

Abstract
The effect of prolonged exposure to nitric oxide on enzymes involved in cell metabolism was investigated in T lymphocyte-derived Jurkat and L929 fibroblast human cell lines using a constant concentration of nitric oxide (1.5 μM) released by the nitric oxide donor DETA-NO (0.5 mM).
Nitric oxide inhibited immediately the respiration of the cells acting reversibly at complex IV. With time, the inhibition became progressively persistent, i.e. not reversed by trapping of nitric oxide with oxyhaemoglobin, and was preceded by a decrease in the concentration of the intracellular reduced glutathione. This persistent effect of nitric oxide on respiration was due to inhibition of complex I activity which could be reversed by addition of reduced glutathione or by cold light, suggesting that it was due to S-nitrosylation of thiols necessary for the activity of the enzyme.
The activity of other enzymes also known to be susceptible to inhibition by S-nitrosylation, i.e. glyceraldehyde-3-phosphate dehydrogenase and glutathione reductase, was progressively decreased by exposure to nitric oxide with a similar time course to that observed for the inhibition of complex I. Furthermore, inhibition of these enzymes only occurred when the concentrations of reduced glutathione had previously fallen and could be prevented by increasing the intracellular concentrations of reduced glutathione.
Our results suggest that S-nitrosylation of different enzymes by nitric oxide may occur only if the reducing potential of the cells is impaired.
Keywords: Nitric oxide, S-nitrosylation, glutathione, oxidative stress, oxygen consumption, complex I, glyceraldehyde-3-phosphate dehydrogenase, glutathione reductase
Introduction
The interaction between nitric oxide (NO) and complex IV in the respiratory chain constitutes a physiological mechanism by which cells regulate their respiration (Clementi et al., 1999; Loke et al., 1999). Moreover, when non-activated J774 murine macrophages are exposed to a constant concentration of NO for prolonged periods, NO induces a progressively persistent inhibition of respiration. This inhibition occurs at the level of complex I and is preceded by a decrease in the concentration of intracellular glutathione (GSH). The inhibition can be reversed by addition of GSH or by cold light, suggesting that it is due to S-nitrosylation of thiols necessary for the activity of the enzyme (Clementi et al., 1998).
S-nitrosylation, which has been reported to affect many cellular components including creatine kinase, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), p21ras as well as the NMDA and the ryanodine receptor (Lipton et al., 1993; Padgett & Whorton, 1995; Lander et al., 1996b; Wolosker et al., 1996; Xu et al., 1998; Yun et al., 1998), results in changes in their activity. These modifications affect crucial biochemical pathways in the cells, such as glycolysis, growth and Ca2+ homeostasis. Furthermore, S-nitrosylation of thiols has been proposed to be a mechanism for signal transduction in cells (Stamler, 1994). In view of this it is important to understand the biological conditions under which S-nitrosylation occurs since this will provide clues as to whether this is a physiological step or may already be a response to stress.
In order to investigate this question we have used human T lymphocyte-derived Jurkat and human fibroblast L929 cells exposed to the slow-releasing NO donor (z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino] diazen-1-ium-1,2 diolate (DETA-NO). We have analysed the function of the respiratory chain, specifically complex I, in these conditions. In addition, we have directly studied the activity of two other enzymes which are known to undergo S-nitrosylation, namely GAPDH (Padgett & Whorton, 1995) and glutathione reductase (GR) (Butzer et al., 1999). Our results show that NO produces a stepwise inhibition of the respiratory chain in both cell lines in a manner similar to that described in the macrophage line J774 (Clementi et al., 1998). Furthermore, direct analysis of complex I, GAPDH and GR showed that the activity of these enzymes is inhibited by NO but only when the reducing potential of the cell is affected.
Methods
Cell culture and preparation
Human T lymphocyte-derived Jurkat cells and human fibroblast L929 cells were grown in suspension using a MCS biological stirrer (Techne, Cambridge, U.K.) in RPMI medium supplemented with foetal bovine serum (FBS; 10% v v−1), L-glutamine (4 mM), penicillin (100 U ml−1), streptomycin (100 μg ml−1) and gentamicin (5 μg ml−1), at 37°C in a humidified atmosphere containing 5% CO2. Only cells in the logarithmic growth phase and with >95% viability (as judged by the trypan blue exclusion test) were used for the experiments. At the time of the experiments the cells were centrifuged, washed and resuspended at a density of 107 cells ml−1 in an incubation medium consisting of (in mM): NaCl 118, KCl 4.8, KH2PO4 1.2, MgSO4 1.2, CaCl2 1, glucose 20, HEPES 25, pH 7.2.
Measurement of oxygen consumption and NO generation
Cells suspended in the incubation medium (pH 7.2, 107 cells ml−1) were incubated at 37°C in the presence or absence of DETA-NO (0.5 and 5 mM) which was dissolved in the incubation medium 20 min before the addition to the cells. Samples (0.7 ml) were analysed at the time-points indicated in the various experiments in a gas-tight vessel maintained at 37°C, equipped with both a Clark type oxygen electrode (Rank Brothers, Bottisham, U.K.) and an NO electrode (Iso-NO, World Precision Instruments, Stevenage, U.K.) connected to a chart recorder. NO release and cellular oxygen consumption were thus measured simultaneously as described (Brown & Cooper, 1994). The oxygen electrode was calibrated with air-saturated medium kept at 37°C, assuming an oxygen concentration of 200 μM; the NO electrode was calibrated by addition of known concentrations of NaNO2 under reducing conditions (KI/H2SO4). In order to investigate the reversibility of NO-induced inhibition of respiration, oxyhaemoglobin (Hb; 8 μM) was added to the cells to remove the NO instantaneously. We define persistent inhibition of respiration as the proportion of this inhibition which is not immediately (<30 s) reversible by the addition of Hb. Data on oxygen consumption by DETA-NO-treated cells are expressed as percentage of that measured in cells subjected to the same treatment in the absence of the NO donor (controls).
In order to assess where the inhibition of respiration occurs in our experimental conditions, the different metabolic steps related to the oxidative phosphorylation activity were dissected pharmacologically, using specific inhibitors and appropriate substrates added to the cells immediately before addition of the NO donor. The addition of the protonophore carbonyl cyanide p-(trifluoro-methoxy) phenylhydrazone (FCCP; 1 μM) to control cells uncouples the flow of electrons from the synthesis of ATP and generates an increase in oxygen consumption. The addition of FCCP was used in some experiments to determine whether NO is acting at a step downstream to the ATP synthase. To study if the impairment of the citric acid cycle was involved in our results, experiments were performed with 3-nitropropionic acid (0.6 mM), an inhibitor of succinate dehydrogenase, in the presence of β-hydroxybutyrate (6 mM), which supplies mitochondria with NADH via its conversion to acetoacetate. In another group of experiments the oxygen consumption sustained by the activity of complex IV was tested in the presence of the complex III inhibitor myxothiazol (0.5 μM) and the supply of electrons to complex IV was maintained using N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD; 80 μM) plus ascorbic acid (4 mM). Finally, the oxygen consumption via complex II was tested using the respiratory substrate succinate (6 mM) in the presence of the complex I inhibitor rotenone (2 μM).
The concentrations of the various compounds mentioned above were selected in preliminary experiments designed as follows: oxygen consumption in untreated cell samples was followed for 4 min, and the various blockers (3nitropropionic acid, myxothiazol, rotenone) added at increasing concentrations to select the minimally effective concentration required to reduce oxygen consumption to less than 10% of control. The corresponding respiratory substrates (β-hydroxybutyrate, TMPD plus ascorbate, succinate) were then added at increasing concentrations to select that which restored respiration to at least 90% of the level of untreated controls. The maintenance of the respiration rate thus obtained was then followed over a 6 h period and compared with that of untreated controls.
In some experiments, cells were incubated with DETA-NO together with GSH-ethyl ester (2 mM). Finally, in some experimental groups, after the cells had been incubated for 5 h with DETA-NO and a persistent inhibition of respiration had been established, cells were divided into two groups. One group received GSH-ethyl ester (2 mM) and the other group was subjected to illumination with high intensity (approx. 8 Mlx) light from a cold light source (KL1500, Schott, Mainz, Germany), for a further incubation period of 1.5 h in both cases.
Measurement of reactive oxygen intermediates
The release of reactive oxygen intermediates from Jurkat and L929 cells was measured using luminol-enhanced chemiluminescence. Measurements were always performed in parallel with the measurement of oxygen consumption. Samples of the cell suspension, (100 μl, 106 cells), with or without DETA-NO, were taken at the indicated time-points and diluted 1 : 10 with incubation medium. Luminol was then added at a final concentration of 0.2 mM and cells analysed in a Wallac-LKB 1250 luminometer equipped with a thermostatically-controlled cuvette at 37°C. Photon counts generated by the chemiluminescent reaction with luminol were documented with a strip chart recorder.
Assay of complex I activity
The activity of complex I was assayed as described (Ragan et al., 1987). Briefly, the cells were centrifuged at 2500×g for 2 min and the pellet was freeze–thawed three times. Cellular homogenate (20 μl, 0.3 mg) was added to 1 ml of 10 mM potassium phosphate buffer, pH 8.0 containing 100 μM NADH in a 1 ml cuvette at 37°C. The rate of NADH oxidation was followed at 340 nm in a UV spectrophotometer. After recording the absorbance for 1 min, 5 μl of 10 mM ubiquinone-1 was added and the stimulated rate of NADH oxidation, which was taken as complex I activity, was followed for further 2 min. The NADH oxidation rate was calculated from the slope of absorbance decrease over time using an extinction coefficient for NADH of 6.81 mM−1 cm−1 at 340 nm.
Assay of GAPDH activity
The activity of GAPDH was determined by following the reduction of NAD+ to NADH at 340 nm. The assay mixture, in a total volume of 1 ml, contained 10 mM sodium pyrophosphate (pH 8.5), 25 mM sodium arsenate, 0.25 mM NAD+, and 1 mM glyceraldehyde-3-phosphate. At the indicated point-times, an aliquot of cells (0.7 ml) was centrifuged at 2500×g for 2 min and the pellet was freeze–thawed three times. Then, 4 μl of the cellular homogenate was added to the assay mixture to start the reaction and the activity was followed spectrophotometrically (340 nm, 25°C).
Assay of GR activity
The activity of GR was determined by following the oxidation of NADPH to NADP+ at 340 nm. The assay mixture, in a total volume of 1 ml, contained 100 mM potassium chloride, 50 mM potassium phosphate, 1 mM EDTA (pH 6.9), 0.1 mM NADPH and 1 mM GSSG. At the indicated point-times, an aliquot of cells (0.7 ml) was centrifuged at 2500×g for 2 min and the pellet was freeze–thawed three times. Then, 4 μl of the cellular homogenate was added to the assay mixture to start the reaction and the activity was followed spectrophotometrically (340 nm, 25°C). The activity of the enzyme was calculated using a molar extinction coefficient of 6.81×103 M cm−1.
Measurement of GSH and proteins
The concentrations of GSH in both cell lines were studied during the time course experiments. At the time indicated, cells were centrifuged at 2500×g for 2 min, the pellet was resuspended in cold metaphosphoric acid solution and the GSH content analysed using the Bioxytech GSH-400 kit. In some experiments, the concentration of intracellular GSH was also measured by HPLC as described by Ramachandran et al. (1999).
The protein content in the samples of all the assays was measured using a bicinchoninic acid based procedure (BCA assay, Pierce, Rockford, IL, U.S.A.).
Materials and drugs
Culture media and FBS were from GIBCO, Paisley, U.K., DETA-NO, a compound known to release NO with a half-life of 20 h at pH 7.4 (Hausladen et al., 1996), was purchased from Alexis Co., Nottingham, U.K. Measurements of GSH were performed using the Bioxytech GSH-400 kit from Oxis Int., Portland, Oregon, U.S.A. Hb was prepared as previously described (Feelisch et al., 1996). Myxothiazol, rotenone, FCCP, TMPD, β-hydroxybutyrate, 3nitropropionic acid, NAD+, NADH, NADPH, glyceraldehyde-3-phosphate (GAP), oxidized glutathione (GSSG), GSH-ethyl ester, ubiquinone 1, luminol and all other reagents were from Sigma-Aldrich, Poole, U.K.
Statistical analysis
The results are expressed as means±standard error of the mean; n represents the number of individual experiments. Statistical analysis was performed by Student's t-test for unpaired variables (two-tailed). Values are considered significant when P<0.05. *Significantly different from control cells. +Significantly different from DETA-NOtreated cells.
Results
DETA-NO dissolved in the incubation medium resulted in an immediate and gradual release of NO, as detected by the NO electrode, which increased constantly for about 20 min and thereafter reached a plateau which was maintained for >7 h. The maximal concentration of NO generated at the concentration of 0.5 mM DETA-NO (used in all the experiments unless otherwise indicated) was 1.5±0.3 μM (n=4) which remained constant for the duration of the experiment.
The addition of DETA-NO to the cells generated an immediate inhibition of respiration (76±1% in Jurkat and 87±1% in L929 cells, n=6). Initially (up to 1 h) this inhibition was fully reversible by addition of Hb but this ability of Hb to reverse the NO effect decreased progressively with time until it became completely ineffective after 5 h of incubation with DETA-NO, in both cell lines (Figure 1). Persistent inhibition of respiration by DETA-NO was due neither to an action of the corresponding amine, DETA, nor to the stable decomposition product of NO, nitrite, since if the compound was allowed to decompose fully under acidic conditions, no effect was observed on the rate of oxygen consumption (not shown). Neither was the inhibition a consequence of a nonspecific toxic effect of NO, since the cells were viable at the end of the experiment (5 h), as assessed by trypan blue exclusion. Viability was 82±5% in Jurkat and 71±3.1% in L929 cells under control conditions and 85±4.2% in Jurkat and 70±4.7% in L929 cells exposed to DETA-NO (n=10, P>0.05).
Figure 1.
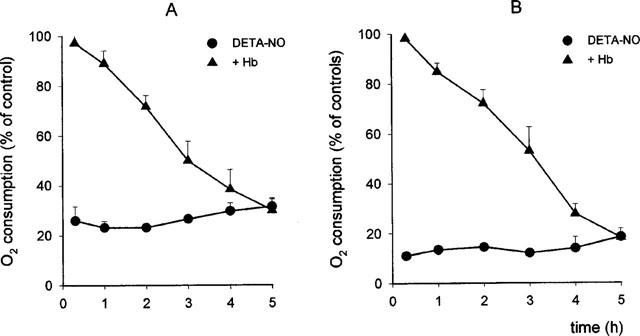
Effects of exposure to DETA-NO on cell respiration in Jurkat (A, n=8) and L929 cells (B, n=5). Cells were incubated with 0.5 mM DETA-NO and oxygen consumption was measured in samples taken at the indicated time points, before and after addition of Hb (8 μM). Oxygen consumption in DETA-NO-treated cells is reported as percentage of that observed in samples treated in the same way but without DETA-NO (controls).
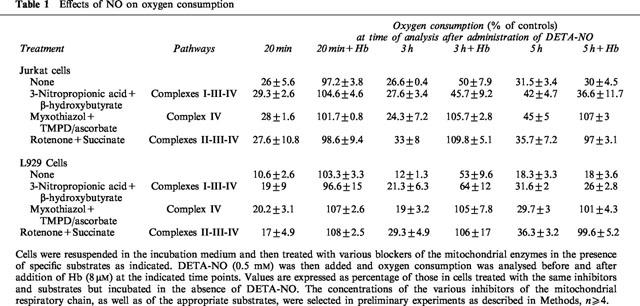
Incubation with DETA-NO did not induce any detectable generation of reactive oxygen intermediates up to 5 h, as assessed by luminol-enhanced chemiluminescence (n=3 in Jurkat and n=2 in L929 cells; not shown). When control cells from both cell lines were exposed to the protonophore FCCP (1 μM; 2 min), which uncouples the flow of electrons from the synthesis of ATP, they increased their oxygen consumption by 39.3±9.8% in Jurkat and 34.6±3.8% in L929 cells (n=3). By contrast, in cells exposed to DETA-NO for 5 h FCCP did not have any effect, indicating that the target of the persistent action of NO was located upstream of ATP synthesis. As summarized in Table 1, when cells were respiring on β-hydroxybutyrate in the presence of citric acid cycle blockade by 3-nitropropionic acid (0.6 mM), no changes in the onset of persistent inhibition by DETA-NO were observed, indicating that inhibition of the citric acid cycle did not play a major role in the inhibition of oxygen consumption observed. Oxygen consumption carried out by the activity of complex IV (in the presence of the complex III inhibitor myxothiazol and TMPD/ascorbate as respiratory substrate) was inhibited by DETA-NO, but this inhibition remained always reversible. Respiration occurring via complex II (in the presence of complex I inhibitor rotenone and succinate as respiratory substrate) was likewise always fully reversible by Hb. All these results suggest that the persistent inhibition of oxygen consumption is confined to complex I activity in both Jurkat and L929 cells.
Table 1.
Effects of NO on oxygen consumption
Complex I activity was then measured directly in homogenates from cells treated with DETA-NO for 20 min, at a point when the inhibition of oxygen consumption was still fully reversible, and then following 5 h of exposure, when the inhibition had become persistent. After 20 min incubation with DETA-NO no significant differences were found between the activities of complex I in control cells and in cells exposed to the NO donor; however, after 5 h of incubation, the activity of complex I was significantly reduced in relation to the respective controls (Figure 2A,B). Figure 2 also shows that rotenone (2 mM) inhibited the activity of complex I already at 20 min and that the extent of inhibition by NO and rotenone was similar after 5 h.
Figure 2.
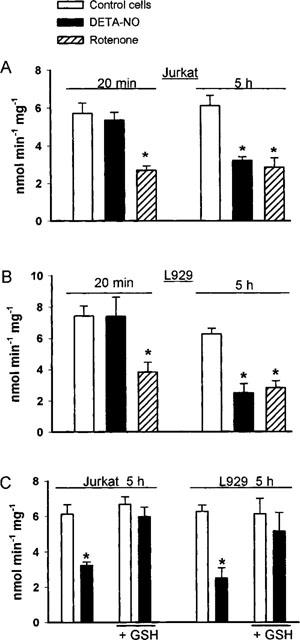
Activity of complex I in cellular extract after cell exposure to DETA-NO, rotenone and GSH-ethyl ester. Jurkat (A) and L929 (B) cells were incubated with or without DETA-NO (0.5 mM) for either 20 min or 5 h, at which times cells were collected and the pellet used to measure complex I activity (n=5). At every time-point studied there was a group of control cells treated with rotenone (2 mM). (C) cells treated for 5 h as in (A) and (B) but in the presence or absence of GSH-ethyl ester (2 mM, n=4).
To investigate the nature of inhibition of complex I by NO, cells exhibiting a persistent inhibition of respiration were either treated with the membrane permeable GSH analogue GSH-ethyl ester (2 mM) or exposed to cold light (8 Mlx), in both cases for a period of 1.5 h. Both treatments restored the reversibility of the persistent inhibition of respiration by Hb. The addition of GSH-ethyl ester restored oxygen consumption to 63±7.5% in Jurkat (n=4) and 61±1.2% in L929 (n=3) cells with respect to control cells. Similarly the recovery after treatment with light was 72.5±17.5% in Jurkat (n=3) and 94.3±2.9% in L929 (n=3). When the incubation was carried out in the presence of GSH-ethyl ester (2 mM) the inhibition of oxygen consumption by DETA-NO was still reversible after 5 h of incubation (Table 2). Direct analysis of the activity of complex I under these conditions showed that the enzyme activity was not significantly inhibited after 5 h (Figure 2C). In experiments in which incubation with DETA-NO was carried out with a doubled concentration of cells in the medium (i.e. 20×106 cells ml−1) the onset of the persistent inhibition was also delayed (Table 2).
Table 2.
Effects of NO and GSH on the oxygen consumption

Exposure of the cells to DETA-NO resulted in the progressive reduction of the intracellular GSH concentration, which was 38±8.9% of the control values in Jurkat (n=7) and 38.5±10.2% of the control values in L929 cells (n=5) after 5 h of incubation with the NO donor (Figure 3A,B). When cells were incubated with ten times the concentration of DETA-NO (5 mM, n=5) the concentrations of intracellular GSH decreased to 45% of the control concentrations in 10 min of incubation (Figure 3A,B). In these experiments the persistent inhibition of respiration was observed after only 1–1.5 h of incubation.
Figure 3.
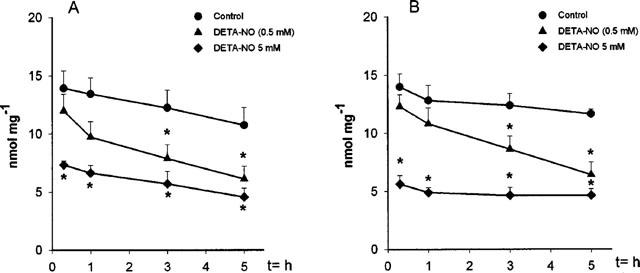
Depletion of intracellular GSH following exposure to DETA-NO of Jurkat (A; n=5) and L929 cells (B; n=7). Cells were incubated with or without DETA-NO (0.5 or 5 mM) for the indicated time points. Cells were centrifuged and the pellet was used to analyse the GSH content as described in the Methods section.
Among the enzymes involved in the metabolic pathways responsible for supplying NADH to complex I, an important role is played by GAPDH, the rate limiting enzyme for glycolysis. This enzyme has been reported to be S-nitrosylated by NO, with consequent inhibition of its activity (Padgett & Whorton, 1995). The activity of this enzyme was not affected after 1 h of incubation with DETA-NO. From then on the enzyme became progressively inhibited in both cell lines, with a time course similar to that observed for inhibition of complex I activity (Figure 4A,B). When a similar experiment was performed in the presence of GSH-ethyl ester (2 mM) the inhibition of the enzyme proceeded at a slower rate than in the cells treated with DETA-NO alone (Figure 5A). When cells exposed to DETA-NO for 5 h, in which inhibition of GAPDH activity was observed, were treated with cold light for 1.5 h the activity was restored to 79.5±0.02% of the control (Figure 5B).
Figure 4.
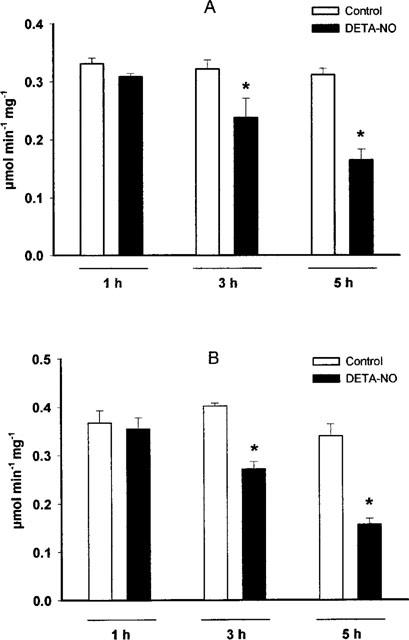
Effects of DETA-NO on the GAPDH activity of Jurkat (A; n=7) and L929 cells (B; n=4). Following continuous exposure in the presence or absence of DETA-NO (0.5 mM) for the indicated time points, cells were centrifuged and the pellet analysed for GAPDH activity as described in the Methods section.
Figure 5.
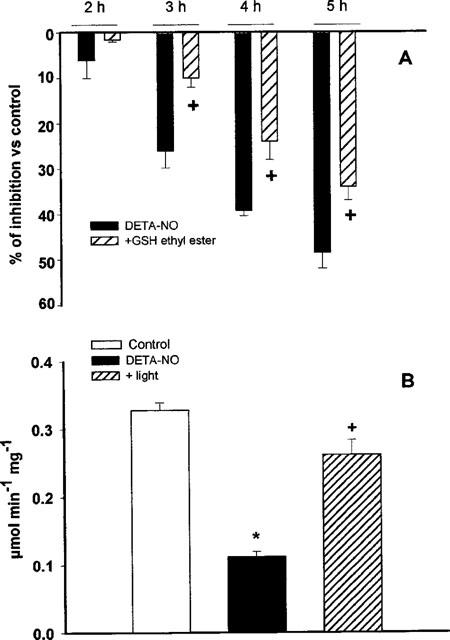
Effects of DETA-NO, GSH and light exposure on GAPDH activity. (A) Cells were treated with DETA-NO (0.5 mM) in the presence or absence of GSH-ethyl ester (2 mM). Cells were collected at the indicated time points and pellets were used for the analysis of the GAPDH activity. Data are expressed as percentages of the activity measured in cells incubated without DETA-NO and represent means±s.e.mean; n=5. (B) Cells were treated with or without DETA-NO for 5 h. An aliquot of the DETA-NOtreated cells was exposed to cold light (8 Mlx) for a further 1.5 h, n=4.
Glutathione reductase is an enzyme of the glutathione redox metabolism (Becker et al., 1995). It contains two thiols at the active site which, when nitrosylated, lead to inhibition of its activity (Butzer et al., 1999). In Jurkat cells incubated with DETA-NO, the activity of the enzyme did not change for up to 1 h of incubation but became progressively affected with time. Consistent with previous observations (Butzer et al., 1999) after 5 h of incubation with DETA an inhibition of 27±6% (Figure 6) was observed. As with the other enzymes, when the experiments were performed in the presence of 2 mM GSH-ethyl ester, inhibition was not observed even after 5 h of incubation.
Figure 6.
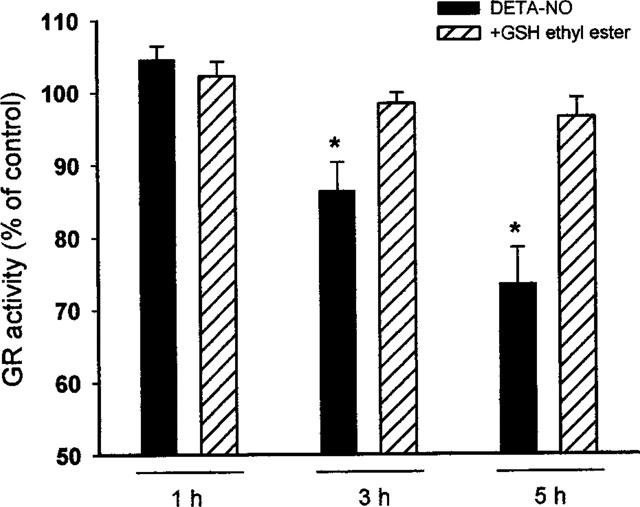
Effects of DETA-NO and GSH on GR activity. Cells were treated with or without DETA-NO (0.5 mM, n=6) in the presence or absence of GSH-ethyl ester (2 mM, n=4). Cells were collected at the indicated time points and pellets were used for the analysis of the GAPDH activity. Data are expressed as percentages of the activity measured in cells incubated without DETA-NO and represent means±s.e.mean. The activity detected for those control cells was 10.8±2.2 nmol min−1 mg−1.
Discussion
Our results show that prolonged exposure of two different human-cell lines (Jurkat and L929) to NO results in inhibition of cell respiration which becomes progressively persistent, with a time course similar to that described in J774 cells (Clementi et al., 1998). Experiments carried out using drugs that inhibit specific enzymes in the citric acid cycle or in the oxidative phosphorylation chain and appropriate substrates to maintain cellular respiration, showed that the persistent inhibition of oxygen consumption was due to inhibition of complex I activity and was preceded by a decrease in the content of GSH of the cells.
Once the persistent inhibition of respiration was established, the addition of GSH-ethyl ester or exposure to light restored the reversibility of the NO-dependent blockade by Hb in both cell lines. Doubling the concentration of the cells, or incubating the cells with GSH-ethyl ester prevented the development of the irreversible inhibition of oxygen consumption. The photolability of the chemical modification, together with its sensitivity to reducing agents, is consistent with the formation of a nitrosothiol (Bauer et al., 1995). These results show that the sequence and characteristics of the events in mitochondria which follow long-term exposure to NO are very similar in diverse cells.
Previous observations on the effects of NO on the mitochondria have been controversial and inhibition or ‘damage' of many enzymes has been reported (Brown & Cooper, 1994). Those results, however, were obtained using NO donors which are either very unstable, release large quantities of NO in short periods, or their breakdown into NO is accompanied by the release of other species which react with NO itself. Our results using DETA-NO at a concentration (0.5 mM) which releases NO in a constant manner (1.5 μM) over long periods (24 h) show that NO, in the mitochondrial respiratory chain, inhibits selectively complexes IV and I. Interestingly, exposure of J774 cells to even larger concentrations of NO (up to 7 μM) released from DETA-NO does not lead to inhibition of any other enzymes in the respiratory chain (not shown). Whether the action of NO on complex I is due to NO itself or to some related species is not yet clear. The mitochondria are a source of superoxide anion (O2−) which can rapidly react with NO to form peroxynitrite (ONOO−) (Beckman et al., 1990; Radi et al., 1991). In our experimental conditions there was not a generalized formation of ONOO− as assessed by the lack of any detectable production of reactive oxygen species during exposure to NO. However, unpublished results (J.J. Poderoso, personal communication) suggest that ONOO− is formed in complex I due to the generation of O2− in complex I itself (Radi & Cassina, 1996; Cadenas, 1997). The persistent impairment of complex I by S-nitrosylation could represent a turning point in the cell leading from the already well-known physiological inhibition of complex IV (Brown & Cooper, 1994) to a pathophysiological inhibition of complex I. Recent experiments indicate that this is indeed the case, since inhibition of complex I has toxic effects and leads to the triggering of an apoptotic programme (Seaton et al., 1998; Barrientos & Moraes, 1999; Loke et al., 1999).
Nitrosylation of thiols has been proposed to be an important signaling event in cells (Stamler, 1994). However, it is not clear whether S-nitrosylation is a physiological mechanism or only occurs in cells exposed to NO during oxidative stress. Therefore, we studied the relationship between the inhibition of two other enzymes known to be S-nitrosylated by NO, i.e. GAPDH and GR, and the redox status of the cell. None of the enzymes were affected by exposure to NO for up to 1 h and significant inhibition of their activity was only observed after 3–5 h. The concentration of GSH was reduced by 20–40% at 1 h depending on the cell type and decreased further with time. With higher concentrations of NO (7 μM), generated by exposure to 5 mM DETA-NO, the drop in GSH was already marked after 20 min and the onset of the inhibition of the enzyme activity was observed at 1–1.5 h. Decreases in GSH always preceded the effects of NO on enzyme activity but interestingly, in every case, there was a time lag between the decrease in GSH and inhibition of the enzyme. This suggests that when GSH decreases, other antioxidant mechanisms such as superoxide dismutase, or the presence of ascorbic acid which can react avidly with ONOO− (Van der Vliet et al., 1994), could act as a second line of antioxidant defense in the cells. Furthermore, induction of an antioxidant mechanism such as thioredoxin (Nakamura et al., 1997) may also occur. Administration of GSH to cells exposed for 5 h to NO restored the activity of the enzymes. When the incubation was carried out in the presence of GSH-ethyl ester (2 mM) the onset of enzyme inhibition was significantly delayed. Thus, inhibition of enzyme activity by S-nitrosylation in Jurkat and L929 cells required a change in the redox status of the cell, its onset being dependent on the degree of this change, and was always prevented by maintaining an antioxidant status.
S-nitrosylation has been proposed to modulate the activity of cell elements, such as the docking/fusion complex (VAMP/SNAP-25/syntaxin 1a) (Meffert et al., 1996), creatine kinase (Wolosker et al., 1996), GAPDH (Padgett & Whorton, 1995), the ryanodine receptor (Xu et al., 1997), and cysteine proteases of the caspase family (Li et al., 1997; Mohr et al., 1997). Those studies have been carried out either with isolated enzymes or with cell-free systems, in which the cellular redox milieu is absent. However, the addition of reducing agents did reverse the S-nitrosylation, indicating that S-nitrosylation indeed depends on the redox environment (Padgett & Whorton, 1995; Li et al., 1997; Mohr et al., 1997). When experiments were performed with whole cells, it appeared that the effect of NO was either independent of S-nitrosylation, as in the case of the ryanodine receptor (Clementi et al., 1996; Galione et al., 1997; Lee et al., 1997), or required high concentrations of NO for S-nitrosylation to occur (Padgett & Whorton, 1997). Interestingly, S-nitrosylation of caspases in the whole cell was demonstrated when apoptosis was triggered by tumour necrosis factor-α and CD95 (Mannick et al., 1994; 1997; Kim et al., 1997), both known to induce apoptosis after lowering of GSH concentrations in the cells (Cossarizza et al., 1995). In neurons, S-nitrosylation of the small G protein p21ras and the NMDA receptor have been observed with concentrations of NMDA which are excitotoxic (Lipton et al., 1993; Yun et al., 1998). Nevertheless, in the case of p21ras, S-nitrosylation was shown to occur in Jurkat cells under what seem to be physiological conditions, i.e. at nanomolar concentrations of NO (Lander et al., 1995a). However, further studies carried out by the same authors showed that depletion of GSH was required for the activation of p21ras (Lander et al., 1995b; 1996a).
In conclusion, whether S-nitrosylation occurs as a physiological regulatory process remains to be determined. Our results suggest that it takes place in the presence of oxidative stress. The progression or reversibility of this process, as well as its relevance in terms of cell survival or death, now require investigation.
Acknowledgments
We thank Dr Tim Bates for his help with the HPLC technique and Annie Higgs for her help in preparing the manuscript. This work was supported by grants from GlaxoWellcome (to S. Moncada), EEC (to A. Orsi), AIRC and MURST (to E. Clementi). Dr Belén Beltrán was supported by a grant from ‘Generalitat Valenciana', 1999, Spain.
Abbreviations
- DETA-NO
(z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino] diazen-1-ium-1,2 diolate
- FCCP
carbonyl cyanide p-(trifluoro-methoxy) phenylhydrazone
- FBS
foetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GR
glutathione reductase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- Hb
oxyhaemoglobin
- NO
nitric oxide
- ONOO−
peroxynitrite
- TMPD
N,N,N′,N′-tetramethyl-p-phenylenediamine
References
- BARRIENTOS A., MORAES C.T. Titrating the effects of mitochondrial complex impairment in the cell physiology. J. Biol. Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- BAUER J.A., BOOTH B.P., FUNG H.L. Nitric oxide donors: biochemical pharmacology and therapeutics. Adv. Pharmacol. 1995;34:362–382. doi: 10.1016/s1054-3589(08)61098-4. [DOI] [PubMed] [Google Scholar]
- BECKER K., GUI M., SCHIRMER H. Inhibition of human glutathione reductase by S-nitrosoglutathione. Eur. J. Biochem. 1995;234:472–478. doi: 10.1111/j.1432-1033.1995.472_b.x. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.M., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN G.C., COOPER C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- BUTZER U., WEIDENBACH H., GANSAUGE S., GANSAUGE F., BEGER H.G., NUSSLER A.K. Increased oxidative stress in the RAW 264.7 macrophage cell line is partially mediated via the S-nitrosothiol-induced inhibition of glutathione reductase. FEBS Lett. 1999;445:274–278. doi: 10.1016/s0014-5793(99)00139-8. [DOI] [PubMed] [Google Scholar]
- CADENAS E. Basic mechanism of antioxidant activity. Biofactors. 1997;6:391–397. doi: 10.1002/biof.5520060404. [DOI] [PubMed] [Google Scholar]
- CLEMENTI E., BROWN G.C., FEELISCH M., MONCADA S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLEMENTI E., BROWN G.C., FOXWELL N., MONCADA S. On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc. Natl. Acad. Sci. U.S.A. 1999;96:1559–1562. doi: 10.1073/pnas.96.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLEMENTI E., RICCIO M., SCIORATI C., NISTICO G., MELDOLESI J. The type 2 ryanodine receptor of neurosecretory PC12 cells is activated by cyclic ADP-ribose. Role of the nitric oxide/cGMP pathway. J. Biol. Chem. 1996;271:17739–17745. doi: 10.1074/jbc.271.30.17739. [DOI] [PubMed] [Google Scholar]
- COSSARIZZA A., FRANCESCHI C., MONTI D., SALVIOLO S., BELLESIA E., RIVABENE R., BIONDO L., RAINALDI T., TINARI A., MALORNI W. Protective effect of N-acetylcysteine in tumor necrosis factor alpha-induced apoptosis in U937 cells: The role of mitochondria. Exp. Cell. Res. 1995;220:232–240. doi: 10.1006/excr.1995.1311. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., KUBITZEK D., WERRINGLOER J. Methods in nitric oxide research 1996John Wiley & Sons, Chichester; 455–478.In: Feelisch, M., Stamler, J.S. (eds) [Google Scholar]
- GALIONE A., JONES K.T., LAI F.A., SWANN S. A cytosolic sperm protein factor mobilizes Ca2+ from intracellular stores by activating multiple Ca2+ release mechanism independently of low molecular weight messengers. J. Biol. Chem. 1997;272:28901–28905. doi: 10.1074/jbc.272.46.28901. [DOI] [PubMed] [Google Scholar]
- HAUSLADEN A., PRIVALLE C.T., KENG T., DEANGELO J., STAMLER J. Nitrosative stress: Activation of the transcription factor OxyR. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- KIM Y.M., TALANIAN R.V., BILLIAR T.R. Nitric oxide inhibits apoptosis by preventing increases in caspase-3 like activity via two distinct mechanisms. J. Biol. Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- LANDER H.M., JACOVINA A.T., DAVIS R.J., TAURAS J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biochem. Chem. 1996a;271:19705–19709. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- LANDER H.M., MILLBANK A.J., TAURAS J.M., HAJJAR D.R., HEMPSTEAD B.L., SCHWARTZ G.D., KRAEMER R.T., MIRZA V.A., CHAIT B.T., BURK S.C., QUILLAM L.A. Redox regulation of cell signalling. Nature. 1996b;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- LANDER H.M., OGISTE J.S., FRIEDA S., PEARCE A., LEVI R., NOVOGRODSKY A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J. Biol. Chem. 1995a;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- LANDER H.M., OGISTE J.S., TENG K.K., NOVOGRODSKY A. p21ras as a common signalling target of reactive free radicals in cellular redox stress. J. Biol. Chem. 1995b;270:21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- LEE M.R., LI L., KITAZAWA T. Cyclic GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J. Biol. Chem. 1997;272:5063–5068. doi: 10.1074/jbc.272.8.5063. [DOI] [PubMed] [Google Scholar]
- LI J., BILLIAR T.R., TALANIAN R.V., KIM Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;240:387–391. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- LIPTON S.A., CHOI Y.B., PAN Z.H., LEI S.A., CHEN H.S., SUCHER N.J, , LOSCALZO J., SINGEL D.J., STAMLER J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- LOKE K.E., MCCONNELL P.I., TUZMAN J.M., SHESELY E.G., SMITH C.J., STACKPOLE C.J., THOMSON C.I., KALEY G., WOLIN M.S., HINTZE T.H. Endogenous endothelial nitric oxide synthase-derived nitric oxide is a physiological regulator of myocardial oxygen consumption. Circ. Res. 1999;84:840–845. doi: 10.1161/01.res.84.7.840. [DOI] [PubMed] [Google Scholar]
- MANNICK J.B., ASANO K., IZUMI K., KIEFF E., STAMLER J.S. Nitric oxide produced by human lymphocytes inhibits apoptosis and Epstein Barr virus reactivation. Cell. 1994;392:274–276. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- MANNICK J.B., MIAO X.Q., STAMLER J.S. Nitric oxide inhibits Fas-induced apoptosis. J. Biol. Chem. 1997;272:24125–24128. doi: 10.1074/jbc.272.39.24125. [DOI] [PubMed] [Google Scholar]
- MEFFERT M.K, , CALAKOS N.C., SCHELLER R.H., SCHULMAN H. Nitric oxide modulates synaptic vesicle docking fusion reactions. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- MOHR S., ZECH B., LAPETINA E.G., BRUNE B. Inhibition of caspase-3 by S-nitrosylation and oxidation caused by nitric oxide. Biochem. Biophys. Res. Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- NAKAMURA H., NAKAMURA K., YODOI J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- PADGETT C.M., WHORTON R. S-nitrosoglutathione reversibly inhibits GAPDH by S-nitrosylation. Am. J. Physiol. 1995;269:C739–C749. doi: 10.1152/ajpcell.1995.269.3.C739. [DOI] [PubMed] [Google Scholar]
- PADGETT C.M., WHORTON R. Glutathione redox cycle regulates nitric oxide-mediated glyceraldehyde-3-phosphate dehydrogenase inhibition. Am. J. Physiol. 1997;272:C99–C108. doi: 10.1152/ajpcell.1997.272.1.C99. [DOI] [PubMed] [Google Scholar]
- RADI R., BECKMAN J.S., BUSH K.M., FREEMAN B.A. Peroxynitrite oxidation of sulfhydrils. J. Biol. Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- RADI R., CASSINA A. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch. Biochem. Biophys. 1996;2:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- RAGAN C.I., WILSON M.T., DARLEY-USMAR V.M., LOWE P.N. Mitochondria: A practical approach 1987IRL Press: Oxford; 79–112.In: Darley-Usmar, V.M., Rickwood, D., Wilson, M.T (eds) [Google Scholar]
- RAMACHANDRAN N., JACOB S., ZIELINSKI B., CURATOLA G., MAZZANTI L., MUTUS B. N-Dansyl-S-nitrosohomocysteine a fluorescent probe for intracellular thiols and S-nitrosothiols. Biochim. Biophys. Acta. 1999;1430:149–154. doi: 10.1016/s0167-4838(98)00286-6. [DOI] [PubMed] [Google Scholar]
- SEATON T.A., COOPER J.M., SCHAPIRA A.H. Cyclosporin reverses the apoptosis stimulated by complex I inhibitors. Brain Res. 1998;809:12–17. doi: 10.1016/s0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- STAMLER J.S. Redox signaling nitrosylation and related target interaction of NO. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- VAN DER VLIET A., SMITH D., O'NEILL C.A., KAUR H., DARLEY-USMAR V.M., CROSS C.E., HALLIWELL B. Interactions of peroxynitrite with human plasma and its constituents: oxidative damage and antioxidant depletion. Biochem. J. 1994;303:295–301. doi: 10.1042/bj3030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLOSKER H., PANIZZUTTI R., ENGELENDER S. Inhibition of creatine kinase by S-nitrosoglutathione. FEBS Lett. 1996;392:274–276. doi: 10.1016/0014-5793(96)00829-0. [DOI] [PubMed] [Google Scholar]
- XU J.P., MEISSNER G., STAMLER J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- YUN H.Y., GONZALEZ-ZULUETA M., DAWSON V.L., DAWSON T.M. Nitric oxide mediates N-methyl-D-aspartate receptor induced activation of p21ras. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]