLER G., KORNDORFER A., KORNAK U., MALAISSE W.J. Porin proteins in mitochondria from rat pancreatic islet cells and white adipocytes: identification and regulation of hexokinase binding by the sulfonylurea glimepiride. Arch. Biochem. Biophys. 1994;308:8–23. doi: 10.1006/abbi.1994.1002. [DOI] [PubMed] [Google Scholar]
- NAKASHIMA R.A., MANGAN P.S., COLOMBINI M., PEDERSEN P.L. Hexokinase receptor complex in hepatoma mitochondria: evidence from N,N′-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry. 1986;25:1015–1021. doi: 10.1021/bi00353a010. [DOI] [PubMed] [Google Scholar]
- O'MEARA N.M., SHAPIRO E.T., VAN CAUTER E., POLONSKY K.S. Effect of glyburide on beta cell responsiveness to glucose in non-insulin dependent diabetes mellitus. Am. J. Med. 1990;89:11S–16S. doi: 10.1016/0002-9343(90)90331-7. [DOI] [PubMed] [Google Scholar]
- PURRELLO F., BUSCEMA M., RABUAZZO A.M., CALTABIANO V., FORTE F., VINCI C., VETRI M., VIGNERI R. Glucose modulates glucose transporter affinity, glucokinase activity and secretory response in rat pancreatic β-cells. Diabetes. 1993;42:199–205. doi: 10.2337/diab.42.1.199. [DOI] [PubMed] [Google Scholar]
- PURRELLO F., VETRI M., GATTA C., GULLO D., VIGNERI R. Effects of high glucose on insulin secretion by isolated rat islets and purified β-cells and possible role of glycosylation. Diabetes. 1989;38:1417–1422. doi: 10.2337/diab.38.11.1417. [DOI] [PubMed] [Google Scholar]
- RABUAZZO A.M., BUSCEMA M., VINCI C., CALTABIANO V., VETRI M., FORTE F., VIGNERI R., PURRELLO F. Glibenclamide and Tolbutamide induce desensitization of insulin release in rat pancreatic islets by different mechanism. Endocrinology. 1992;131:1815–1820. doi: 10.1210/endo.131.4.1396327. [DOI] [PubMed] [Google Scholar]
- RABUAZZO A.M., PATANÈ G., ANELLO M., PIRO S., VIGNERI R., PURRELLO F. Hexokinase shift to mitochondria is associated to an increased sensitivity to glucose in rat pancreatic islets. Diabetes. 1997;46:1148–1152. doi: 10.2337/diab.46.7.1148. [DOI] [PubMed] [Google Scholar]
- RASSCHAERT J., MALAISSE W.J. Hexose metabolism in pancreatic islets: preferential utilization of mitochondrial ATP for glucose phosphorylation. Biochim. Biophys Acta. 1990;1015:353–360. doi: 10.1016/0005-2728(90)90040-b. [DOI] [PubMed] [Google Scholar]
- SENER A., MALAISSE-LAGAE F., GIROIX M.H., MALAISSE W.J. Hexose metabolism in pancreatic islets: compartmentation of hexokinase in islet cells. Arch. Biochem. Biophys. 1986;251:61–67. doi: 10.1016/0003-9861(86)90051-2. [DOI] [PubMed] [Google Scholar]
- SHIMIZU T., KNOWLES B.B., MATSCHINSKY F.M. Control of glucose phosphorylation and glucose usage in clonal insulinoma cells. Diabetes. 1988;37:563–568. doi: 10.2337/diab.37.5.563. [DOI] [PubMed] [Google Scholar]
- SZEWCZYK A., WOJCIK G., LOBANOV N.A., NALECZ M.J. The mitochondrial sulfanylurea receptor: identification and characterization. Biochem. Biophys. Res. Commun. 1997;230:611–615. doi: 10.1006/bbrc.1996.6023. [DOI] [PubMed] [Google Scholar]
- TRUS M.D., ZAWALICH W.S., BURCH P.T., BERNER D.K., WEILL V.A., MATSCHINSKY F.M. Regulation of glucose metabolism in pancreatic islets. Diabetes. 1981;30:911–922. doi: 10.2337/diab.30.11.911. [DOI] [PubMed] [Google Scholar]
- VOSS-MCCOWAN M.E., XU B., EPSTEIN P.N. Insulin synthesis, secretory competence, and glucose utilization are sensitized by transgenic yeast hexokinase. J. Biol. Chem. 1994;269:15814–15818. [PubMed] [Google Scholar]
- XIE G., WILSON J.E. Tetrameric structure of mitochondrially bound rat brain hexokinase: a crosslink study. Arch. Biochem. Biophys. 1990;276:285–293. doi: 10.1016/0003-9861(90)90040-6. [DOI] [PubMed] [Google Scholar]
- ZAWALICH W.S. Phosphoinoside hydrolysis and insulin secretion in response to glucose stimulation are impaired in isolated rat islets by prolonged exposure to the sulfonylurea tolbutamide. Endocrinology. 1989;125:281–286. doi: 10.1210/endo-125-1-281. [DOI] [PubMed] [Google Scholar]
- ZAWALICH W.S. Glyburide priming of beta cells: possible involvement of phosphoinoside hydrolysis. Biochem. Pharmacol. 1991;41:807–813. doi: 10.1016/0006-2952(91)90084-i. [DOI] [PubMed] [Google Scholar]