

Abstract
The mechanism of endothelium-dependent regulation of vascular tone of bradykinin was investigated by simultaneously monitoring the changes in the cytosolic Ca2+ concentration and the force of smooth muscle in fura-2-loaded strips of the porcine renal artery with endothelium.
During phenylephrine-induced sustained contraction, bradykinin (>3×10−9 M) caused endothelium-dependent triphasic changes in the force of the strips, composed of an initial relaxation, a subsequent transient contraction and a late sustained relaxation.
At low concentrations (10−10–10−9 M), bradykinin caused an endothelium-dependent biphasic relaxation with no contraction.
A thromboxane A2 (TXA2)/prostaglandin H2 (PGH2) receptor antagonist (10−5 M ONO-3708) completely inhibited, while a TXA2 synthase inhibitor (10−5 M OKY-046) only partially inhibited, the transient contraction induced by bradykinin.
Under conditions where the bradykinin-induced contraction was inhibited by ONO-3708 during the phenylephrine-induced contraction, bradykinin induced only a transient relaxation in the presence of NΩ-nitro-L-arginine methyl ester (L-NAME). This transient relaxation was inhibited when the precontraction was initiated by phenylephrine plus 40 mM extracellular K+. The removal of L-NAME from this condition caused a partial reappearance of the initial relaxation and a complete reappearance of the sustained relaxation.
In conclusion, bradykinin caused the endothelium-dependent triphasic regulation of vascular tone in the porcine renal artery. The concentrations of bradykinin required to induce a contraction was higher than that required to induce relaxation. Both TXA2 and PGH2 were involved in the bradykinin-induced contraction. The initial relaxation was mediated by nitric oxide and hyperpolarizing factors while the sustained relaxation depended on nitric oxide.
Keywords: Renal artery, endothelial cells, Ca2+, bradykinin, prostaglandin H2, endothelium-dependent contraction, endothelium-dependent relaxation
Introduction
Endothelial cells can modulate the tone of underlying vascular smooth muscle by producing potent vasorelaxing and vasocontracting substances (Furchgott & Vanhoutte, 1989). Endothelium-dependent relaxation has been well reported in many kinds of species and types of blood vessels. Mediators for vasodilation include endothelium-derived relaxing factor (EDRF), which proved to be nitric oxide (NO) (Palmer et al., 1987), endothelium-derived hyperpolarizing factor (EDHF) (Chen et al., 1988), and prostacyclin. On the other hand, reports on endothelium-dependent contraction are limited to specific combinations of agonist and types of blood vessels (Katusic & Shepherd, 1991). In the cerebral artery, A23187 (Shirahase et al., 1988), acetylcholine (Usui et al., 1993) and somatostatin (Shirahase et al., 1993) caused endothelium-dependent contractions. In the pulmonary artery, arachidonic acid (Buzzard et al., 1993) and substance P (Shirahase et al., 1995) caused endothelium-dependent contractions. In the renal artery, acetylcholine induced endothelium-dependent contractions (Nishimura et al, 1995). The precise mechanism and physiological role of endothelium-dependent contraction in these blood vessels thus remains to be understood.
Bradykinin is a potent endogenous vasoactive polypeptide released when kallikrein hydrolyzes the substrate kininogen (Regoli & Barabe, 1980). Vascular smooth muscle cells have kallikrein and produce kinins (Oza et al., 1990), which contribute to the basal release of endothelium derived vasorelaxing mediators. This supports a paracrine/autocrine role for kinins in the regulation of vascular tone (Mombouli & Vanhoutte, 1995). Bradykinin has been reported to cause endothelium-dependent relaxation in several species and types of vessels (Ignarro et al., 1987; Ohlmann et al., 1997; Zhu et al., 1997). On the other hand, bradykinin induces endothelium-independent contractions in such veins as the guinea-pig anterior mesenteric vein (Gaudreau et al., 1981), rabbit jugular vein (Calixto & Medeiros, 1992), rabbit mesenteric vein (Regoli et al., 1977) and rabbit saphenous vein (Eguchi et al., 1997). A recent report in the porcine iliac artery demonstrated that bradykinin induced endothelium-dependent relaxation by activating B2 receptor at low concentration (10−10–10−8 M) whereas it caused an endothelium-independent contraction by activating B1 receptor at 10−7 M and higher concentrations (Persson & Andersson, 1998). However, there has been no report on the bradykinin-induced endothelium-dependent contraction.
In the present study, we demonstrated that bradykinin caused only an endothelium-dependent biphasic relaxation at lower concentrations whereas it caused both endothelium-dependent triphasic contraction and relaxation at higher concentrations in the porcine interlobar renal artery. We investigated the mechanism of these bradykinin-induced endothelium-dependent responses, by simultaneously monitoring the changes in the cytosolic Ca2+ concentration ([Ca2+]i) and the force of smooth muscle in fura-2-loaded strips with an intact endothelium.
Methods
Tissue preparation for front-surface fluorometry
Porcine kidneys were freshly obtained from a local slaughterhouse and transported to the laboratory in the aerated ice-cold physiological salt solution (PSS). Interlobar arteries were dissected from the kidney and the adventitia was mechanically removed under a binocular scope. The arterial segments thus obtained were opened longitudinally and cut into circular strips (approximately 1 mm wide, 3 mm long, 0.05 mm thick). Care was taken to avoid damaging the endothelium. To obtain strips without an endothelium, the inner surface was rubbed off with a cotton swab.
Fura-2 loading of renal arterial strips
The smooth muscle of the strips with and without an endothelium were loaded with fura-2, by incubating in Dulbecco's modified Eagle medium (DMEM) containing 25 μM fura-2/AM (an acetoxymethylester form of fura-2) and 5% foetal bovine serum for 4 h at 37°C, as previously described (Ihara et al., 1999). After loading with fura-2, the strips were rinsed with normal PSS to remove the dye in the extracellular space and then were equilibrated for at least 60 min at 37°C before starting the experimental protocols.
Simultaneous measurement of [Ca2+]i and force of arterial strips
Measurements of changes in fura-2 fluorescence and force of arterial strips were carried out at 37°C. The fura-2 loaded strips were mounted vertically to a force transducer TB-612T (Nihon Koden, Japan) in a quartz organ bath filled with normal PSS. Any changes in the level of [Ca2+]i of the smooth muscle in the arterial strips were monitored, using a front-surface fluorometer CAM-OF-3 (JASCO, Tokyo, Japan) as previously described (Ihara et al., 1999). The strips were illuminated by alternating (400 Hz) 340 and 380 nm excitation light from a xenon light source through quartz optic fibres. The surface fluorescence of the strips was collected by glass optic fibres and introduced through a 500 nm band pass filter (full width at half maximum transmission=10 nm) into a photomultiplier. The quartz and glass optic fibres were arranged in a concentric inner circle (3 mm diameter) and an outer circle (7 mm diameter), respectively, at one end of the optic fibres facing to the strips. The fluorescence intensities (500 nm) at 340 nm (F340) and 380 nm (F380) excitation and their ratio (Ratio=F340/F380), which indicated [Ca2+]i of smooth muscle, were continuously monitored. We previously showed that the fura-2 signal arose exclusively from the smooth muscle in the strips with an endothelium and that the signal derived from endothelial cells, if any, was negligible (Ihara et al., 1999).
During the 60 min equilibration periods, the strips were stimulated with 118 mM K+ every 10 min, and the resting load was increased in a stepwise manner and finally adjusted to 100 mg. When the renal artery was exposed to 10−6 M phenylephrine in normal PSS, [Ca2+]i and force rose rapidly and reached a peak within 1–3 min, and then slightly declined to reach a sustained level within 15 min (Figure 1a). This level was maintained for more than 30 min. After the [Ca2+]i level and force returned to the resting level upon the removal of phenylephrine, the strip was stimulated once with 118 mM K+ in order to refill the intracellular Ca2+ stores. The second stimulation with phenylephrine induced a similar contraction to that observed with the first stimulation. The levels of [Ca2+]i and force obtained with the second stimulation was 99.2±4.6 and 99.7±4.2% (n=6) of those obtained with the first stimulation, respectively. As a result, the response to the first stimulation with 10−6M phenylephrine was used as a reference response in the renal artery. The level of [Ca2+]i and force at rest and at the sustained phase of the 10−6 M phenylephrine-induced contraction were designated as 0 and 100%, respectively. The extent of the relaxation and the inhibition were evaluated by how much of values of the [Ca2+]i and force remained.
Figure 1.
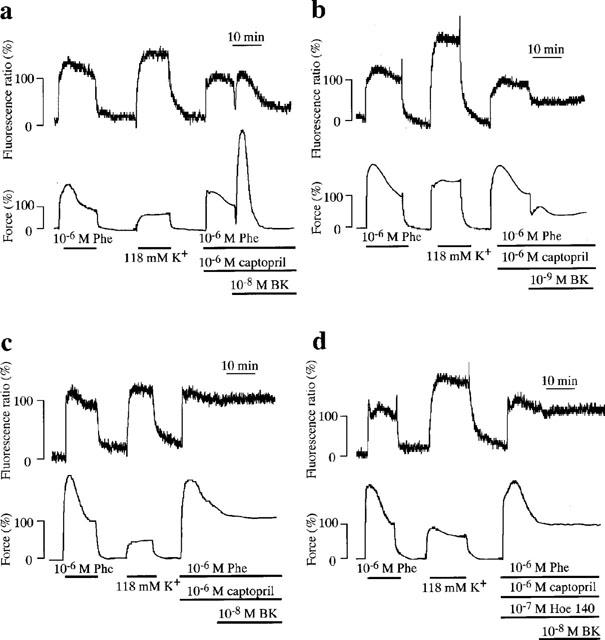
Bradykinin (BK)-induced endothelium-dependent triphasic changes in [Ca2+]i and force during phenylephrine (Phe)-induced sustained contraction in porcine renal artery. (a–c) The representative recordings of changes in [Ca2+]i (ratio) and force induced by 10−8 M (a) and 10−9 M (b) BK in the strips with endothelium, and by 10−8 M BK in the strips without endothelium (c). (d) 10−7 M Hoe 140 (D-Arg-[Hyp3, Thi5, D-Tic7, Oic8] bradykinin), a specific bradykinin B2 receptor antagonist completely abolished the 10−8 M BK-induced triphasic responses. Hoe 140 (10−7 M) were applied just prior to the initiation of contraction by Phe. BK was applied 15 min after the initiation of the precontraction by 10−6 M Phe. The levels of [Ca2+]i and force at rest and during the first Phe-induced sustained contraction were designated to be 0 and 100%, respectively.
Drugs and solutions
The composition of normal PSS was (in mM): NaCl 123, KCl 4.7, NaHCO3 15.5, KH2PO4 1.2, MgCl2 1.2, CaCl2 1.25 and D-glucose 11.5. PSS was aerated with 95% O2 and 5% CO2, with the resulting pH to be 7.4. PSS containing high K+ was prepared by replacing NaCl with equimolar KCl.
Fura-2/AM was purchased from Dojindo Laboratories (Kumamoto, Japan), Indomethacin was purchased from Wako (Osaka, Japan). DMEM was purchased from GIBCO (Grand Island, NY, U.S.A.). ONO-3708, a thromboxane A2 (TXA2)/prostaglandin H2 (PGH2) receptor antagonist and OKY-046, a TXA2 synthase inhibitor, were kindly donated by the Ono pharmaceutical Co. (Osaka, Japan). Captopril, a kininase inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME) and phenylephrine were purchased from Sigma (St. Louis, MO, U.S.A.). Bradykinin was purchased from the Peptide Institute, Inc (Osaka, Japan). PGH2 was purchased from Cayman Chemical (Ann Arbor, MI, U.S.A.). Hoe 140 (D-Arg-[Hyp3, Thi5, D-Tic7, Oic8] bradykinin), a specific bradykinin B2 receptor antagonist, was from Research Biochemicals International (Natick, MA, U.S.A.).
Data analysis
All data were expressed as the mean±s.e.mean. One strip obtained from one animal was used for each experiment, therefore the number of experiments (n value) also indicates the number of animals. Student's t-test was used to determine statistical significance between the two groups. An analysis of variance (ANOVA) followed by Scheffe's test was used to determine statistical significance between three groups. P values of less than 0.05 were considered to be significant. All data were collected using a computerized data acquisition system (MacLab; Analog Digital Instruments, Australia, Macintosh; Apple Computer, U.S.A.).
Results
Effect of bradykinin on the [Ca2+]i level and force of smooth muscle in renal arterial strips with an intact endothelium
Figure 1a shows the representative recordings of the changes in the [Ca2+]i level and force induced by 10−8 M bradykinin in the presence of 10−6 M captopril during the 10−6 M phenylephrine-induced sustained contraction of the strips with an endothelium. Captopril was used to prevent a breakdown of bradykinin (Hirano et al., 1993). In fact, captopril augmented the response to 3×10−9 M bradykinin as shown in Figure 2d. Bradykinin induced an initial rapid relaxation and a subsequent transient contraction, followed by a sustained relaxation within 20 min after the application of bradykinin. Then the level of [Ca2+]i and force gradually and slowly recovered to the sustained level induced by 10−6 M phenylephrine. The initial and sustained relaxations were associated with decreases in [Ca2+]i. The force development of a transient contraction was about three times as large as that of the phenylephrine-induced sustained contraction, although the [Ca2+]i level observed at a transient contraction was almost the same as that observed during the phenylephrine-induced sustained contraction. At 10−9 M, however, bradykinin induced only a biphasic relaxation composed of an initial relaxation and a sustained relaxation with a decrease in [Ca2+]i, and no apparent contraction was observed (Figure 1b). The endothelium-dependent triphasic responses induced by 10−8 M bradykinin were completely abolished either by the removal of the endothelium (Figure 1c) or pretreatment with 10−7 M Hoe 140 (D-Arg-[Hyp3, Thi5, D-Tic7, Oic8] bradykinin), a highly specific bradykinin B2 receptor antagonist (Figure 1d).
Figure 2.
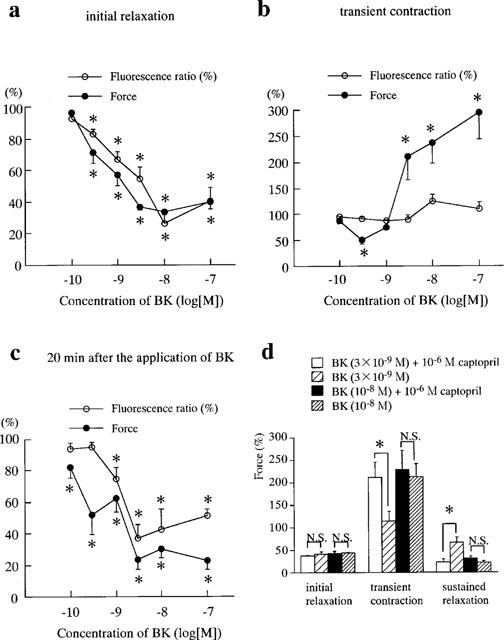
Concentration-dependent triphasic effects of bradykinin (BK) on the phenylephrine-induced sustained contraction and the effects of captopril on these BK-induced responses in the porcine renal artery. (a–c) The levels of [Ca2+]i (ratio) and force at the initial relaxation (a), the peak of transient contraction (b) and 20 min after application of BK (late sustained relaxation) (c) are shown. At 10−10–10−9 M BK, which induce no apparent transient contraction, the levels of [Ca2+]i and force of the initial relaxation, transient contraction and sustained relaxation were measured at 1, 4 and 20 min after the application of BK, respectively. *Significantly different from the levels of [Ca2+]i and force seen during the sustained phase of control contraction induced by 10−6 Phe (Student's t-test, P<0.05). N.S., not significantly different (Student's t-test, P>0.05). All data are the mean±s.e.mean (n=4). (d) Effects of captopril on the triphasic responses to 3×10−9 M and 10−8 M BK. Captopril was applied just prior to the initiation of contraction by Phe. *Significantly different (Student's t-test, P<0.05); N.S., not significantly different (Student's t-test, P>0.05). The levels of [Ca2+]i and force at rest and during Phe-induced sustained contraction were designated to be 0 and 100%, respectively. All data are the mean±s.e.mean (n=4).
Figure 2a–c summarize the dose-dependent changes in the endothelium-dependent initial relaxation (a), transient contraction (b) and sustained relaxation (c) induced by bradykinin in the presence of 10−6 M captopril, respectively. Bradykinin caused endothelium-dependent biphasic relaxation at a lower concentration (10−10–10−9 M) whereas it caused an endothelium-dependent triphasic response at 3×10−9 M and higher concentrations. In the case of triphasic response, the initial relaxation and transient contraction were evaluated at the time of the lowest point and peak, respectively. The sustained relaxation was evaluated at 20 min after the application of bradykinin. The initial relaxation reached the lowest point at 1.0±0.1 min (3×10−9 M), 1.2±0.1 min (10−8 M), 1.0±0.2 min (10−7 M) (n=4). The transient contraction reached its peak at 3.1±0.2 min (3×10−9 M), 3.5±0.3 min (10−8 M) and 3.6±0.5 min (10−7 M) (n=4). No significant differences were observed among the three values. Therefore, in the case of biphasic relaxation the initial relaxation, transient contraction and sustained relaxation were examined at 1, 4 and 20 min, respectively. The initial relaxation was observed at 3×10−10 M and higher concentrations (Figure 2a) (P<0.05). The maximal relaxation (26.6±6.5%, n=4) was obtained with 10−8 M bradykinin accompanied by the maximal decrease in [Ca2+]i (33.6±1.6%, n=4). The transient contraction was observed at 3×10−9 M and higher concentrations (P<0.05). The levels of force of the transient contraction obtained with 3×10−9, 10−8 and 10−7 M bradykinin were 212.5±30.2, 237.0±39.8 and 294.0±49.0%, respectively (n=4). The [Ca2+]i levels obtained with these concentrations of bradykinin were not statistically different from those obtained during the phenylephrine-induced control contraction just prior to the application of bradykinin. Thus, the contraction induced by bradykinin was considered to mainly be due to an increase in the Ca2+ sensitivity of the contractile apparatus. A sustained relaxation was observed with 10−10 M and higher concentrations (Figure 2c) (P<0.05). The sustained relaxation seen at concentrations higher than 10−9 M were accompanied by a decrease in [Ca2+]i. The maximal decrease in [Ca2+]i (36.8±8.7%, n=4) and force (23.7±6.3%, n=4) were observed at 3×10−9 M bradykinin.
Figure 2d shows the effects of captopril on the bradykinin-induced endothelium-dependent responses. Captopril had no effects on the initial relaxation, transient contraction and sustained relaxation induced by 10−8 M bradykinin, whereas it augmented the endothelium-dependent transient contraction and sustained relaxation induced by 3×10−9 M bradykinin. The initial relaxation induced by 3×10−9 M bradykinin was not influenced by captopril. The angiotensin converting enzyme expressed in the porcine renal arterial strips with an endothelium was observed to degrade the exogenously added bradykinin. In the present study, we therefore investigated the mechanisms of the bradykinin-induced endothelium-dependent contraction and relaxation in the presence of 10−6 M captopril.
The effects of a TXA2/PGH2 receptor antagonist and a TXA2 synthase inhibitor on bradykinin-induced endothelium-dependent contraction
To determine whether PGH2 or TXA2 was involved in the bradykinin-induced endothelium-dependent contraction, the effects of ONO-3708 (a TXA2/PGH2 receptor antagonist) and OKY-046 (a TXA2 synthase inhibitor) were examined. Figure 3a shows the time courses of the endothelium-dependent responses induced by 10−8 M bradykinin in the presence of 10−5 M ONO-3708. Treatment with 10−5 M ONO-3708, per se, had no effect on the phenylephrine-induced contraction. The addition of 10−8 M bradykinin during the phenylephrine-induced sustained contraction caused a rapid transient relaxation followed by a sustained relaxation with a decrease in [Ca2+]i, and no apparent contraction was observed. Since the initial relaxation and transient contraction reached their peaks at about 1 min and 4 min, respectively, after the application of bradykinin in the absence of inhibitors, the effects of such inhibitors on the bradykinin-induced initial relaxation and transient contraction were evaluated at 1 and 4 min, respectively. The effects on the bradykinin-induced sustained relaxation was evaluated at 20 min. The level of [Ca2+]i at 1, 4 and 20 min after the application of bradykinin in the presence of 10−5 M ONO-3708 were 32.1±14.1, 47.9±7.1 and 46.0±1.8% (n=4), respectively (Figure 3c), and the level of force at 1, 4 and 20 min were 42.6±8.6, 35.1±4.8 and 24.5±2.4% (n=4), respectively (Figure 3c). ONO-3708 significantly inhibited the bradykinin-induced contraction (P<0.05). These observations indicate that the TXA2 and/or PGH2 are involved in the bradykinin-induced contraction.
Figure 3.
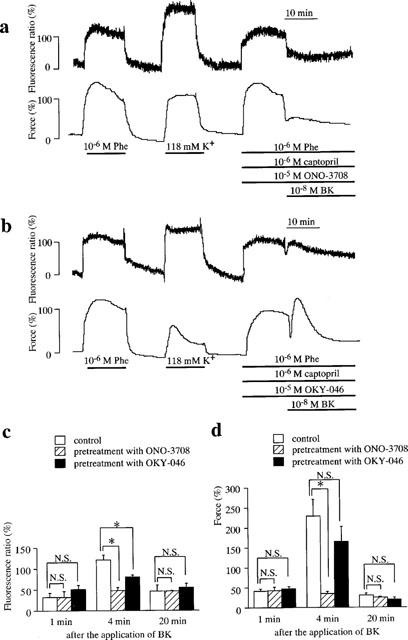
Effects of ONO-3708 (a thromboxane A2/prostaglandin H2 receptor antagonist), and OKY-046 (a thromboxane A2 synthase inhibitor) on bradykinin (BK)-induced endothelium-dependent contraction. (a, b) The representative recordings of BK-induced changes in [Ca2+]i and force during the phenylephrine (Phe)-induced contraction in the presence of 10−5 M ONO-3708 (a) and 10−5 M OKY-046 (b) in the porcine renal artery. After recording the reference response to 10−6 M Phe, each inhibitor was applied just prior to the initiation of the precontraction. BK was applied at 15 min after the initiation of the precontraction. The levels of [Ca2+]i and force at rest and during the Phe-induced sustained contraction were designated to be 0 and 100%, respectively. (c, d) Summary of the effects of ONO-3708 and OKY-046 on BK-induced endothelium-dependent contraction. The levels of [Ca2+]i (c) and force (d) at 1 min (initial relaxation), 4 min (subsequent contraction), and 20 min (sustained relaxation) after the application of BK were shown. All data are the mean±s.e.mean (n=4). *Significantly different from control level (Student's t-test, P<0.05).
To determine the relative contribution of TXA2 and PGH2 in the bradykinin-induced contraction, the effects of a TXA2 synthase inhibitor (OKY-046) was examined (Figure 3b). Treatment with 10−5 M OKY-046 had no direct effect on the phenylephrine-induced contraction. In the presence of OKY-046, bradykinin induced a transient initial relaxation with decrease in [Ca2+]i, a subsequent transient contraction with small increase in [Ca2+]i and a sustained relaxation with decrease in [Ca2+]i. The levels of [Ca2+]i at 1, 4 and 20 min after the stimulation of bradykinin were 50.9±9.8, 80.5±4.5, and 54.5±10.3% (n=4), respectively (Figure 3c), and the level of force at 1, 4 and 20 min were 47.7±5.7, 165.7±37.7 and 18.9±5.7% (n=4), respectively (Figure 3d). OKY-046 significantly (P<0.05) decreased [Ca2+]i level during the bradykinin-induced contraction. However, the force development was not significantly inhibited. OKY-046 had no effect on [Ca2+]i level and force during the initial and sustained relaxation induced by bradykinin.
Relative contribution of NO and EDHF to the bradykinin-induced endothelium-dependent relaxation
To examine the contribution of NO and EDHF to the bradykinin-induced relaxation, the effects of L-NAME and the elevation of external K+ concentration were examined under conditions in which the bradykinin-induced contraction was inhibited by 10−5 M ONO-3708 (Figure 4). In the presence of 10−4 M L-NAME and 10−5 M ONO-3708, bradykinin induced only a transient relaxation with a transient decrease in [Ca2+]i during the phenylephrine-induced contraction (Figure 4a). L-NAME thus markedly inhibited the sustained relaxation seen in Figure 3a. This transient relaxation became smaller with the increment of external K+ concentration. Eventually, no transient relaxation was observed when the concentration of external K+ reached 40 mM K+ (Figure 4b). The force levels of the transient relaxation during the contraction induced by phenylephrine plus 5.9, 20, 30 and 40 mM K+ were 30.0±7.8, 77.1±6.7, 86.6±4.2 and 99.2±0.6% (n=4), respectively, designating the sustained level just prior to the application of bradykinin to be 100%. When L-NAME was removed (Figure 4c), bradykinin induced an initial relaxation followed by a sustained relaxation with a decrease in [Ca2+]i during the contraction induced by phenylephrine plus 40 mM K+ in the presence of ONO-3708.
Figure 4.
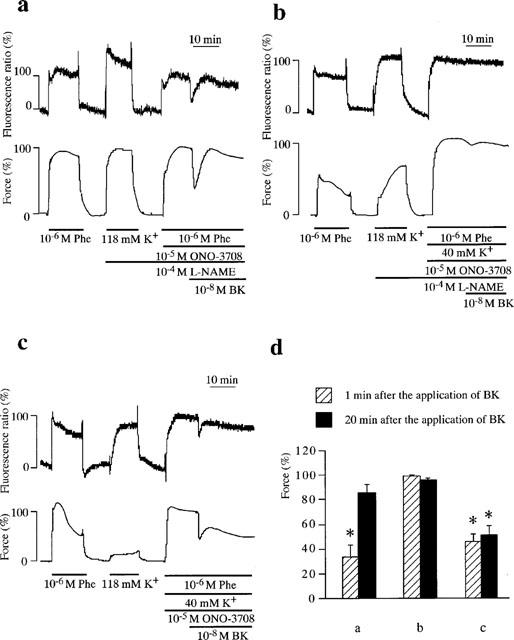
Involvement of endothelium-derived hyperpolarizing factor and nitric oxide in bradykinin (BK)-induced relaxation in the porcine renal artery. (a, b) Representative recordings of changes in [Ca2+]i and force induced by BK during the contraction induced by 10−6 M phenylephrine (Phe) (a) and Phe plus 40 mM K+ (b) in the presence of 10−5 M ONO-3708 and 10−4 M NΩ-nitro-L-arginine methylester (L-NAME). (c) Representative recordings of changes in [Ca2+]i and force induced by BK during the contraction initiated by Phe plus 40 mM K+ in the presence of 10−5 M ONO-3708. (d) Summary of the changes in the levels of force obtained at 1 and 20 min after the application of BK in protocol a, b and c. The level of force at rest was designated to be 0% and the level of force during the sustained contraction just prior to the application of BK under each experimental condition was designated as 100%. All data are the mean±s.e.mean (n=4). *Significantly different from the level of force seen just prior to the application of BK (100% level). (ANOVA, Scheffe's F test, P<0.05).
Treating strips with L-NAME augmented the phenyle-phrine-induced force development (163.4±12.2%, n=4) in the presence of 10−5 M ONO-3708 (Figure 4a), while it had little effect on the [Ca2+]i elevation (98.8±1.9%, n=4). An increase of external K+ to 40 mM augmented [Ca2+]i elevation (217.5±55.1%, n=4) and the force development (201.4±20.8%, n=4) induced by phenylephrine in the presence of ONO-3708 (Figure 4b). Therefore, the effects of L-NAME and an elevation of K+ were evaluated by assigning the level of force obtained just before the application of bradykinin to be 100% in each experimental condition (Figure 4d). In the presence of L-NAME and ONO-3708, the force decreased to 34.5±9.0% (n=4) at 1 min after the application of bradykinin during the phenylephrine-induced contraction, while it returned to the level obtained before the application of bradykinin at 20 min (85.6±7.1%, n=4). In the presence of L-NAME, ONO-3708 and 40 mM K+, no significant decrease in force was obtained either at 1 min (99.2±0.6%, n=4) or at 20 min (96.1±1.2%, n=4). In the presence of ONO-3708 and 40 mM K+, the force levels at 1 and 20 min were 45.5±6.2 and 51.1±7.9%, n=4, respectively.
Discussion
We presented the first evidence that bradykinin induced not only relaxation but also contraction in an endothelium-dependent manner in the porcine interlobar renal artery. Bradykinin caused a triphasic regulation of vascular tone in the porcine renal artery. The bradykinin-induced endothelium-dependent relaxation was reported in many species and types of blood vessels (Ignarro et al., 1987; Ohlmann et al., 1997; Zhu et al., 1997). Bradykinin was also reported to induce contraction directly acting on smooth muscle. This type of contractile response was demonstrated mainly in veins (Calixto & Medeiros, 1992; Gaudreau et al., 1981; Regoli et al., 1977) including rabbit saphenous vein (Eguchi et al., 1997) and in some arteries such as sheep femoral artery (Feletou et al., 1994) and porcine iliac artery (Persson & Andersson, 1998). However, no bradykinin-induced endothelium-dependent contraction has ever been reported. The observation that the bradykinin-induced contraction as well as relaxation was abolished by the removal of the endothelium clearly indicated that both contraction and relaxation depended on the endothelium. Bradykinin was also suggested to have no direct effect on smooth muscle in porcine renal artery. The type of bradykinin receptor involved in bradykinin-induced endothelium-dependent relaxations and endothelium-independent contraction was reported to vary with the species and type of tissue (Regoli et al., 1996). The bradykinin-induced endothelium-dependent relaxations were mediated by B2 receptor of the endothelial cells in many types of artery (Mombouli et al., 1992; Ohlmann et al., 1997; Persson & Andersson, 1998; Rhaleb et al., 1989), and by B1 receptor in rabbit carotid artery (Pruneau & Belichard, 1993). The bradykinin-induced direct contractions are mediated by B1 receptor of smooth muscle cells in rabbit artery (Audet et al., 1994), human umbilical vein (Sardi et al., 1997) and porcine iliac artery (Persson & Andersson, 1998), and by B2 receptor in the rabbit jugular vein (Feletou et al., 1994) and rabbit saphenous vein (Eguchi et al., 1997). The present study suggests that not only endothelium-dependent relaxation but also the endothelium-dependent contraction induced by bradykinin were mediated by B2 receptor of endothelial cells in porcine renal artery.
The bradykinin-induced endothelium-dependent contraction was inhibited by a TXA2/PGH2 antagonist. This observation indicated that the bradykinin-induced contraction was mediated by the production of TXA2 and/or PGH2 in the endothelium. Indomethacin, a cyclo-oxygenase inhibitor, also abolished the bradykinin-induced endothelium-dependent contraction (data not shown). This observation also supports the involvement of TXA2 and PGH2 in the bradykinin-induced contraction. Furthermore, a TXA2 synthase inhibitor only partially inhibited the bradykinin-induced contraction. This observation suggests that PGH2 plays a major role in the bradykinin-induced endothelium-dependent contractile response. Most of the literature reported either TXA2 or PGH2 to be a mediator of the endothelium-dependent contraction. However, the relative contribution of TXA2 and PGH2 varied with the type of stimulation (Buzzard et al., 1993; Kato et al., 1990; Shirahase et al., 1995). We reported that thapsigargin induced the triphasic responses similar to those observed in the present study (Ihara et al., 1999). The thapsigargin-induced endothelium-dependent contraction was completely inhibited by either indomethacin or ONO-3708 as observed with bradykinin in the present study. However, the TXA2 synthase inhibitor strongly inhibited the thapsigargin-induced contractile response. Therefore, we concluded that TXA2 played a major role in the thapsigargin-induced endothelium-dependent contraction, and the involvement of PGH2 was considered to be minor. This observation also indicates that the concentration of OKY-046 used in the present study was high enough to substantially inhibit the TXA2 synthase activity, and also rules out the possibility that the partial inhibition of the bradykinin-induced contraction was due to an insufficient inhibition of the TXA2 synthase. We therefore suggest that PGH2 plays a more important role in the bradykinin-induced endothelium-dependent contraction than in the thapsigargin-induced contraction.
Since the TXA2 synthase-mediated conversion of PGH2 to TXA2 is the only pathway in the synthesis of TXA2, the activity of TXA2 synthase is the primary determinant of the amount of PGH2 and TXA2. It is thus considered that the TXA2 synthase is either activated in the endothelial cells stimulated by thapsigargin or inhibited in the cells stimulated by bradykinin. The differences in the intracellular signals activated between thapsigargin and bradykinin may correlate with the difference in the activity of the enzyme enrolled. Thapsigargin induced a large sustained [Ca2+]i elevation in porcine renal arterial endothelial cells in primary culture (Ihara et al., 1999). Ca2+-ATPase inhibitors such as thapsigargin (Thastrup et al., 1990) and cyclopiazonic acid (Seidler et al., 1989) have been reported to induce a large sustained elevation of [Ca2+]i due to the activation of Ca2+ entry in many types of cells including endothelial cells (Kawasaki et al., 1999; Schilling et al., 1992). The thapsigargin-induced [Ca2+]i elevation sustained for more than 20 min (Ihara et al., 1999; Kawasaki et al., 1999). On the other hand, bradykinin induced a transient [Ca2+]i elevation, reaching its peak at 1–2 min, followed by a little or small sustained elevation in both in situ and cultured endothelial cells (Aoki et al., 1994; Hirano et al., 1993). It is thus considered that the thapsigargin-induced [Ca2+]i elevation in endothelial cells remained high above the resting level while the bradykinin induced [Ca2+]i elevation returned close to the prestimulation level, when the endothelium-dependent contractions were obtained. As a result, the difference in Ca2+ signal between thapsigargin and bradykinin may contribute to the difference in the relative contribution of TXA2 and PGH2. On the other hand, bradykinin was shown to activate divergent intracellular signal transduction pathways in addition to the Ca2+ signal. It includes the activation of tyrosine phosphorylation (Fleming & Busse, 1997), phospholipase C γ (Venema et al., 1998), MAP kinase (Naraba et al., 1998), inositol 1,4,5-trisphosphate (Rabito et al., 1996). The bradykinin-specific intracellular signals other than Ca2+ may also contribute to an inhibition of the conversion of PGH2 to TXA2.
Since bradykinin was reported to stimulate endothelial cells and caused the release of endothelin (Marsden et al., 1991), it is possible that endothelin was involved in the bradykinin-induced endothelium-dependent contraction in the porcine renal artery. However, BQ-123, a selective ETA receptor antagonist, had no effect on the bradykinin-induced contraction (data not shown). The involvement of endothelin was thus considered to be negligible.
There are two phases in the bradykinin-induced relaxations; the initial relaxation and the late sustained relaxation. The initial relaxation was observed even in the presence of L-NAME, indicating that relaxing factor(s) other than NO were involved in the initial relaxation. In fact, the initial relaxation was abolished in the presence of 40 mM K+. It was reported that the elevation of external K+ concentration inhibited the hyperpolarization of smooth muscle by the EDHF (Chen & Suzuki, 1989). Our findings thus suggest that EDHF is involved in the initial relaxation. However, bradykinin induced the initial relaxation in the presence of 40 mM K+ but the absence of L-NAME. It suggests that NO was also involved in this relaxation. On the other hand, the sustained relaxation was completely inhibited by L-NAME and reappeared by its removal, thus suggesting that the sustained relaxation was mainly mediated by NO. In terms of Ca2+ signal induced by bradykinin, it is considered that the production of NO induced by bradykinin was sustained longer than the bradykinin-induced [Ca2+]i elevation in endothelial cells. Nitric oxide synthase is Ca2+ dependent enzyme and activated by Ca2+ and calmodulin (Michel et al., 1997; Schmidt et al., 1992). The Ca2+-sensitivity of NO production is thus considered to be potentiated during the bradykinin-induced sustained relaxation. We reported that the production of NO for a given [Ca2+]i elevation varied with the type of stimulation, namely, thrombin induced greater production of NO with a smaller increase in [Ca2+]i than ATP (Mizuno et al., 1998). Recently, it has been reported that phosphorylation of NO synthase induced by Akt/protein kinase B increased the enzymatic activity in the absence of Ca2+ (Fulton et al., 1999; Dimmeler et al., 1999). Akt/protein kinase B is shown to be activated by phosphatidylinositol 3-kinase-mediated signalling (Burgering & Coffer, 1995), which is activated in tyrosine-phosphorylated-receptor kinase (Otsu et al., 1991). This mode of activation of NO synthesis may contribute to the bradykinin-induced sustained production of NO. Bradykinin was shown to activate tyrosine phosphorylation in endothelial cells (Fleming & Busse, 1997). It is thus possible that bradykinin activates phosphatidylinositol 3-kinase and Akt/protein kinase B. However, this possibility remains to be evaluated.
One of the important findings of the present study is that the concentrations of bradykinin required to induce endothelium-dependent contractions were higher than those required to induce relaxation. Relaxation was observed at 10−10 M and the higher concentrations, while contractions were observed at concentrations higher than 3×10−9 M. Namely, the production of contracting factors and that of relaxing factors are differentially regulated. It is suggested that the production of TXA2 and PGH2 require either a higher [Ca2+]i elevation or the signals other than Ca2+, both of which are only induced by high concentrations of bradykinin. The key enzymes for the production of TXA2 and PGH2 are phospholipase A2 and cyclo-oxygenase. Phospholipase A2 is a Ca2+-dependent enzyme (Chen et al., 1996). It is possible that the activation of phospholipase A2 requires a higher [Ca2+]i elevation than that of NO synthase. This quantitative difference in the Ca2+ sensitivity of the activation of the two enzymes may cause a difference in the concentrations of bradykinin required to induce relaxation and contraction.
Most of the reports on the endothelium-dependent contraction under physiological conditions are limited to the cerebral artery and renal artery. These arteries are known to show an autoregulation of the blood flow (Aukland, 1989) and endothelial cells may be involved in such autoregulation (Faraci et al., 1989). On the other hand, kinins were reported to play an important role in the autoregulation of the glomerular filtration rate in dogs (Beierwaltes et al., 1988) and of the renal medullary flow in rats (Nafz et al., 1998). Therefore, the physiological role of endothelium-dependent biphasic and triphasic regulation induced by bradykinin may be linked to the autoregulation of the renal blood flow.
In conclusion, this is the first report demonstrating that bradykinin caused not only relaxation but also contraction in an endothelium-dependent manner in the porcine interlobar renal artery. Both contraction and relaxation were mediated by B2 receptor of the endothelial cells. The bradykinin-induced relaxation was composed of the two components; the initial and sustained relaxations. Relaxation was observed at 10−10 M and higher concentrations, while contraction was observed only at concentrations higher than 3×10−9 M. The production of the contracting factors is thus suggested to require additional signals, such as either higher elevations of [Ca2+]i or the intracellular signals other than the Ca2+ signal. The bradykinin-induced endothelium-dependent contractions were mediated by the production of TXA2 and PGH2. Both NO and hyperpolarizing factors are involved in the initial relaxation, while NO is the major mediator of the sustained relaxation. The bradykinin-induced endothelium dependent regulation of vascular tone may therefore play an important role in the autoregulation of the renal blood flow.
Acknowledgments
We thank Mr Brian Quinn for comments and help with the manuscript. This study was supported in part by Grants-in-Aid for Scientific Research (No. 10557072, 11838013, 11670687), for the Encouragement of Young Scientists (No. 10770308) from the Ministry of Education, Science, Sports and Culture, Japan, Yokoyama Rinshoyakuri Foundation, Mochida Memorial Foundation for Medical and Pharmaceutical Research and Foundation for the Promotion of Clinical Medicine.
Abbreviations
- ANOVA
analysis of variance
- ATP
adenosine 5′-triphosphate
- [Ca2+]i
cytosolic Ca2+ concentration
- DMEM
Dulbecco's modified Eagle medium
- EDHF
endothelium-derived hyperpolarizing factor
- EDRF
endothelium-derived relaxing factor
- fura-2/AM
acetoxymethylester form of fura-2
- L-NAME
NΩ-nitro-L-arginine methylester
- NO
nitric oxide
- PGH2
prostaglandin H2
- PSS
physiological salt solution
- TXA2
thromboxane A2
References
- AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Sensitivity of G-protein involved in endothelin-1-induced Ca2+ influx to pertussis toxin in porcine endothelial cells in situ. Br. J. Pharmacol. 1994;111:989–996. doi: 10.1111/j.1476-5381.1994.tb14841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AUDET R., PETITCLERC E., DRAPEAU G., RIOUX F., MARCEAU F. Further analysis of the upregulation of bradykinin B1 receptors in isolated rabbit aorta by using metabolic inhibitors. Eur. J. Pharmacol. 1994;271:551–555. doi: 10.1016/0014-2999(94)90819-2. [DOI] [PubMed] [Google Scholar]
- AUKLAND K. Myogenic mechanisms in the kidney. J. Hypertens. 1989;7:S71–S77. [PubMed] [Google Scholar]
- BEIERWALTES W.H., CARRETERO O.A., SCICLI A.G. Renal hemodynamics in response to a kinin analogue antagonist. Am. J. Physiol. 1988;255:F408–F414. doi: 10.1152/ajprenal.1988.255.3.F408. [DOI] [PubMed] [Google Scholar]
- BURGERING B.M., COFFER P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- BUZZARD C.J., PFISTER S.L., CAMPBELL W.B. Endothelium-dependent contractions in rabbit pulmonary artery are mediated by thromboxaneA2. Circ. Res. 1993;72:1023–1034. doi: 10.1161/01.res.72.5.1023. [DOI] [PubMed] [Google Scholar]
- CALIXTO J.B., MEDEIROS Y.S. Effect of protein kinase C and calcium on bradykinin-mediated contractions of rabbit vessels. Hypertension. 1992;19:I187–I193. doi: 10.1161/01.hyp.19.2_suppl.ii87. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. J. Physiol. 1989;410:91–106. doi: 10.1113/jphysiol.1989.sp017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN M., YANG Z., NAJI A., WOLF B.A. Identification of calcium-dependent phospholipase A2 isoforms in human and rat pancreatic islets and insulin secreting β-cell lines. Endocrinology. 1996;137:2901–2909. doi: 10.1210/endo.137.7.8770912. [DOI] [PubMed] [Google Scholar]
- DIMMELER S., FLEMING I., FISSLTHALER B., HERMANN C., BUSSE R., ZEIHER A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- EGUCHI D., NISHIMURA J., KOBAYASHI S., KOMORI K., SUGIMACHI K., KANAIDE H. Mechanism of contraction induced by bradykinin in the rabbit saphenous vein. Br. J. Pharmacol. 1997;120:371–378. doi: 10.1038/sj.bjp.0700902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARACI F.M., BAUMBACH G.L., HEISTAD D.D. Myogenic mechanisms in the cerebral circulation. J. Hypertens. 1989;7:S61–S65. [PubMed] [Google Scholar]
- FELETOU M., GERMAIN M., THURIEAU C., FAUCHERE J.L., CANET E. Agonistic and antagonistic properties of the bradykinin B2 receptor antagonist, Hoe 140, in isolated blood vessels from different species. Br. J. Pharmacol. 1994;112:683–689. doi: 10.1111/j.1476-5381.1994.tb13130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEMING I., BUSSE R. Tyrosine phosphorylation and bradykinin-induced signaling in endothelial cells. Am. J. Cardiol. 1997;80:102A–109A. doi: 10.1016/s0002-9149(97)00464-5. [DOI] [PubMed] [Google Scholar]
- FULTON D., GRATTON J.P., MCCABE T.J., FONTANA J., FUJIO Y., WALSH K., FRANKE T.F., PAPAPETROPOULOS A., SESSA W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F., VANHOUTTE P.M. Endothelium-derived relaxing and contracting factors. FASEB. J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- GAUDREAU P., BARABE J., ST-PIERRE S., REGOLI D. Pharmacological studies of kinins in venous smooth muscles. Can. J. Physiol. Pharmacol. 1981;59:371–379. doi: 10.1139/y81-059. [DOI] [PubMed] [Google Scholar]
- HIRANO K., HIRANO M., KANAIDE H. Enhancement by captopril of bradykinin-induced calcium transients in cultured endothelial cells of the bovine aorta. Eur. J. Pharmacol. 1993;244:133–137. doi: 10.1016/0922-4106(93)90018-5. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., BYRNS R.E., BUGA G.M., WOOD K.S. Mechanisms of endothelium-dependent vascular smooth muscle relaxation elicited by bradykinin and VIP. Am. J. Physiol. 1987;253:H1074–H1082. doi: 10.1152/ajpheart.1987.253.5.H1074. [DOI] [PubMed] [Google Scholar]
- IHARA E., HIRANO K., NISHIMURA J., NAWATA H., KANAIDE H. Thapsigargin-induced endothelium-dependent triphasic regulation of vascular tone in the porcine renal artery. Br. J. Pharmacol. 1999;128:689–699. doi: 10.1038/sj.bjp.0702821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATO T., IWAMA Y., OKUMURA K., HASHIMOTO H., ITO T., SATAKE T. Prostaglandin H2 may be the endothelium-derived contracting factor released by acetylcholine in the aorta of the rat. Hypertension. 1990;15:475–481. doi: 10.1161/01.hyp.15.5.475. [DOI] [PubMed] [Google Scholar]
- KATUSIC Z.S., SHEPHERD J.T. Endothelium-derived vasoactive factors: II. Endothelium-dependent contraction. Hypertension. 1991;18:III86–III92. doi: 10.1161/01.hyp.18.5_suppl.iii86. [DOI] [PubMed] [Google Scholar]
- KAWASAKI J., HIRANO K., HIRANO M., NISHIMURA J., FUJISHIMA M., KANAIDE H. Troglitazone inhibits the capacitative Ca2+ entry in endothelial cells. Eur. J. Pharmacol. 1999;373:111–120. doi: 10.1016/s0014-2999(99)00257-5. [DOI] [PubMed] [Google Scholar]
- MARSDEN P.A., DORFMAN D.M., COLLINS T., BRENNER B.M., ORKIN S.H., BALLERMANN B.J. Regulated expression of endothelin 1 in glomerular capillary endothelial cells. Am. J. Physiol. 1991;261:F117–F125. doi: 10.1152/ajprenal.1991.261.1.F117. [DOI] [PubMed] [Google Scholar]
- MICHEL J.B., FERON O., SACKS D., MICHEL T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 1997;272:15583–15586. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- MIZUNO O., HIRANO K., NISHIMURA J., KUBO C., KANAIDE H. Mechanism of endothelium-dependent relaxation induced by thrombin in the pig coronary artery. Eur. J. Pharmacol. 1998;351:67–77. doi: 10.1016/s0014-2999(98)00292-1. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., ILLIANO S., NAGAO T., SCOTT-BURDEN T., VANHOUTTE P.M. Potentiation of endothelium-dependent relaxations to bradykinin by angiotensin I converting enzyme inhibitors in canine coronary artery involves both endothelium-derived relaxing and hyperpolarizing factors. Circ. Res. 1992;71:137–144. doi: 10.1161/01.res.71.1.137. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Kinins and endothelial control of vascular smooth muscle. Ann. Rev. Pharmacol. Toxicol. 1995;35:679–705. doi: 10.1146/annurev.pa.35.040195.003335. [DOI] [PubMed] [Google Scholar]
- NAFZ B., BERGER K., ROSLER C., PERSSON P.B. Kinins modulate the sodium-dependent autoregulation of renal medullary blood flow. Cardiovasc. Res. 1998;40:573–579. doi: 10.1016/s0008-6363(98)00194-1. [DOI] [PubMed] [Google Scholar]
- NARABA H., UENO A., KOSUGI Y., YOSHIMURA M., MURAKAMI M., KUDO I., OH-ISHI S. Agonist stimulation of B1 and B2 kinin receptors causes activation of the MAP kinase signaling pathway, resulting in the translocation of AP-1 in HEK 293 cells. FEBS Lett. 1998;435:96–100. doi: 10.1016/s0014-5793(98)01045-x. [DOI] [PubMed] [Google Scholar]
- NISHIMURA Y., USUI H., KURAHASHI K., SUZUKI A. Endothelium-dependent contraction induced by acetylcholine in isolated rat renal arteries. Eur. J. Pharmacol. 1995;275:217–221. doi: 10.1016/0014-2999(95)00023-e. [DOI] [PubMed] [Google Scholar]
- OHLMANN P., MARTINEZ M.C., SCHNEIDER F., STOCLET J.C., ANDRIANTSITOHAINA R. Characterization of endothelium-derived relaxing factors released by bradykinin in human resistance arteries. Br. J. Pharmacol. 1997;121:657–664. doi: 10.1038/sj.bjp.0701169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OTSU M., HILES I., GOUT I., FRY M.J., RUIZ-LARREA F., PANAYOTOU G., THOMPSON A., DHAND R., HSUAN J., TOTTY N., SMITH A.D., MORGAN S.J., COURTNEIDGE S.A., PARKER P.J. , WATERFIELD M.D. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- OZA N.B., SCHWARTZ J.H., GOUD H.D., LEVINSKY N.G. Rat aortic smooth muscle cells in culture express kallikrein, kininogen, and bradykininase activity. J. Clin. Invest. 1990;85:597–600. doi: 10.1172/JCI114479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAMLER R.M., FERRIGE A.G., MONCADA S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- PERSSON K., ANDERSSON R.G. Biphasic response to bradykinin in isolated porcine iliac arteries is mediated by bradykinin B1 and B2 receptors. J. Cardiovasc. Pharmacol. 1998;31:306–313. doi: 10.1097/00005344-199802000-00018. [DOI] [PubMed] [Google Scholar]
- PRUNEAU D., BELICHARD P. Induction of bradykinin B1 receptor-mediated relaxation in the isolated rabbit carotid artery. Eur. J. Pharmacol. 1993;239:63–67. doi: 10.1016/0014-2999(93)90976-o. [DOI] [PubMed] [Google Scholar]
- RABITO S.F., MINSHALL R.D., NAKAMURA F., WANG L.X. Bradykinin B2 receptors on skeletal muscle are coupled to inositol 1,4,5-trisphosphate formation. Diabetes. 1996;45:S29–S33. doi: 10.2337/diab.45.1.s29. [DOI] [PubMed] [Google Scholar]
- REGOLI D., BARABE J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- REGOLI D., BARABE J., PARK W.K. Receptors for bradykinin in rabbit aortae. Can. J. Physiol. Pharmacol. 1977;55:855–867. doi: 10.1139/y77-115. [DOI] [PubMed] [Google Scholar]
- REGOLI D., PHENG L.H., ALLOGHO S.N., NGUYEN-LE X.K., GOBEIL F. Receptors for kinins: from classical pharmacology to molecular biology. Immunopharmacology. 1996;33:24–31. doi: 10.1016/0162-3109(96)00077-x. [DOI] [PubMed] [Google Scholar]
- RHALEB N.E., DION S., BARABE J., ROUISSI N., JUKIC D., DRAPEAU G., REGOLI D. Receptors for kinins in isolated arterial vessels of dogs. Eur. J. Pharmacol. 1989;162:419–427. doi: 10.1016/0014-2999(89)90332-4. [DOI] [PubMed] [Google Scholar]
- SARDI S.P., PEREZ H., ANTUNEZ P., ROTHLIN R.P. Bradykinin B1 receptors in human umbilical vein. Eur. J. Pharmacol. 1997;321:33–38. doi: 10.1016/s0014-2999(96)00927-2. [DOI] [PubMed] [Google Scholar]
- SCHILLING W.P., CABELLO O.A., RAJAN L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca2+-influx pathway. Biochem. J. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMIDT H.H., POLLOCK J.S., NAKANE M., FORSTERMANN U., MURAD F. Ca2+/calmodulin-regulated nitric oxide synthases. Cell. Calcium. 1992;13:427–434. doi: 10.1016/0143-4160(92)90055-w. [DOI] [PubMed] [Google Scholar]
- SEIDLER N.W., JONA I., VEGH M., MARTONOSI A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- SHIRAHASE H., KANDA M., KURAHASHI K., NAKAMURA S., USUI H., SHIMIZU Y. Endothelium-dependent contraction in intrapulmonary arteries: mediation by endothelial NK1 receptors and TXA2. Br. J. Pharmacol. 1995;115:1215–1220. doi: 10.1111/j.1476-5381.1995.tb15028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIRAHASE H., KANDA M., SHIMAJI H., USUI H., RORSTAD O.P., KURAHASHI K. Somatostatin-induced contraction mediated by endothelial TXA2 production in canine cerebral arteries. Life Sci. 1993;53:1539–1544. doi: 10.1016/0024-3205(93)90562-h. [DOI] [PubMed] [Google Scholar]
- SHIRAHASE H., USUI H., MANABE K., KURAHASHI K., FUJIWARA M. An endothelium-dependent contraction induced by A-23187, a Ca2+ ionophore in canine basilar artery. J. Pharmacol. Exp. Ther. 1988;247:701–705. [PubMed] [Google Scholar]
- THASTRUP O., CULLEN P.J., DROBAK B.K., HANLEY M.R., DAWSON A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USUI H., KURAHASHI K., SHIRAHASE H., JINO H., FUJIWARA M. Endothelium-dependent contraction produced by acetylcholine and relaxation produced by histamine in monkey basilar arteries. Life Sci. 1993;52:377–387. doi: 10.1016/0024-3205(93)90151-r. [DOI] [PubMed] [Google Scholar]
- VENEMA V.J., JU H., SUN J., EATON D.C., MARRERO M.B., VENEMA R.C. Bradykinin stimulates the tyrosine phosphorylation and bradykinin B2 receptor association of phospholipase C gamma 1 in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998;246:70–75. doi: 10.1006/bbrc.1998.8574. [DOI] [PubMed] [Google Scholar]
- ZHU P., BENY J.L., FLAMMER J., LUSCHER T.F., HAEFLIGER I.O. Relaxation by bradykinin in porcine ciliary artery. Role of nitric oxide and K+-channels. Invest. Ophthalmol. Vis. Sci. 1997;38:1761–1767. [PubMed] [Google Scholar]