

Abstract
We examined the effects of the novel hypoglycaemic agent JTT-608 [trans-4-(4-methylcyclohexyl)-4-oxobutyric acid] on insulin secretion using rat pancreatic islets, and analysed the mechanism of its effect.
JTT-608 augmented 8.3 mM glucose-induced insulin secretion dose-dependently, and there was a stimulatory effect of 100 μM JTT-608 at both moderate and high concentrations (8.3, 11.1 and 16.7 mM) of glucose, but not at low concentrations (3.3 and 5.5 mM). In perifusion experiments, both phases of insulin release were enhanced, and the effect was eliminated 10 min after withdrawal of the agent.
In the presence of 200 μM diazoxide and a depolarizing concentration (30 mM) of K+, there was an augmentation of insulin secretion by 100 μM JTT-608, not only under high levels of glucose but also under low levels, and the effects were abolished by 10 μM nitrendipine.
JTT-608 augmented insulin secretion from electrically permeabilized islets in the presence of stimulatory concentrations (0.3 and 1.0 μM) of Ca2+, and the intracellular Ca2+ concentration ([Ca2+]i) response under 16.7 mM glucose, 200 μM diazoxide, and 30 mM K+ was also increased.
The cyclic AMP content in the islets was increased by 100 μM JTT-608, and an additive effect to 1 μM forskolin was observed, but not to 50 μM 3-isobutyl-1-methylxanthine (IBMX). JTT-608 inhibited phosphodiesterase (PDE) activity dose-dependently.
We conclude that JTT-608 augments insulin secretion by enhancing Ca2+ efficacy and by increasing Ca2+ influx. This appears to be a result of the increased intracellular cyclic AMP concentration due to PDE inhibition.
Keywords: JTT-608, pancreatic islets, insulin secretion, Ca2+ sensitivity, Ca2+ influx, cyclic AMP, phosphodiesterase inhibitor
Introduction
An impaired β-cell insulin response to glucose makes an important contribution to the metabolic derangement in non-insulin-dependent diabetes mellitus (NIDDM) (Ward et al., 1984), so an improvement of insulin secretion is effective in the treatment for patients with NIDDM who have sufficient functional β-cell reserve. To evaluate therapeutic strategies, it is essential to understand the mechanism of insulin secretion induced by glucose and of its enhancement by physiological factors. Glucose metabolism raises the ATP/ADP ratio in the β-cell and closes the ATP-sensitive K+ channels (KATP channels), leading to membrane depolarization and subsequent activation of the voltage-dependent Ca2+ channels (VDCCs). Elevation of the intracellular Ca2+ concentration ([Ca2+]i) due to Ca2+ influx through the VDCCs triggers exocytosis of insulin granules (Ashcroft & Rorsman, 1989). Circulating peptides such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) secreted from the intestine, as well as glucagon from pancreatic α-cells, are well known to be important physiological potentiators of insulin secretion. The insulinotropic effect of these peptides is due to the increase of the cyclic AMP concentration in the β-cells and subsequent activation of protein kinase A, which increases Ca2+ influx (Grapengiesser et al., 1991; Lu et al., 1993; Yada et al., 1993) through the activated VDCCs (Britsch et al., 1995; Ding & Gromada, 1997; Gromada et al., 1998) and enhances Ca2+ efficacy in the exocytotic system (Ding & Gromada, 1997; Gromada et al., 1998).
There are several ways to stimulate insulin secretion: potentiation of glucose metabolism, inhibition of KATP channel activity, increase of Ca2+ influx by VDCC activation, and enhancement of Ca2+ efficacy in insulin exocytosis. However, the only method clinically used presently in NIDDM treatment is blocking the KATP channels by sulphonylureas and other agents. A novel oral insulinotropic agent which acts on a site distinct from that of the sulphonylureas, therefore, might be a helpful supplemental clinical modality.
We have recently identified such a novel hypoglycemic agent, JTT-608 [trans-4-(4-methylcyclohexyl)-4-oxobutyric acid] (Shinkai et al., 1998). An in vivo study shows that the agent increases plasma insulin and lowers plasma glucose in both normal and NIDDM rat models. Interestingly, these effects were not observed in the basal state, but only at a stimulatory concentration of glucose, in contrast to the effects of the sulphonylureas, which also affect the basal glucose level (Ohta et al., 1999a, 1999b). JTT-608, therefore, acts at a site distinct from the sulphonylureas to stimulate insulin release, by a mechanism which remains to be clarified.
In the present study, we have analysed the mechanism of the augmentation of insulin release by JTT-608. Insulin secretory capacity was examined in both static incubation and perifusion experiments. The mechanism distal to Ca2+ influx was investigated using the [Ca2+]i response in fluorescence measurements and insulin release from electrically permeabilized islets in which [Ca2+]i can be manipulated.
Methods
Animals
Male Wistar rats were obtained from Shimizu Co. (Kyoto, Japan). The animals were fed standard lab chow ad libitum and allowed free access to water in an air-conditioned room with a 12 h light, 12 h dark cycle until used for experiments. All experiments were carried out with rats of age 8–10 weeks.
Measurement of insulin release from intact islets
Islets of Langerhans were isolated from rats by collagenase digestion (Sutton et al., 1986). Insulin secretory capacity was assessed by static incubation and in perifusion conditions using freshly isolated islets. For static incubation experiments, islets were preincubated for 30 min at 37°C in Krebs-Ringer bicarbonate buffer (KRBB) supplemented with 3.3 mM glucose and 0.2% BSA and gassed with 95% O2 and 5% CO2. Five or six islets were placed in each batch (five or eight batches per one group) and were incubated for 30 min in 0.7 ml KRBB containing 0.2% BSA and a selected concentration of glucose with test materials. At the end of the incubation period, they were placed on ice and pelleted by centrifugation (5000×g 60 s). Aliquots of the incubation buffer were diluted for insulin assay. For perifusion experiments, 20 islets were placed in each of the parallel chambers (400 μl each) of a perifusion apparatus and perifused at a continuous flow rate of 0.7 ml min−1 with a peristaltic pump at 37°C (Fujimoto et al., 1998). The buffer was continuously gassed with 95% O2 and 5% CO2. Usually, islets were perifused for 30 min with KRBB containing 5.5 mM glucose and 0.2% BSA and were then exposed to the same buffer supplemented with the indicated level of glucose and test materials. Perifusion samples were collected at the times indicated in the figures. The amount of released insulin was determined by RIA using rat insulin (Novo Nordisk, Bagsvaerd, Denmark) as the standard (Tsuji et al., 1988). Static experiments using the same protocol were repeated at least three times to ascertain reproducibility.
Measurement of insulin release from permeabilized islets
After preincubation as described above, the islets were washed twice in cold potassium aspartate buffer (KA buffer in mM) containing potassium aspartate 140, MgSO4 7, ATP 5, glucose 3.3, EGTA 2.5, HEPES 30 and 0.5% BSA (pH 7.0), with CaCl2 added to give a Ca2+ concentration of 30 nM. The islets were then permeabilized by high voltage discharge (four exposures each of 450-μs duration to an electrical field of 4.0 kV cm−1) in KA buffer and washed once with the same buffer. Five electrically permeabilized islets were then each batch-incubated for 30 min at 37°C in 0.4 ml KA buffer containing various concentrations of Ca2+ with test materials. Aliquots of the buffer after centrifugation were assayed for insulin as described above. The Ca2+ concentrations in the KA buffer were determined as previously reported (Okamoto et al., 1995). Experiments using the same protocol were repeated at least three times to ascertain reproducibility.
Measurement of [Ca2+]i
Freshly isolated islets were dispersed using 0.25% trypsin and 1 mM EDTA solution (GIBCO BRL, Grand Island, NY, U.S.A.) (Kato et al., 1996). Dispersed islet cells were suspended in KRBB supplemented with 5.5 mM glucose and 0.2% BSA and cultured on small glass coverslips (15×4 mm) coated with Cell-Tak (Collaborative Biomedical Products, Bedford, MA, U.S.A.) for more than 30 min at 37°C in a humidified incubator gassed with 95% O2 and 5% CO2. Fura-2/acetoxymethyl ester (fura-2/AM; 1 μM) (Molecular Probes, Eugene, OR, U.S.A.) was then loaded in dispersed islet cells for 20 min at 37°C. Individual coverslips were transferred to a heat-controlled chamber on the stage of an inverted microscope kept at 36±1°C and superfused with KRBB supplemented with 3.3 mM glucose. Ratiometry of the emission light (510 nm) elicited by dual wave excitation (340 and 380 nm) was performed on an ARGUS-50 image analysing system (Hamamatsu Photonics, Hamamatsu, Japan). The 340 nm (F340) and 380 nm (F380) fluorescence signals were detected every 10 s and the ratios (F340/F380) were calculated. In vivo calibration was performed as previously described (Fujimoto et al., 1998).
Measurement of cyclic AMP contents
Twenty preincubated islets were each batch-incubated for 30 min at 37°C in 0.4 ml KRBB containing 0.2% BSA with test materials. They were placed on ice after the incubation and then mixed with 0.1 ml of 30% trichloroacetic acid. After removing trichloroacetic acid by water-saturated diethylether, cyclic AMP contents were determined by RIA (Yamasa Shoyu Co., Chiba, Japan) following the succinylation step. Experiments using the same protocol were repeated at least three times to ascertain reproducibility.
Phosphodiesterase assay
Phosphodiesterase (PDE) activity was determined using a two-step radiometric assay according to the procedure of Thompson & Appleman (1971). Freshly isolated islets and liver were washed once in Tris-imidazole buffer containing (mM): Tris-HCl 40, MgCl2 5, imidazole 4, 2-mercaptoethanol 1, and CaCl2 0.08 (pH 7.5) and homogenized in the same buffer. 0.5 ml of the islet homogenates (100–200 islets) and liver homogenates (∼1 mg) were incubated for 60 min at 37°C with 0.1 mM cyclic AMP and 8 nM [3H]-cyclic AMP (NEN, Boston, MA, U.S.A.) and test materials. The reaction was terminated by heating for 2 min at 100°C. After being cooled down, 0.05 ml of 10 u ml−1 5′-nucleotidase was added to the reaction homogenates, and the mixtures were incubated for 15 min at 37°C to convert the formed [3H]-AMP into [3H]-adenosine. Unchanged [3H]-nucleotides were separated by mixing with 1 ml of AG1-X2 resin (200–400 mesh, Bio-Rad, Richmond, CA, U.S.A.) as a 1 : 3 slurry in water and centrifuging (3000×g, 5 min). [3H]-adenosine in the supernatants was quantified by liquid scintillation counting. The results are expressed as percentage of control activity without test materials.
Materials
Diazoxide, 3-isobutyl-1-methylxanthine (IBMX), forskolin, 12-O-tetradecanoyl-phorbol-13-acetate (TPA), potassium aspartate, cyclic AMP, and 5′-nucleotidase were purchased from Sigma Chemicals (St. Louis, MO, U.S.A.). Glibenclamide was obtained from Hoechst Japan (Tokyo, Japan). ATP was purchased from Kohjin (Tokyo, Japan), and the other chemicals from Nacalai Tesque (Kyoto, Japan). Test materials were prepared as stock solutions in dimethyl sulphoxide, and further diluted to given final concentrations with the buffers.
Statistical Analysis
Results are expressed as mean±s.e.mean. Statistical significance was evaluated by ANOVA and unpaired Students t-test, and P<0.05 was considered significant.
Results
Augmentation of glucose-induced insulin secretion by JTT-608
JTT-608 augmented 8.3 mM glucose-induced insulin secretion from rat islets in a dose-dependent manner. Significant augmentation was observed at concentrations above 30 μm of JTT-608. Three hundred μM JTT-608 increased insulin release about 2.3 fold from 1.21±0.12 to 2.75±0.25 ng islet−1 30 min−1 (n=5, P<0.01) (Figure 1). JTT-608 did not affect insulin release in the presence of 3.5 or 5.5 mM glucose, but the 8.3, 11.1 or 16.7 mM glucose-induced insulin secretions were augmented significantly by JTT-608 (Table 1). JTT-608, however, did not augment insulin secretion from islets which were hyperpolarized by 200 μM diazoxide, a KATP channel opener, even in the presence of 8.3 mM glucose (Table 3).
Figure 1.
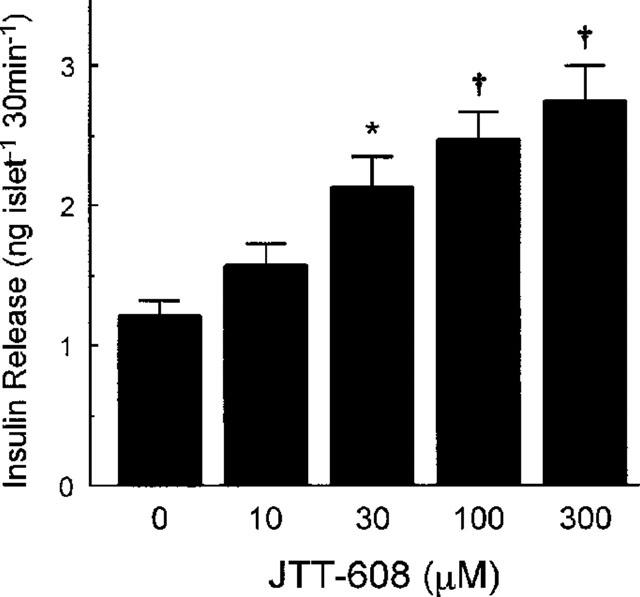
Dose-dependent effect of 100 μM JTT-608 on 8.3 mM glucose-induced insulin release from pancreatic islets. Values are mean±s.e.mean of five observations in the same experiment. *P<0.05, †P<0.01 vs control.
Table 1.
Effects of JTT-608 on insulin release in the presence of various concentrations of glucose

Table 3.
Abolishment of insulin release augmentation of JTT-608 in the presence of 8.3 mM glucose, diazoxide, and 30 mM K+ by nitrendipine
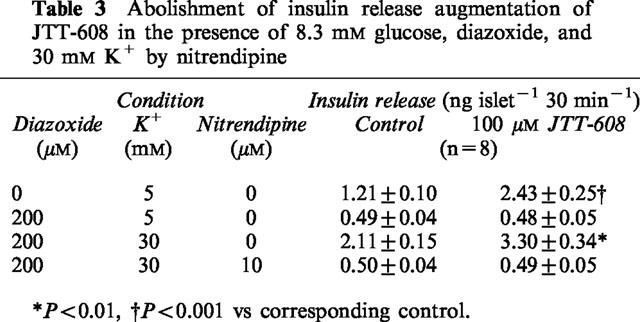
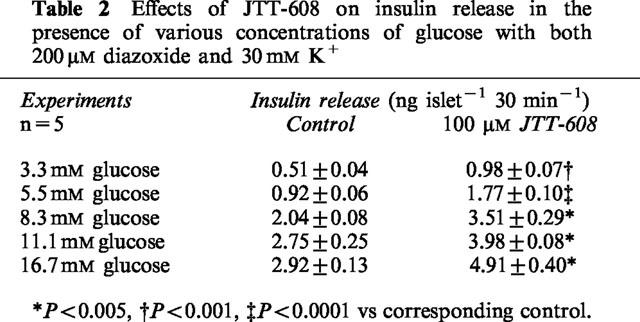
To analyse the stimulatory effect of JTT-608 on KATP channel-independent insulin secretion (Gembal et al., 1992; Sato et al., 1992), augmentation of insulin secretion was measured in the presence of 200 μM diazoxide and a depolarizing high concentration (30 mM) of K+. JTT-608 augmented KATP channel-independent insulin secretion in the presence of both high concentrations and low concentrations of glucose (Table 2). Moreover, the augmentation was completely abolished by 10 μM nitrendipine, a VDCC blocker (Table 3).
Table 2.
Effects of JTT-608 on insulin release in the presence of various concentrations of glucose with both 200 μM diazoxide and 30 mM K+
Enhancements of insulin release by JTT-608 were also found in the perifusion experiments. Insulin release in the presence of 3.3 mM glucose was not altered by 100 μM JTT-608. However, 100 μM JTT-608 significantly enhanced the 8.3 mM glucose-induced biphasic insulin release during both the first and second phase. Its effect was reversible 10 min after cessation of JTT-608 stimulation (Figure 2a). JTT-608 also significantly enhanced the high K+-induced monophasic insulin release in the presence of 200 μM diazoxide and 3.3 mM glucose (Figure 2b). Insulin release in the presence of 16.7 mM glucose, 200 μM diazoxide, and 30 mM K+ was significantly enhanced 5 min after application of JTT-608 (Figure 2c).
Figure 2.
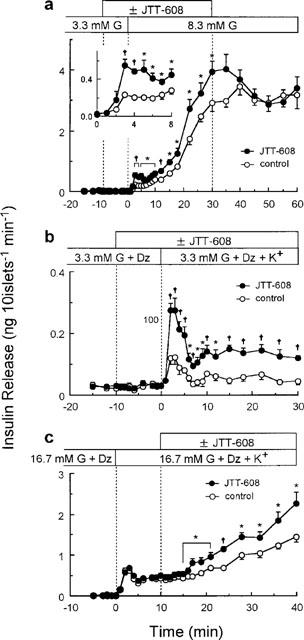
Effects of 100 μM JTT-608 on the time courses of insulin release under different conditions. (a) Effect of JTT-608 on 8.3 mM glucose-induced biphasic insulin release. Two groups of islets were perifused with 5.5 mM glucose for 30 min, and then with 3.3 mM glucose for 30 min to establish a stable rate of secretion. For an additional 30 min, they were stimulated by 8.3 mM glucose with or without 100 μM JTT-608. JTT-608 was applied from 10 min before 8.3 mM glucose stimulation. Withdrawal effect of JTT-608 was also determined for 30 min in the presence of 8.3 mM glucose alone. Inset shows the time course of insulin secretion during the first 8 min after 8.3 mM glucose stimulation in the same experiment. Values are mean±s.e.mean of six observations in the same experiment. (b) Effect of JTT-608 on a depolarizing high K+-stimulated monophasic insulin release. Two groups of islets were perifused with 5.5 mM glucose for 30 min, and then with 3.3 mM glucose and 200 μM diazoxide for 30 min to establish a stable rate of secretion. For an additional 30 min, they were stimulated by 30 mM K+ in the presence of 3.3 mM glucose and 200 μM diazoxide with or without 100 μM JTT-608. JTT-608 was applied from 10 min before 30 mM K+ stimulation. Values are mean±s.e.mean of six observations in the same experiment. (c) Effect of JTT-608 on a sustained insulin release in the presence of 16.7 mM glucose, 200 μM diazoxide, and 30 mM K+. Two groups of islets were perifused with 16.7 mM glucose and 200 μM diazoxide for 30 min, and then stimulated by 30 mM K+ under the same condition. The peak elevation of insulin release was observed after 2–3 min, and was sustained or gradually increased. They were then perifused with or without 100 μM JTT-608 from 10 min after application of 30 mM K+. Values are mean±s.e.mean of seven observations in the same experiment. *P<0.05, †P<0.01 vs corresponding control, respectively. G, glucose; Dz, diazoxide.
Additive effects of JTT-608 on insulin secretion stimulated by insulinotropic agents
Figure 3 shows the additive effects of JTT-608 on insulin secretion stimulated by various insulinotropic agents through different mechanisms. TPA, which activates protein kinase C; forskolin and IBMX, which activate protein kinase A by increasing the intracellular cyclic AMP concentration through activation of adenylyl cyclase and inhibition of PDE; and glibenclamide, which blocks KATP channels, all stimulated insulin secretion in the presence of 8.3 mM glucose dose-dependently. One hundred μM JTT-608 further increased insulin secretion stimulated by the various concentrations of TPA, forskolin, and glibenclamide (n=5, P<0.01 vs control, respectively). In contrast, JTT-608 failed to increase the insulin secretion stimulated by various concentrations of IBMX (n=5, not significant vs control) (Figure 3a).
Figure 3.
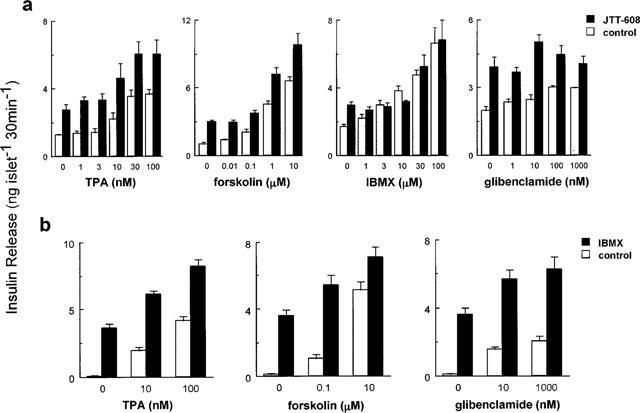
Additive effects of JTT-608 (a) or IBMX (b) on insulin release stimulated by different insulinotropic agents in the presence of 8.3 mM glucose. Values are mean±s.e.mean of five observations.
We also examined to find out if IBMX further increases the TPA, forskolin, or glibenclamide-stimulated insulin secretion, as shown in the case of JTT-608. Fifty μM IBMX increased such insulin secretion stimulated by various concentrations of each agent (n=5, P<0.01 vs control, respectively) (Figure 3b).
Effect of JTT-608 on Ca2+-induced insulin secretion from permeabilized islets
To examine the direct effect of JTT-608 on the Ca2+ sensitive exocytotic apparatus, islets were electrically permeabilized to manipulate [Ca2+]i by the extracellular Ca2+ concentration, and the insulin secretion in the presence of various concentrations of Ca2+ was measured. As shown in Figure 4, 100 μM JTT-608 augmented insulin release in the presence of stimulatory concentrations (0.3 or 1.0 μM) of Ca2+ (1.11±0.12 and 2.62±0.12 vs 0.40±0.02 and 1.70±0.18 ng islet−1 30 min−1 in control, n=5; P<0.01, respectively), but not in the presence of sub-stimulatory concentrations (0.03 or 0.1 μM) of Ca2+ (0.33±0.02 and 0.39±0.06 vs 0.29±0.04 and 0.31±0.02 ng islet−1 30 min−1 in control, n=5; not significant, respectively).
Figure 4.
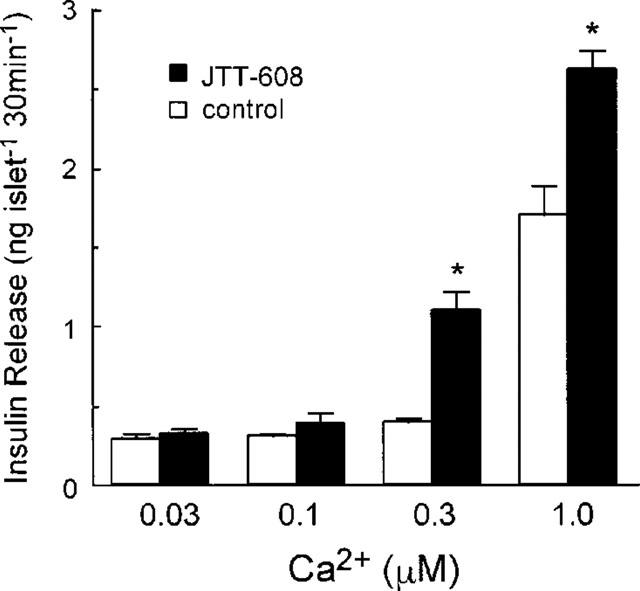
Effect of 100 μM JTT-608 on the Ca2+ dose response of insulin release from electrically permeabilized islets. Values are mean±s.e.mean of five observations in the same experiment. *P<0.01 vs corresponding control.
Effect of JTT-608 on the [Ca2+]i response in dispersed β-cells
Because the [Ca2+]i in pancreatic β-cell responds very heterogeneously to elevated glucose, it is difficult to evaluate the effect of JTT-608 on the [Ca2+]i response in the presence of glucose alone. Its effect was examined, therefore, in the presence of 16.7 mM glucose with 200 μM diazoxide and 30 mM K+, in which a relatively stabilized [Ca2+]i response can be obtained (Fujimoto et al., 1998), and control cells showed a moderate and stable elevation of the [Ca2+]i response (around 230 nM). After application of 100 μM JTT-608, no significant increase in [Ca2+]i response was observed for the first 8 min, however, a gradual increase was apparent after 9 min (about 280 nM min−1) (P<0.05 vs control) (Figure 5). The [Ca2+]i response to the basal level of 3.3 mM glucose was not affected by 100 μM JTT-608.
Figure 5.
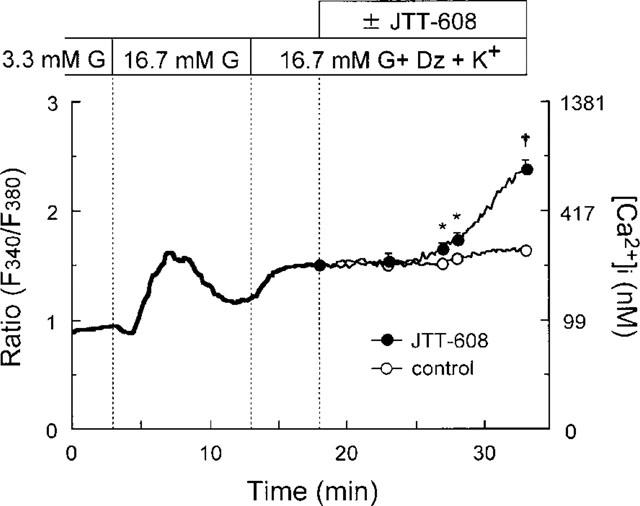
[Ca2+]i response to 100 μM JTT-608 in the presence of 16.7 mM glucose, 200 μM diazoxide, and 30 mM K+ in dispersed islet cells by fluorescence measurement. The glucose concentration was raised from 3.3 to 16.7 mM at 3 min, and cells which responded to high glucose and showed an initial decrease and subsequently a peak elevation of [Ca2+]i were regarded as β-cells. Two hundred μM diazoxide and 30 mM K+ were then applied at 13 min. The trace during the first 18 min indicates the average ratio of 21 such cells, and those from 18 to 33 min the average ratio of ten cells to which JTT-608 was applied and 11 control cells. Vertical bars show±s.e.mean at 23, 27, 28, and 33 min, respectively. *P<0.05, †P<0.01 vs corresponding control. G, glucose; Dz, diazoxide.
Increase of cyclic AMP content in islets by JTT-608
To analyse the mechanism of the enhancement of Ca2+-induced insulin secretion and of the [Ca2+]i response to JTT-608, cyclic AMP contents in islets were measured. As shown in Figure 6, 100 μM JTT-608 increased cyclic AMP content in the presence of 8.3 mM glucose alone (50.6±2.6 vs 38.1±0.6 fmol islet−1 in control, n=5; P<0.05). JTT-608 also increased cyclic AMP content in the presence of 3.3 mM glucose, 200 μM diazoxide, and 30 mM K+ (23.7±2.3 vs 16.5±1.2 fmol islet−1 in control, n=5; P<0.05). Such increase of cyclic AMP content was also elicited by 1 μM forskolin or 50 μM IBMX in the presence of 8.3 mM glucose (88.1±3.5 and 75.7±4.8 fmol islet−1 vs control, n=5; P<0.01, respectively). The cyclic AMP increase by 1 μM forskolin was further increased by 100 μM JTT-608 (127.3±4.3 fmol islet−1 in JTT-608 plus forskolin vs the value in forskolin alone, n=5; P<0.01). Such further increase was also produced by 50 μM IBMX (168.9±4.5 fmol islet−1 in IBMX plus forskolin vs the value in forskolin alone, n=5; P<0.01). However, JTT-608 did not affect the cyclic AMP increase by 50 μM IBMX (77.6±6.4 fmol islet−1 in JTT-608 plus IBMX vs the value in IBMX alone, n=5; not significant).
Figure 6.
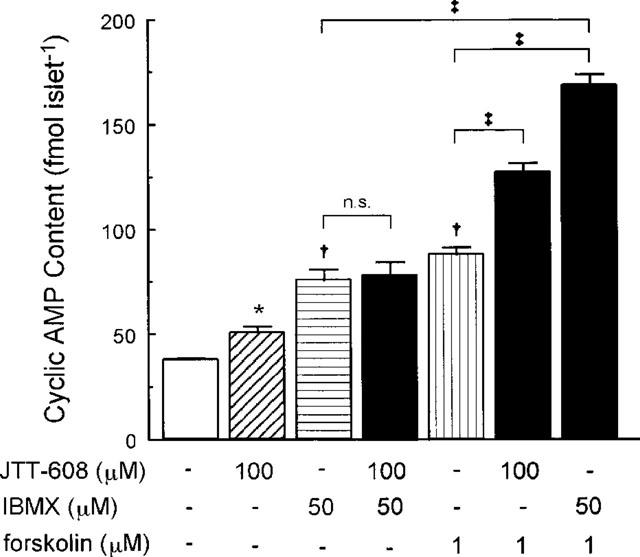
Effect of 100 μM JTT-608 on cyclic AMP content of pancreatic islet compared with that of 50 μM IBMX or 1 μM forskolin. Additive effect of JTT-608 to 50 μM IBMX or 1 μM forskolin is also shown. Values are mean±s.e.mean of five observations in the same experiment. *P<0.05, †P<0.01 vs control; ‡P<0.01; N.S., no significance.
Inhibition of phosphodiesterase activity by JTT-608
IBMX, a nonselective inhibitor of PDE, inhibited PDE activity in pancreatic islets in a dose-dependent manner. Maximum inhibition of 63.4±0.7% was produced by 500 μM IBMX. JTT-608 also inhibited islet cell PDE activity dose-dependently, and the maximum inhibition at 300 μM was 35.4±0.7%. The maximum inhibition due to JTT-608 was 55.8% of that produced by 500 μM IBMX (Figure 7a). PDE activity in hepatocytes was also inhibited by IBMX dose-dependently, with maximum inhibition of 74.3±0.4% at 500 μM. However, JTT-608 was capable of weakly inhibiting hepatocyte PDE activity, because maximum inhibition at 300 μM was 21.0±1.4%, and it was found in only 28.3% of the case of 500 μM IBMX (Figure 7b).
Figure 7.
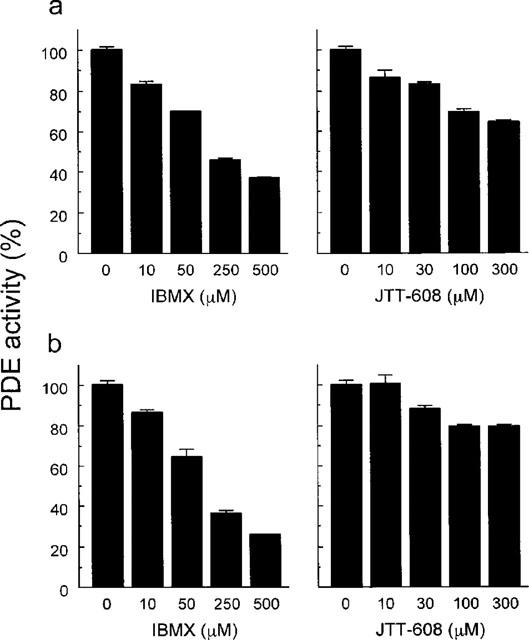
Dose-dependent inhibition of PDE activity in pancreatic islet (a) and in liver (b). Values are mean±s.e.mean of three observations in the same experiment, respectively.
Discussion
The present study shows that the novel hypoglycaemic agent JTT-608 augments glucose-induced insulin secretion from pancreatic islets at middle and high levels of glucose but not at a low level. In addition, the [Ca2+]i response in the presence of a low concentration of glucose was found not to be increased by the agent. Moreover, 100 μM JTT-608 elicited no change in KATP channel unitary amplitude or open probability (data not shown). JTT-608, therefore, unlike sulphonylureas is not a depolarizing agent.
Once depolarization was induced by a high concentration of K+, JTT-608, even in the presence of a low concentration of glucose, enhanced insulin secretion from diazoxide-treated islets. The enhancement was abolished by the VDCC blocker nitrendipine, indicating that Ca2+ influx is required for the insulinotropic effect of this agent. Thus, it is likely that augmentation of Ca2+ influx under the depolarized condition and enhancement of Ca2+ efficacy in the exocytotic system distal to the [Ca2+]i rise are involved in the insulinotropic mechanism of this agent.
Cyclic AMP is important as an intracellular messenger that activates protein kinase A and potentiates insulin release (Sharp, 1979). It enhances VDCC activity in a depolarized condition (Ämmälä et al., 1993), which is followed by increased Ca2+ influx (Grapengiesser et al., 1991; Yada et al., 1993; Yaekura et al., 1996), and directly enhances Ca2+ efficacy in the exocytotic mechanism (Tamagawa et al., 1985; Jones et al., 1986; Ämmälä et al., 1993). The cyclic AMP content was increased by 100 μM JTT-608 and, interestingly, JTT-608 further increased the cyclic AMP increase by 1 μM forskolin. In contrast, the cyclic AMP increase by 50 μM IBMX was not affected by the addition of JTT-608, suggesting that the agent affects PDE activity. A simple addition might be expected even if JTT-608 has same effects as IBMX since the concentration of IBMX is submaximal. JTT-608, however, did not increase the cyclic AMP content in the presence of IBMX. Such effects of JTT-608 also were observed in the case of insulin secretion, where JTT-608 did not affect the insulin secretion stimulated by even low concentrations of IBMX. No simple addition by JTT-608 seems to be due to inhibition of multiple PDE isoforms (Beavo, 1995; Manganiello et al., 1995) by IBMX that is a non-selective inhibitor, but the details are unknown. A further increase of forskolin-induced cyclic AMP increase by JTT-608 seems not to be a simple addition since such increase was similarly observed in the case of IBMX. The increase by JTT-608 was little less than that by IBMX, but which is consistent with the cyclic AMP increase by each agent alone. The present study clearly shows that JTT-608 inhibits PDE activity by direct enzyme assay.
PDE isoforms exist in pancreatic β-cells and play an important role in insulin secretion (Shafiee-Nick et al., 1995; Parker et al., 1995). Indeed, it has been demonstrated that PDE could be involved in the modulation of insulin release by biological factors such as insulin-like growth factor 1 (Zhao et al., 1997) and leptin (Zhao et al., 1998). It is possible, however, that JTT-608 also affects PDE activity in other tissues. The present data show JTT-608 to weakly inhibit PDE activity in hepatocytes. This effect could have some influence on the regulation of glucose homeostasis, but hypoglycaemia and an increase in plasma insulin by JTT-608 occurred simultaneously in our previous in vivo study (Ohta et al., 1999a, 1999b), consistent with the present in vitro study. The PDE inhibition by JTT-608 was not fully effective compared with IBMX. The agent might have other effects than PDE inhibition. However, the inhibition of multiple PDEs by IBMX could induce a substantial effect, even though IBMX itself might have other sites of action. It is important to identify the PDE isoform that is the target protein of JTT-608 by further investigations.
It is noteworthy that the significant elevation of [Ca2+]i and the augmentation of insulin secretion were observed in this study later than 5 min after application of the agent. Conversely, the potent PDE inhibitor IBMX augments [Ca2+]i and insulin secretion within a few minutes (Siegel et al., 1980; Yaekura et al., 1996). The reason for this interesting discrepancy is still to be determined, but the difference in intracellular distribution of JTT-608 in β-cells could take a longer time to affect the inhibition of PDE activities than IBMX.
Leibowitz et al. (1995) reported that an oral hypoglycaemic agent, arylpiperazine, an agent structurally distinct from JTT-608, stimulates insulin release by inhibiting PDEs and subsequently increasing cyclic AMP. However, although they showed the importance of the increased [Ca2+]i response in the mechanism, the effect on Ca2+ efficacy was not indicated. Our data demonstrate that JTT-608 augments insulin secretion from pancreatic β-cells both by increasing Ca2+ influx and enhancing Ca2+ efficacy in the exocytotic system of insulin granules.
Acknowledgments
This study was supported in part by Grants-in-Aids for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan; Grant-in-Aid for Creative Basic Research (10NP0201) from the Ministry of Education, Science, Sports and Culture of Japan; by a grant from ‘Research for the Future' Program of the Japan Society for the Promotion of Science (JSPS-RFTF97I00201); and by a grant for Diabetic Research from Tsumura & Co., Japan. The first author (E. Mukai) is a research fellow of the Japan Society for the Promotion of Science.
Abbreviations
- [Ca2+]i
intracellular Ca2+ concentration
- GLP-1
glucagon-like peptide-1
- IBMX
3-isobutyl-1-methylxanthine
- KA buffer
potassium aspartate buffer
- KATP channel
ATP-sensitive K+ channel
- KRBB
Krebs-Ringer bicarbonate buffer
- NIDDM
non-insulin-dependent diabetes mellitus
- PDE
phosphodiesterase
- TPA
12-O-tetradecanoyl-phorbol-13-acetate
- VDCC
voltage-dependent Ca2+ channel
References
- ÄMMÄLÄ C., ASHCROFT F.M., RORSMAN P. Calcium-independent potentiation of insulin release by cyclic AMP in single β-cells. Nature. 1993;363:356–358. doi: 10.1038/363356a0. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. Properties and functions of ATP-sensitive K-channel. Prog. Biophys. Molec. Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- BEAVO J.A. Cyclic nucleotide phosphodiesterases: functional implication of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- BRITSCH S., KRIPPEIT-DREWS P., LANG F., GREGOR M., DREWS G. Glucagon-like peptide-1 modulates Ca2+ current but not K+ATP current in intact mouse pancreatic B-cells. Biochem. Biophys. Res. Commun. 1995;207:33–39. doi: 10.1006/bbrc.1995.1149. [DOI] [PubMed] [Google Scholar]
- DING W.-G., GROMADA J. Protein kinase A-dependent stimulation of exocytosis in mouse pancreatic β-cells by glucose-dependent insulinotropic polypeptide. Diabetes. 1997;46:615–621. doi: 10.2337/diab.46.4.615. [DOI] [PubMed] [Google Scholar]
- FUJIMOTO S., ISHIDA H., KATO S., OKAMOTO Y., TSUJI K., MIZUNO N., UEDA S., MUKAI E., SEINO Y. The novel insulinotropic mechanism of pimobendan: direct enhancement of the exocytotic process of insulin secretory granules by increased Ca2+ sensitivity in β-cells. Endocrinology. 1998;139:1133–1140. doi: 10.1210/endo.139.3.5771. [DOI] [PubMed] [Google Scholar]
- GEMBAL M., GILON P., HENQUIN J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse β-cells. J. Clin. Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAPENGIESSER E., GYLFE E., HELLMAN B. Cyclic AMP as a determinant for glucose induction of fast Ca2+ oscillations in isolated pancreatic β-cells. J. Biol. Chem. 1991;266:12207–12210. [PubMed] [Google Scholar]
- GROMADA J., BOKVIST K., DING W.-G., HOLST J.J., NIELSEN J.H., RORSMAN P. Glucagon-like peptide 1(7-36) amide stimulates exocytosis in human pancreatic β-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes. 1998;47:57–65. doi: 10.2337/diab.47.1.57. [DOI] [PubMed] [Google Scholar]
- JONES P.M., FYLES J.M., HOWELL S. Regulation of insulin secretion by cAMP in rat islets of Langerhans permeabilised by high-voltage discharge. FEBS Letters. 1986;205:205–209. doi: 10.1016/0014-5793(86)80898-5. [DOI] [PubMed] [Google Scholar]
- KATO S., ISHIDA H., TSUURA Y., TSUJI K., NISHIMURA M., HORIE M., TAMINATO T., IKEHARA S., OKADA H., IKEDA H., OKADA Y., SEINO Y. Alterations in basal and glucose-stimulated voltage-dependent Ca2+ channel activities in pancreatic β cells of non-insulin-dependent diabetes mellitus GK rats. J. Clin. Invest. 1996;97:2417–2425. doi: 10.1172/JCI118688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEIBOWITZ M.D., BISWAS C., BRADY E.J., CONTI M., CULLINAN C.A., HAYES N.S., MANGANIELLO V.C., SAPERSTEIN R., WANG L., ZAFIAN P.T., BERGER J. A novel insulin secretagogue is a phosphodiesterase inhibitor. Diabetes. 1995;44:67–74. doi: 10.2337/diab.44.1.67. [DOI] [PubMed] [Google Scholar]
- LU M., WHEELER M.B., LENG X.-H., BOYD A.E., III The role of the free cytosolic calcium level in β-cell signal transduction by gastric inhibitory polypeptide and glucagon-like peptide I (7-37) Endocrinology. 1993;132:94–100. doi: 10.1210/endo.132.1.8380389. [DOI] [PubMed] [Google Scholar]
- MANGANIELLO V.C., MURATA T., TAIRA M., BELFRAGE P., DEGERMAN E. Diversity in cyclic nucleotide phosphodiesterase isoenzyme families. Arch. Biochem. Biophys. 1995;322:1–13. doi: 10.1006/abbi.1995.1429. [DOI] [PubMed] [Google Scholar]
- OHTA T., FURUKAWA N., KOMURO G., YONEMORI F., WAKITANI K. JTT-608 restores impaired early insulin secretion in diabetic Goto-Kakizaki rats. Br. J. Pharmacol. 1999a;126:1674–1680. doi: 10.1038/sj.bjp.0702481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHTA T., FURUKAWA N., YONEMORI F., WAKITANI K. JTT-608 controls blood glucose by enhancement of glucose-stimulated insulin secretion in normal and diabetes mellitus rats. Eur. J. Pharmacol. 1999b;367:91–99. doi: 10.1016/s0014-2999(98)00952-2. [DOI] [PubMed] [Google Scholar]
- OKAMOTO Y., ISHIDA H., TSUURA Y., YASUDA K., MATSUBARA H., NISHIMURA M., MIZUNO N., IKEDA H., SEINO Y. Hyperresponse in calcium-induced insulin release from electrically permeabilized pancreatic islets of diabetic GK rats and its defective augmentation by glucose. Diabetologia. 1995;38:772–778. doi: 10.1007/s001250050351. [DOI] [PubMed] [Google Scholar]
- PARKER J.C., VANVOLKENBURG M.A., KETCHUM R.J., BRAYMAN K.L., ANDREWS K.M. Cyclic phosphodiesterases of human and rat islets of Langerhans: contribution of types III and IV to the modulation of insulin secretion. Biochem. Biophys. Res. Commun. 1995;217:916–923. doi: 10.1006/bbrc.1995.2858. [DOI] [PubMed] [Google Scholar]
- SAITO Y., AIZAWA T., KOMATSU M., OKADA N., YAMADA T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic β-cell. Diabetes. 1992;41:438–443. doi: 10.2337/diab.41.4.438. [DOI] [PubMed] [Google Scholar]
- SHAFIEE-NICK R., PYNE N.J., FURMAN B.L. Effects of type-selective phosphodiesterase inhibitors on glucose-induced insulin secretion and islet phosphodiesterase activity. Br. J. Pharmacol. 1995;115:1486–1492. doi: 10.1111/j.1476-5381.1995.tb16641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARP G.W.G. The adenylate cyclase-cyclic AMP system in islets of Langerhans and its role in the control of insulin release. Diabetologia. 1979;16:287–296. doi: 10.1007/BF01223617. [DOI] [PubMed] [Google Scholar]
- SHINKAI H., OZEKI H., MOTOMURA T., OHTA T., FURUKAWA N., UCHIDA T. 4-(trans-4-methylcyclohexyl)-4-oxobutyric acid (JTT-608). A new class of antidiabetic agent. J. Med. Chem. 1998;41:5420–5428. doi: 10.1021/jm9804228. [DOI] [PubMed] [Google Scholar]
- SIEGEL E.G., WOLLHEIM C.B., KIKUCHI M., RENOLD A.E., SHARP G.W.G. Dependency of cyclic AMP-induced insulin release on intra- and extracellular calcium in rat islets of Langerhans. J. Clin. Invest. 1980;65:233–241. doi: 10.1172/JCI109665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUTTON R., PETERS M., MCSHANE P., GRAY D.W.R., MORRIS P.J. Isolation of rat pancreatic islets by ductal injection of collagenase. Transplantation. 1986;42:689–691. doi: 10.1097/00007890-198612000-00022. [DOI] [PubMed] [Google Scholar]
- TAMAGAWA T., NIKI H., NIKI A. Insulin release independent of a rise in cytosolic free Ca2+ by forskolin and phorbol ester. FEBS Letters. 1985;183:430–432. doi: 10.1016/0014-5793(85)80825-5. [DOI] [PubMed] [Google Scholar]
- THOMPSON W.J., APPLEMAN M.M. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochem J. 1971;10:311–316. [PubMed] [Google Scholar]
- TSUJI K., TAMINATO T., USAMI M., ISHIDA H., KITANO N., FUKUMOTO H., KOH G., KUROSE T., YAMADA Y., YANO H., SEINO Y., IMURA H. Characteristic features of insulin secretion in the streptozotocin-induced NIDDM rat model. Metabolism. 1988;37:1040–1044. doi: 10.1016/0026-0495(88)90064-9. [DOI] [PubMed] [Google Scholar]
- WARD W.K., BEARD J.C., HALTER J.B., PFEIFFER M.A., PORTE D. Pathophysiology of insulin secretion in non-insulin dependent diabetes mellitus. Diabetes Care. 1984;7:491–502. doi: 10.2337/diacare.7.5.491. [DOI] [PubMed] [Google Scholar]
- YADA T., ITOH K., NAKATA M. Glucagon-like peptide-1-(7-36) amide and a rise in cyclic adenosine 3′,5′-monophosphate increase cytosolic free Ca2+ in rat pancreatic β-cells by enhancing Ca2+channel activity. Endocrinology. 1993;133:1685–1692. doi: 10.1210/endo.133.4.8404610. [DOI] [PubMed] [Google Scholar]
- YAEKURA K., KAKEI M., YADA T. CAMP-signaling pathway acts in selective synergism with glucose or tolbutamide to increase cytosolic Ca2+ in rat pancreatic β-cells. Diabetes. 1996;45:295–301. doi: 10.2337/diab.45.3.295. [DOI] [PubMed] [Google Scholar]
- ZHAO A.Z., BORNFELD K.E., BEAVO J.A. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J. Clin. Invest. 1998;102:869–873. doi: 10.1172/JCI3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO A.Z., ZHAO H., TEAGUE J., FUJIMOTO W., BEAVO J.A. Attenuation of insulin secretion by insulin-like growth factor 1 is mediated through activation of phosphodiesterase 3B. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3223–3228. doi: 10.1073/pnas.94.7.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]